
Abstract
Introduction
Hemoglobin (Hb) is the dominant oxygen transport protein in the mammalian circulatory system. It is released from the erythrocyte into the plasma following hemolysis as a result of physiological stress, such as exercise (40), or pathological stress, such as sickle-cell disease (24). Outside the protective environment of the erythrocyte, Hb can be toxic (1); hemolysis is therefore a causal factor in a wide range of pathological processes with heme chemistry being a major contributing factor (2). What is the mechanism for this toxicity? Two theories have been suggested. Free Hb can bind nitric oxide and hence interfere with blood pressure and flow directly (12) or it can react with peroxides and initiate free-radical-catalyzed lipid peroxidation (27) and vasoconstriction via isoprostane formation (25). Hb itself can be the initiator of oxidative stress via the autoxidation of oxyhemoglobin (oxyHb), releasing superoxide, which subsequently dismutates to form hydrogen peroxide. Once in the plasma, Hb rapidly forms a tight complex with the protein haptoglobin (Hp) (18), Hb dimers binding to Hp dimers. This prevents the Hb dimer from being filtered through the kidney, where it can undergo a range of deleterious reactions. Instead, the Hb:Hp complex is removed via binding to the CD163 receptor and subsequent degradation by heme oxygenase in the macrophage (39). The clearance time is potentially quite slow with the t 1/2 in the plasma—at least for the externally added complex—being over 12 h (14). Given this long half time and the potential toxicity of free Hb, the binding of Hp to Hb must significantly attenuate heme reactivity. What are these protective mechanisms likely to be? Although the Hp:Hb complex has unaltered nitric oxide reactivity (4), it has been shown to prevent lipid peroxidation in vitro (17) and vasoconstriction in vivo (6); this suggests that the removal of the oxidative reactivity may be key to preventing heme toxicity in the vasculature.
Innovation
Following erythrocyte lysis, hemoglobin's (Hb) ability to oxidize lipids and proteins is dramatically decreased when it binds to the plasma protein haptoglobin (Hp). The mechanism for this protection is unknown. This work addresses two long-standing unanswered questions. First, how does the tight binding of Hp prevent heme toxicity and second, where is the site of free radical reactivity on Hb? We demonstrate that the answers to these two questions are linked by the novel finding that Hp binding stabilizes both the ferryl iron and a specific free radical (βTyr145) on the Hb protein, in the process decreasing the oxidative ability of both species.
In peroxidases the ferric (Fe3+) form of the heme group reacts with the two-electron oxidant hydrogen peroxide to form the higher oxidation ferryl (Fe4+=O2−) form and an associated free radical. In some enzymes (e.g., catalase and horseradish peroxidase) this radical resides on the porphyrin itself. In others it migrates to an amino acid elsewhere in the protein, for example, prostaglandin H synthase (23) or cytochrome c peroxidase (42). In all cases the two strong oxidizing equivalents (ferryl and free radical) are used to catalyze further oxidative chemistry.
The situation is not so clear in the respiratory globins, Hb and myoglobin (Fig. 1). Met (Fe3+) Hb reacts with the two-electron oxidant hydrogen peroxide to form the higher oxidation ferryl (Fe4+=O2−) and, presumably, a cation radical on the porphyrin ring. The ferryl Hb species is readily detectable and quantifiable to one iron per peroxide equivalent added (30). However, although it is possible to capture two electrons by the addition of an external reductant, such as ascorbate (10), in the absence of such reductants, the fate of the second oxidizing equivalent is uncertain. In myoglobin, transient formation of an oxidized porphyrin cation radical has been detected (13) consistent with the compound I ferryl/porphyrin radical species seen in catalase and horseradish peroxidase. In both myoglobin (16) and Hb (26) protein free radicals are detectable following peroxide addition. However, unlike the case of cytochrome c peroxidase or prostaglandin H synthase, these radicals—despite over 50 years of study (16)—have never been detected at a population of more than 10% of the total protein even when using rapid kinetic methods. The difficulty in capturing the second oxidizing equivalent is presumably due to rapid and uncontrolled side reactions. As well as oxidizing nearby proteins and lipids, Hb also oxidizes its own amino acids in a self-destructive process (21).
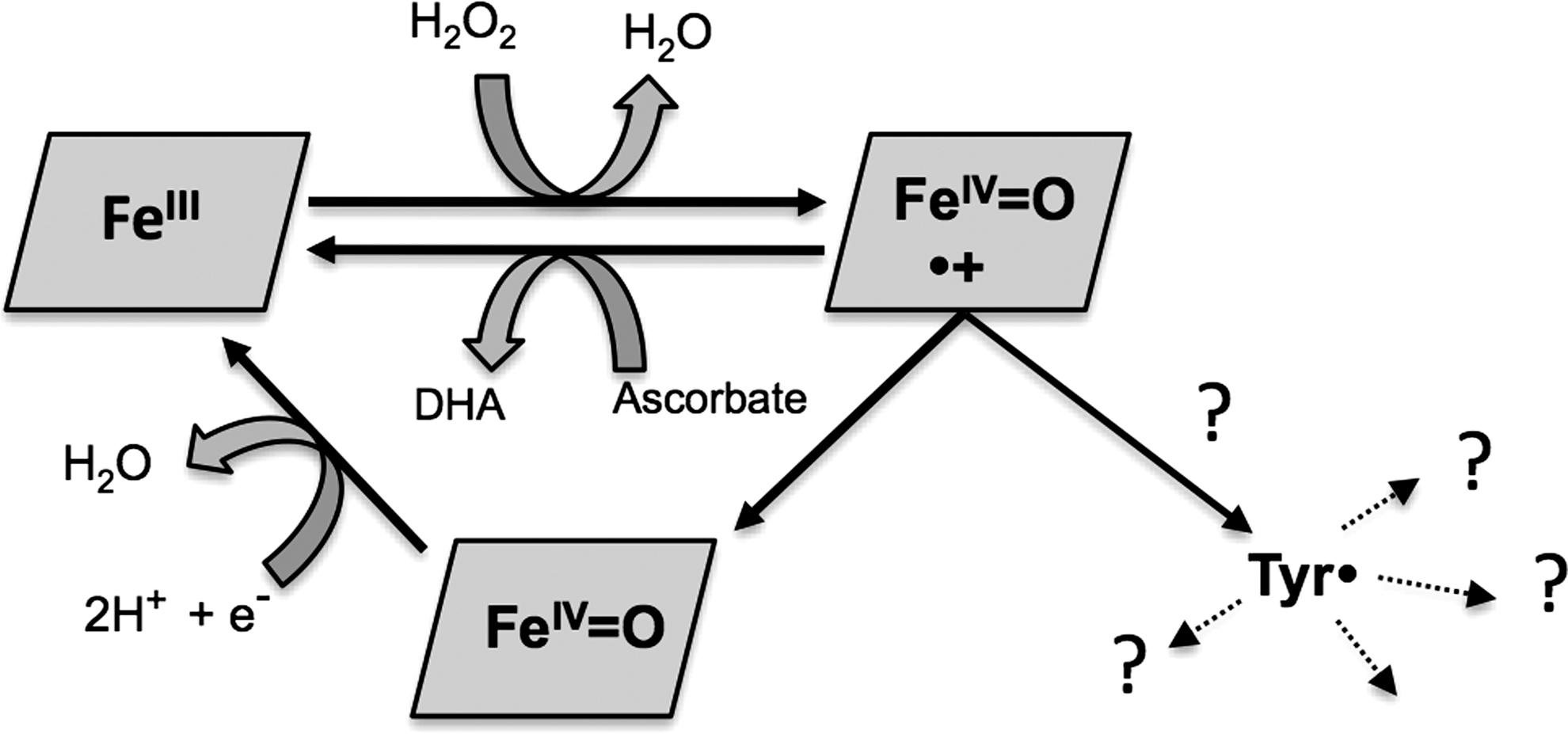
Like Hb, Hp is a dimer of dimers, the tetrameric protein comprising of two α-β dimers (33). Hp binds Hb with very high affinity (K D=10−12 M). There is so far no high resolution structure available of Hp or the Hb:Hp complex. Although several groups have tried to crystallize the complex, the inherent flexibility between the different Hp subunits and/or micro-heterogeneity in the glycosylation pattern of Hp has hindered crystallography (34). The known structural details are, therefore, mostly indirect and have been provided by biochemical studies of ligand interactions (18, 29), mass spectrometry (32), electron microscopy (46), and molecular modeling (33). These data predict that each Hp β subunit can bind one Hb αβ dimer. This complex is considered irreversible under physiologic conditions. Both Hb subunits participate in Hp binding, suggesting that a rather extensive area of the Hb dimer is covered by the corresponding Hp binding sites.
The evolutionary rationale for the presence of a distinct, but lowly populated, globin free radical in Hb is unclear; as is the molecular mechanism by which Hp binding can prevent heme-initiated toxicity. In this article we propose a novel mechanism that links these apparently distinct phenomena, involving stabilization of a tyrosine amino acid free radical in the Hb:Hp complex.
Results
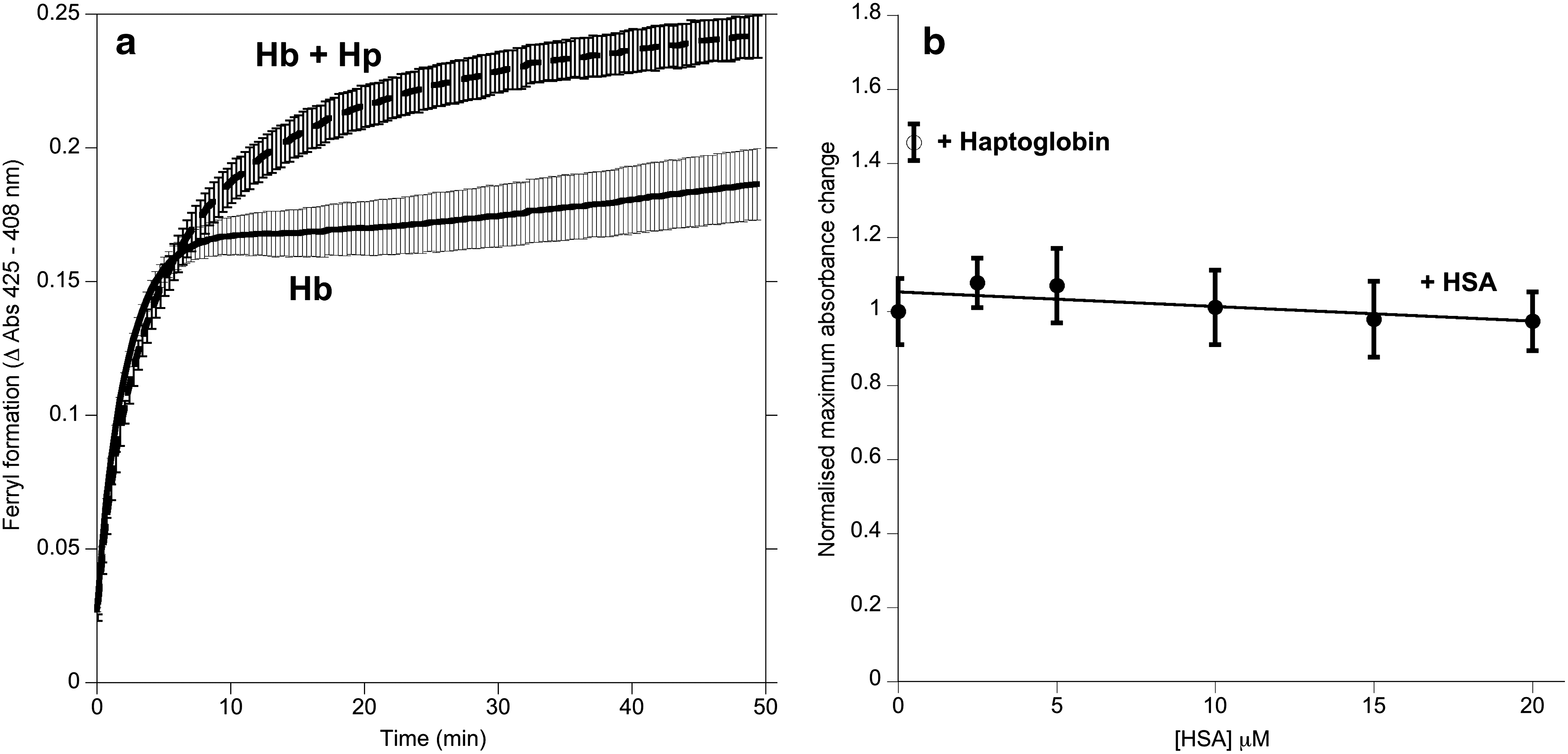
Ferryl Hb was used to initiate lipid oxidation in liposomes (Fig. 2). In the first 5 min, the presence of Hp stabilized the ferryl species compared with controls lacking Hp or containing equal concentrations of human serum albumin (HSA) (Fig. 2a). Over the subsequent 2 h, both controls showed significant lipid oxidation (conjugated diene formation detected in the UV) and heme degradation (bleaching of the Soret peak). Hp prevented these phenomena (Fig. 2b, c).

The direct addition of the lipid peroxide 13-S 9-cis, 11-trans octadecadienoic acid (HPODE) to ferric Hb induces redox cycling between the ferric and ferryl states. Hp binding caused a significant decrease in this HPODE consumption from 0.0088±0.0009 s−1 to 0.0031±0.0007 s−1 (mean±standard deviation, n=4). Analysis of the heme optical spectra revealed that this was accompanied not by a change in the initial rate of HPODE-catalyzed ferryl Hb formation, but by an increase in the final steady state concentration (Fig. 3a). No effects were seen (Fig. 3b) following the addition of a control protein (HSA).
Hp stabilization of the ferryl intermediate is most clearly seen at low pH where ferryl Hb becomes highly reactive. Preformed ferryl Hb was mixed with buffers of differing pH in a stopped flow experiment (Fig. 4). The protonation of the ferryl ion is rapid and occurs in the dead time of the mixing; the first spectrum observed therefore monitors the equilibrium associated with proton binding to the heme (41). The changes can be monitored at 580 nm. The spectral changes on the initial pH jump in the presence of Hp (Fig. 4a) were identical to those reported in its absence (41). The pK was also unchanged at 4.27 (Fig. 4b). However, the stability of the ferryl species formed differed significantly following the addition of Hp. In the control (Fig. 4c) there was a rapid removal of the ferryl (spectral changes between 500 and 600 nm) and heme damage (loss of heme spectra between 400 and 440 nm). These changes were much less noticeable in the presence of Hp (Fig. 4c, d). Even at pH 2, where in the absence of Hp, heme bleaching occurred in under a second; ferryl heme was stabilized in the presence of Hp. This rate of heme bleaching was independent of protein concentration in the range from 0.4 to 40 μM (results not shown).

The detailed interpretation of the spectral changes at low pH is complicated by events secondary to ferryl reactivity, such as protein unfolding. We therefore explored the stability of ferryl heme and its reactivity to external reductants at higher pH values where the dominant reaction is the conversion of ferryl to the ferric(met) protein. Measurements of the intrinsic decay of the Hb ferryl species (in the absence of added reductants) showed a >4-fold decrease following Hp binding. The effect was seen in both the α and β subunits (Fig. 5a) and was consistently observed at all pH values from pH 5 to pH 10. The pH dependence of this intrinsic ferryl autoreduction rate agreed with the ferryl protonation pK measured in Figure 4 (although it is not possible to measure a pK accurately given the limited pH range, forcing a curve fit using a pK of 4.27 was consistent with the spread of data points). Reactivity was also decreased to external reductants. Figure 5b shows the reduction of ferryl to ferric heme by phenol, a small reductant able to readily access the heme pocket. Hp caused a threefold decrease in the rate of reduction in both the α and β subunits.

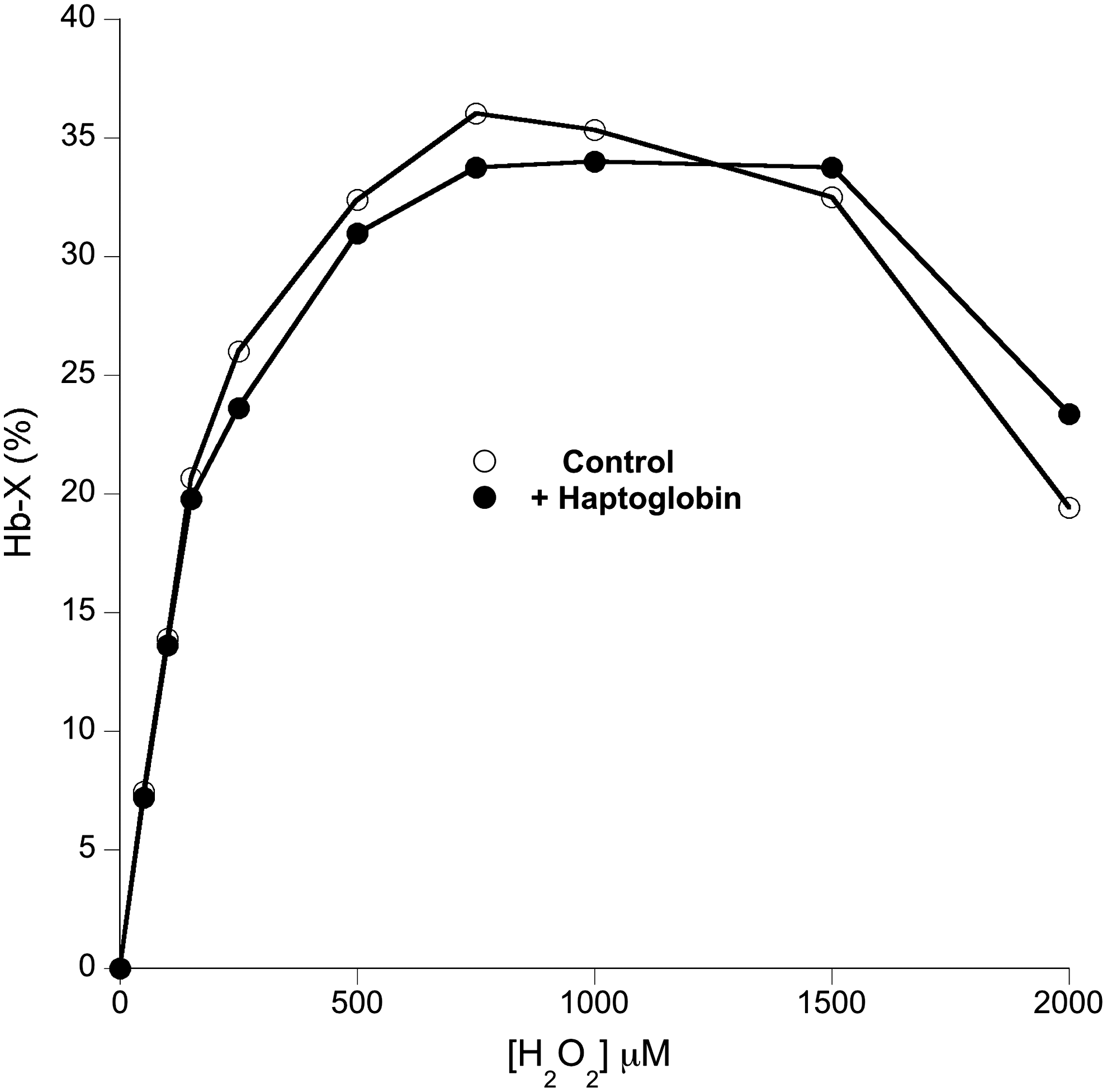
While the oxidative reactivity to exogenous reductants was affected by Hp binding, direct oxidative reactions within the heme pocket seemed unaffected. For example, Figure 6 shows that the extent of the formation of a heme:protein cross-link between the heme and the distal histidine residue (36) was unaffected by Hp binding.
Low-temperature electron paramagnetic resonance (EPR) spectroscopy was used to look at the amino acid free radicals in the protein formed following hydrogen peroxide addition to methemoglobin (metHb) in the absence and presence of Hp (Fig. 7). The signal at g=6 represents high-spin ferric heme, that is the fraction of metHb that has not yet been converted to the EPR undetectable ferryl state. The signal at g=2 represents the tyrosine free radical. There is a significant increase in the amount of this free radical detected in the presence of Hp. The similarity in the EPR spectra indicates that Hp is almost certainly stabilizing the same free radical that is detected in its absence. The extent of high-spin ferric heme loss and free radical formation can be quantitated as a function of time. Hydrogen peroxide converts the ferric heme quantitatively to ferryl heme; therefore, the ratio of the radical increase to the ferric heme decrease indicates the ratio of free radical: ferryl in the Hb molecule. At the earliest time points this ratio is not significantly different from one (Fig. 7, inset); Hp binding enables, for the first time, quantitative capture of the globin radical formed in the reaction of metHb and hydrogen peroxide.

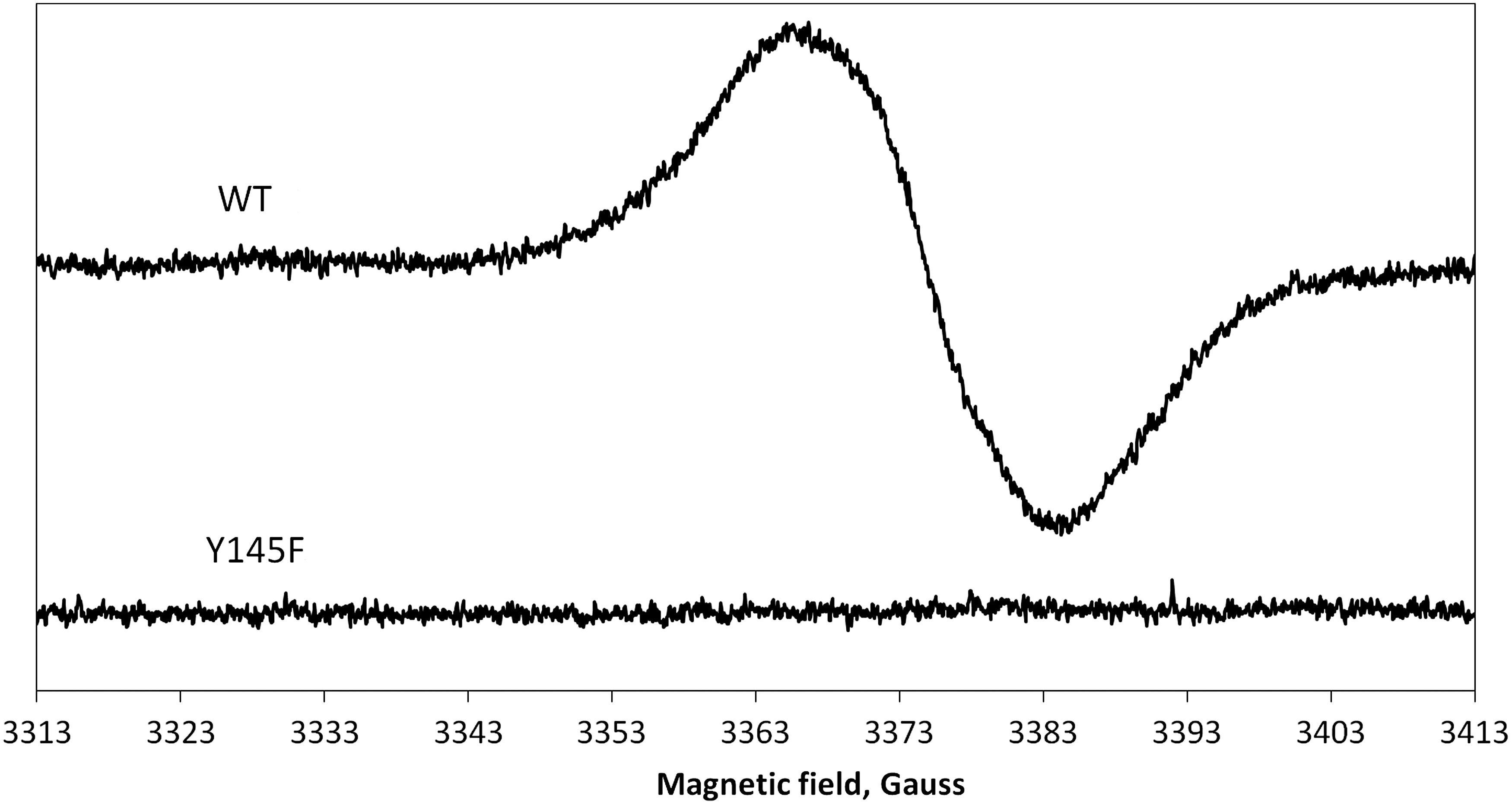
A simulation (Fig. 8) of the EPR spectrum of the tyrosyl free radical in the Hb:Hp complex was used to obtain information about the spin density on atom C1 at the possible rotation angles around the Cβ-C1 bond (44). There was a significant increase in the C1 spin density following Hp binding (from 0.375±0.003 to 0.41±0.003). The possible angles around the Cβ-C1 bond (47° and 73°) were consistent with a tyrosine radical residing on α42, β35, β130, or β145. Site-directed mutagenesis showed that the mutant βTyr145Phe has no detectable free radical following peroxide addition (Fig. 9), confirming βTyr145 as the site of stable radical formation in Hb.

Discussion
The strong oxidative nature of ferryl Hb, especially in its protonated state (41), suggests that compounds like Hp that decrease Hb's pro-oxidative chemistry might be expected to act on this ferryl intermediate. Miller et al. (27) explored this possibility looking at Hb reactivity with low-density lipoprotein (LDL). They showed that Hp increased the steady state concentration of ferryl Hb following peroxide addition; this was accompanied by decreased LDL oxidation. Our results with liposome oxidation confirm these findings.
Miller et al. went on to conclude that, as ferryl Hb is still formed during lipid peroxidation, the ferryl iron is not required for LDL oxidation. They attributed Hp's effects solely to globin radical chemistry rather than any direct effect on ferryl iron, suggesting that the ferryl iron itself cannot be primarily responsible for lipid peroxidation in LDL as ferryl Hb is not consumed during the lipid peroxidation process (27). This argument, however, ignores the fact that lipid peroxides themselves are able to re-convert ferric back to ferryl Hb. Ferryl can therefore enter a true steady state during lipid peroxidation, rather than merely being consumed. In this article we show a number of direct effects of Hp on the reactivity of ferryl iron. These include a decrease in the ability to oxidize lipid hydroperoxides such as HPODE and a decrease in the ability of internal and external reductants to re-reduce the ferryl iron to the ferric state. The latter occurs in ferryl Hb even at times when all the radicals have long-since decayed; Hp therefore makes ferryl itself less reactive, independent of the presence of protein free radicals. Contrary to Miller et al. (27) we therefore propose that Hp affects the oxidative reactivity of both the globin ferryl iron and the protein free radical oxidative activity.
What is the molecular mechanism for the increased stability of the ferryl state? By preferentially bonding strongly to the dimeric form, Hp induces the conversions of Hb from a tetramer into a dimer. This is the explanation for the decrease in the ferric/ferrous redox potential (7) and the decrease in the oxygen dissociation rate (9), with a consequent increase in oxygen affinity (38). However, in the absence of Hp, the autoreduction (35) and pH stability of ferryl Hb (this article) are unaffected by protein concentration changes that convert Hb from a predominantly tetrameric to dimeric state. Hp does not, therefore, prevent oxidative damage by merely converting Hb tetramers into dimers.
We have shown that lowering the pH enhances ferryl reactivity by favoring the formation of the more reactive protonated ferryl intermediate (41). An obvious mechanism for Hp to decrease ferryl reactivity at physiological or pathophysiological pH values is the modification of this pK. However, we show that this is not the case. Heme:protein cross-linking is a reaction catalyzed inside the heme pocket between the protonated ferryl and a nearby protein radical, probably the distal histidine (36). Consistent with the lack of effect on the ferryl pK, Hp does not alter the extent of this peroxide-induced cross-linking across a range of pH values. In line with previous work, the presence of Hp did not affect peroxide-induced ferryl iron formation (8, 27). The binding rates for other small heme ligands, such as NO (4) and O2 (9), are also unaltered in the complex.
Therefore, both the initial peroxide reaction and subsequent chemical reactivity within the heme pocket are largely unaffected by Hp binding. However, events that require electron transfer to/from the ferryl heme appear to be specifically perturbed by Hp binding. These include both autoreduction and external reduction of ferryl Hb. Although we cannot completely rule out large conformational changes that stearically hinder the ability of external lipids to access the oxidative reactivity of ferryl iron and free radicals, we feel that, taken in combination, this data suggest that Hp inhibits lipid peroxidation via a thermodynamic stabilization of the ferryl form of the protein.
What is the molecular mechanism for the increased stability of the protein free radical? The globin free radical was the first ever to be discovered on a protein. However, its precise chemical nature and location has been difficult to elucidate. An EPR-detectable Hb radical species was first discovered in the 1950s (15), but characterization of the species proved difficult because the radical is unstable and migrates, forming a range of differentially populated redox intermediates. Following peroxide addition there is a wide variety of candidate amino acid sites that can harbor free radicals with various degrees of stability (43). Mason and coworkers have (11) shown that it is possible to spin trap a radical at αTyr42 and that this site can be involved in electron transfer pathways. We have shown that αTyr42 can donate electrons to reduce ferryl Hb, presumably via a radical intermediate. This appears to facilitate the detoxification of ferryl by endogenous antioxidants, such as ascorbate (37).
However, mutagenesis studies rule out αTyr42 as the final destination of the endogenous nonspin trapped EPR-detectable tyrosyl radical (37). In this article we have looked at the effect of Hp on radical concentrations. We show that Hp acts as a physiological spin trap. Instead of trapping the radical itself it alters the structure of Hb to stabilize a radical on the βTyr145. The data are consistent with the full capture of the oxidizing equivalent, with a >85% yield of the free radical on the protein as opposed to the maximum 10%–15% from previous studies (43). Hb now behaves like cytochrome c peroxidase or prostaglandin H synthase, with quantitative capture of peroxide equivalents on ferryl iron and a protein free radical.
We have previously shown that oxidative modifications of amino acids on Hb are restricted to peptides containing αTyr42, βTyr145, and βCys93 (6, 31). Hp binding prevents this damage. Of the other three tyrosine residues on the β subunit that could harbor the radical, only βTyr145 is in a region that is oxidatively modified, providing further evidence of its importance.
How does the radical move to βTyr145 from the heme groups? The distance between the β heme and βTyr145 is 7.5 Å. In the tetramer a possible route from the α heme can be mapped via αTyr42 (5.3 Å) to βTyr145 (8.9 Å). In the dimer the αTyr42 (5.3 Å) to βTyr145 distance is very long (30Å), but an alternative route can be mapped from the heme via βTyr35. Therefore, although the three-dimensional structure of the Hb:Hp complex is not known, an analysis of the known protein structures suggests that, whether formed initially on the α subunit or the β subunit, the radical is likely to be able to migrate to βTyr145, via redox active tyrosine residues with no distance exceeding 19 Å; this enables efficient electron transfer on the ms time scale (28).
Tyr145 is the penultimate C-terminal amino acid in the β chain. In its normal (non-radical) state it makes a hydrogen bond to the carbonyl group in βVal98. The loss of this bond at βTyr145Phe seems to be compensated in part by other interactions involving the aromatic ring (20). βTyr145Phe is therefore a relatively normal Hb thermodynamically (20). How then can Hp binding stabilize the Tyr145 radical?
Protonated tyrosine radicals are highly reactive, rapidly capturing an electron from a neighboring residue(s) leading to radical migration away from the tyrosine. Our previous work (44, 45) demonstrated that spin densities above 0.38 indicate hydrogen bonding between the oxygen of a neutral tyrosine radical and a nearby hydrogen atom. Therefore, our data (spin density increasing from 0.375 to 0.41) demonstrate that Hp binding induces a new hydrogen bond (or possibly strengthens an existing one). We postulate that this could provide a route to deprotonate the reactive tyrosine radical safely (e.g., via a histidine relay), resulting in the higher concentrations of the stable neutral tyrosine radical that we detect.
The final molecular fate of the oxidizing equivalents is still unknown. However, it has recently been shown that, while Hp binding to Hb prevents oxidative damage to the globin (21), covalent cross-links can still be formed between the two proteins (22). This suggests the possibility that Hp not only stabilizes Hb ferryl iron and free radicals, but can act as a quasi-sacrificial electrode by becoming the final electron donor in the system.
In summary, a compelling picture now emerges about the molecular mechanism of Hp's protective function. Following Hp binding the heme active site has unaltered reactivity with small ferrous (oxygen and nitric oxide) and ferric (peroxide) ligands. However, while peroxides can still cause direct oxidative reactions at the heme, transfer of this oxidizing potential to neighboring molecules is prevented by the stabilization of both the ferryl iron and the free radical on βTyr145. This results in less-redox active oxidized species that do not initiate undesirable free radical chain reactions.
Materials and Methods
Hb was prepared from human volunteer blood using the method of Antonini and Brunori (3) with the addition of ion exchange chromatography on DEAE–Sephadex A50 (5) to remove contaminating catalase. Wild type and βTyr145Phe mutants of human Hb were constructed, expressed, and purified as described previously (37). Hb was stored as the CO-bound form at 77 K to prevent autoxidation. metHb was made by first converting the CO form to oxyHb by shining cold light (20 W energy-saving light bulb; GE Healthcare) on the sample during gentle agitation under a stream of oxygen gas. OxyHb was then oxidized to the ferric (met) form by the addition of a 1.5 excess (mole oxidant: mole heme) of potassium ferricyanide. The excess ferricyanide was then removed by filtration through a Sephadex G25 column. Ferryl Hb was made by the addition of H2O2 to metHb in a 2:1 ratio; after 2 min trace, catalase (10 nM) was then added to remove excess peroxide and the solution was used immediately. Equivalent ferryl formation was detectable for both the recombinant wild-type protein and the βTyr145Phe mutant.
Human Hp Phenotype 1-1 (H0138) was purchased from Sigma-Aldrich. It was either added directly to Hb in the experiment at the concentrations indicated or was used as a preformed purified complex, prepared as described previously (8). Liposomes (∼100 nm diameter) were made using an extruder (Northern Lipids) on 2 mg/ml L-alpha-phosphatidylcholine (soybean, Type II-S; Sigma-Aldrich). UV/Visible optical spectroscopy used either an HP diode array 8453 or a Cary 5E spectrophotometer. Ferryl Hb was incubated with liposomes by mixing it initially in the metHb form. A fourfold excess of hydrogen peroxide was then added and optical spectra were taken to confirm the conversion to the ferryl state (this took from 7 to 10 min). The excess peroxide was then removed by the addition of trace amounts of catalase (10 nM). The percentage of ferryl remaining at a specific time was calculated from the optical changes observed at 417–475 nm using an extinction of 119,600 M −1·cm−1.
Stopped-flow spectroscopy was carried out using an Applied Photophysics SX-20 instrument with a diode array attachment. For pH jump experiments, preformed ferryl Hb in 20 mM Triz. HCl was mixed with buffers of varying values (100 mM sodium acetate or 100 mM phosphoric acid/dihydrogen phosphate) to obtain the pH titration. All kinetic studies were carried out at 22°C. HPODE consumption was monitored by the addition of ferric Hb or ferric Hb:Hp complex (both 10 μM heme) to 40 μM HPODE and the reaction was monitored by loss of lipid conjugation at 234 nm. Conversely, liposome oxidation was measured by the increase in lipid conjugation at 234 nm. Ferryl Hb reduction kinetics were monitored at 406 and 425 nm and the time courses were fitted to a double exponential function, each exponential representing the kinetics of either α or β chain reduction. The separate rate constants were calculated as described previously (37). The formation of covalent heme:protein cross-links (Hb-X) was determined by reversed phase HPLC as described previously (36).
All EPR spectra were measured using a Bruker EMX EPR spectrometer (X-band) equipped with a spherical high-quality resonator SP9703 and an Oxford Instruments liquid helium system. Spectra were recorded at 10K. The instrumental settings (Fig. 7) were as follows: microwave frequency νMW=9.46 GHz; microwave power P MW=3.18 mW; modulation amplitude A M=5 G; modulation frequency νM=100 kHz; time constant τ=81.92 ms; and scan rate v=22.6 G/s. For Figures 8 and 9, P MW was reduced to 0.0504 mW and the spectra were corrected to remove the small contribution of the high-spin ferric heme signal at g=1.99. For other details see Reeder et al. (37). MetHb concentration was determined by comparison to the sample preperoxide addition. The free radical concentration was determined by double integration of the free radical EPR signal, after subtraction of the background signal recorded at the same instrumental conditions on a blank (frozen water) sample. A Cu2+ concentration standard (98 μM) was used, and both the standard and the sample EPR signals were recorded in the absence of microwave power saturation.
Note in all cases the concentrations of proteins are quoted on a per subunit basis, that is, 1 μM Hb is 1 μM heme or 0.25 μM tetramer. Hp concentration is similarly quantified such that 1 μM Hp would form a tight complex with 1 μM Hb (as the stable complex consists of an Hp dimer binding to an Hb dimer). When HSA was used as a control, it was added at 0.25 times the concentration of heme protein subunits so as to equate on a protein molecule per protein molecule basis (which also equates to a similar protein mass given their similar molecular weights).
Statistics
The use of the term significant in the text relates to comparison between experiments using unpaired t-tests (p<0.05). Errors on rate constants (Fig. 4) represent the standard error of the regression curve fit. Other statistical tests are as indicated in figure legends.
Footnotes
Acknowledgments
The authors thank Kristian Kallberg and Khuanpiroon Ratanasopa (Lund University) for assistance with the mutagenesis and purification of proteins. The authors would like to thank the UK Biotechnology and Biological Sciences Research Council (BBSRC BB/F007663/1 and BB/FOF/209) and the Swiss National Science Foundation (Grant 31-120658) for financial support. P.W.B. and A.I.A. acknowledge CBER's Critical Path Funding.
Author Disclosure Statement
C.E.C., B.J.R., and M.T.W. have a patent pending relating to modification of Hb amino acids designed to render a blood substitute less toxic. P.W.B. and A.I.A. are employees of the U.S. Food and Drug Administration and note that the findings and conclusions in this article have not been formally disseminated by the Food and Drug Administration and should not be construed to represent any agency determination or policy.