
Abstract
Introduction
NADPH Oxidases
Several potential sources of ROS are present in the cell, including mitochondrial respiratory chain enzymes, xanthine oxidases (XOs), lipoxygenases, myeloperoxidases, uncoupled nitric oxide synthases (NOSs), and Nox proteins (68). Noxs are multi-subunit transmembrane enzymes that utilize NADPH as an electron donor to reduce oxygen to superoxide anion (O2 −) and hydrogen peroxide (H2O2). They were initially discovered in phagocytes several decades ago, with the characterization of the Nox2 isoform (also referred to as gp91phox) (117). More recently, 6 other family members each encoded by separate genes have been identified, namely, Nox1, Nox3, Nox4, Nox5, dual oxidase 1 (Duox1), and Duox2 (Fig. 1) (5, 23, 65). These Nox proteins show 21%–59% identity to Nox2, with Nox3 being the most similar and Nox5 the most divergent. Nox5 contains EF-hand calcium-binding motifs not found in Nox1–4 and is absent in rodents. Duox1 and Duox2 show 53% and 47% homology, respectively, to Nox2 within their C-terminal region, but they also contain two N-terminal EF-hand calcium-binding motifs and a peroxidase-like domain (65).

Although Nox2 was discovered as a central regulator of microbicidal activity in neutrophils, recent evidence uncovers an important role for Nox2 and other Nox proteins in the cardiovascular system. Noxs regulate a variety of physiological processes, including endothelial cell (EC) migration, angiogenesis, vascular tone, smooth muscle growth, and inflammatory responses, where the tightly regulated generation of ROS is needed to mediate these functions (31). They have also been implicated in pathological processes, such as hypertension, atherosclerosis, restenosis, diabetic vascular disease, cardiac hypertrophy, fibrosis, and heart failure (23, 31, 183). Under these pathological stresses, the quantity of ROS generated by Noxs may increase several fold upon specific stimulation, in turn altering the redox status and the function of macromolecules within the cell.
Cellular distribution of Noxs in the heart
Nox1, Nox2, Nox4, and Nox5 are expressed in the cardiovascular system (with Nox5 only found in higher mammals). These isoforms are found in specific cell types within the heart, including cardiomyocytes, cardiac fibroblasts, vascular smooth muscle cells (VSMCs), ECs, and resident and infiltrating leukocytes.
Nox2 and Nox4 are the predominant isoforms expressed in cardiomyocytes and are involved in regulating several processes as discussed in later sections. The subcellular localization of the two isoforms in cardiomyocytes is distinct, as in other cell types. Activated Nox2 is predominantly expressed at the plasma membrane (88), whereas Nox4 is found intracellularly although the precise location remains controversial. It has been reported to be in a perinuclear location associated with the endoplasmic reticulum (ER) or mitochondria as well as intranuclear (4, 214). The role of Nox4 in cardiomyocytes is still not clear, as discussed later, and it has been suggested that it is only expressed at significant levels in response to cardiac stresses, such as hypoxia, myocardial ischemia, and pressure overload (113, 214).
Freshly cultured cardiac fibroblasts express low levels of Nox4, Nox2, and Nox1, but Nox2 is higher in the adventitial layer of human coronary arteries (184). The latter study showed that Nox5 and Nox4 mRNA are abundantly expressed in cultured human cardiac fibroblasts, whereas Nox1 and Nox2 are barely detectable (39). In general, adventitial fibroblast Nox activity is increased in various vascular diseases, including diabetes mellitus, hypertension, atherosclerosis, and vascular injury (38), but the in vivo contribution of Nox in cardiac fibroblasts to heart function and failure is still unclear.
The predominant Nox isoforms in ECs are also Nox2 and Nox4 (3). Activated Nox2 is again mainly found at the plasma membrane and at perinuclear membranes (126) while Nox4 is localized to the ER (9, 27, 32, 206) and nucleus (114, 156). In contrast, VSMCs express Nox1 and Nox4 (119). While Nox1 is localized to caveolae at the cell surface, Nox4 is found in focal adhesions and inside the nucleus (89). This led to the hypothesis that these two enzymes play different roles within the cell. Nox1 is highly expressed in proliferating VSMCs and its mRNA as well as enzyme activity are further upregulated by agonists, such as angiotensin II (AngII) and platelet-derived growth factor (119). On the contrary, Nox4 is downregulated by the same agonists and has been suggested to be involved in maintaining the quiescent phenotype (118, 119). Another major function of Noxs in VSMCs is the regulation of expression and activity of matrix metalloproteinases (MMPs), thus facilitating vessel remodeling (76).
Leukocytes are the major players of the innate immune response. An essential component of this process is the ability of these cells to generate ROS at a high concentration, crucial for the clearance of invading pathogens. Increased activation of Nox2 in granulocytes has been implicated in a large number of inflammatory pathologies (45). Inflammation is also triggered during the response of the heart to pathological stresses, and Nox2-derived ROS generation within inflammatory cells may be an important contributor to cardiac remodeling (see later sections).
Biochemical regulation of Nox enzymes
The activity of Nox enzyme complexes is regulated through different mechanisms depending on the isoform. Here, we discuss the regulation of Nox2 and Nox4, the main isoforms expressed in cardiomyocytes. Although the regulation of Nox1 is not considered, it is broadly similar to that of Nox2 (23, 65).
Tight control of Nox2 activation in phagocytes is essential in order to facilitate the killing of ingested pathogens while avoiding the potentially damaging effects of ROS on the host cell. Under resting conditions, Nox2 forms an inactive transmembrane complex with a smaller p22phox subunit (collectively referred to as flavocytochrome b558) that stabilizes both proteins (166). Activation of the enzyme depends upon the binding to the flavocytochrome of four regulatory subunits (p47phox, p67phox, p40phox, and Rac2) that normally reside in the cytoplasm of unstimulated leukocytes (12). Nox2 activation is triggered by different cell surface receptors and their downstream transduction pathways (Fig. 2). Central among these are protein kinase C (PKC)–, phosphoinositide-3-kinase (PI3K)–, protein kinase D (PKD)–, phospholipase D (PLD)–, phospholipase A2 (PLA2)–, and mitogen-activated protein kinase (MAPK)–dependent signaling (23). These kinases phosphorylate p47phox at the C-terminus and relieve an intramolecular Src homology 3 (SH3) domain/C-terminus constraint, allowing the interaction of p47phox with the other cytosolic subunits and with p22phox at the plasma membrane (59, 75). p47phox acts as an “organizer” subunit in this process while p67phox is the “activator” subunit that triggers Nox2 activation upon binding to a specific domain in the protein (196). Homologues of p47phox and p67phox perform similar functions in the activation of Nox1. The recruitment of GTP-bound Rac to the Nox2 complex is a critical step required for full enzyme activation (1, 110). GTP/GDP exchange on Rac promotes a conformational change that regulates its interaction with p67phox and Nox2 (43, 69). The activation of Rac in the vicinity of Nox2 is regulated by the production of 3-phosphorylated phosphoinositides by members of the PI3K enzyme family. Among these lipids, phosphatidylinositol(3, 4, 5)trisphosphate (PIP3) recruits to the plasma membrane several PH-domain containing activators of Rac, such as the guanine-nucleotide-exchange factors (GEFs) P-Rex and Vav1 (105, 201). PI3K products different from PIP3 are also involved in the membrane anchoring of cytosolic subunits, in response to appropriate stimuli. In particular, p40phox specifically binds to PI(3)P through its PX domain (24, 54, 55), while the PX domain of p47phox binds preferentially to PI(3, 4)P2 (102, 104).

Although these mechanisms of Nox2 regulation have been mainly studied in leukocytes, they appear to be generally valid in other cell types, such as ECs and cardiomyocytes (72). However, there may be subtle differences in Nox2 activation between phagocytes and nonphagocytes. For example, the phagocyte Nox2 complex interacts with Rac2 whereas Rac1 is the regulator of endothelial and cardiomyocyte Nox2. There may also be differences in the specific GEFs that are involved. Although definitive comparative data on enzyme kinetics among different cell types are lacking, it appears that nonphagocytic Nox2 enzymes are low-output enzymes that produce substantially less ROS than the neutrophil complex (37, 71). Moreover, in response to cellular stimulation, Nox enzyme activation in VSMCs, ECs, cardiomyocytes, and fibroblasts is slower than in neutrophils (16, 71, 152, 200). The main agonists and stimuli of Nox2 activation in cardiomyocytes and ECs include G-protein coupled receptor agonists (GPCRs), such as AngII and endothelin-1; growth factors; cytokines, such as tumor necrosis factor-α (TNF-α); mechanical forces; and metabolic factors, for example, glucose, insulin, glycated proteins (6, 125), and oxidized low-density lipoprotein (ox-LDL)(137).
Nox4 also exists in a complex with p22phox that stabilizes both proteins but is otherwise a very differently regulated enzyme compared with Nox2. The activity of Nox4 does not require binding with either Rac or other cytosolic regulatory subunits, and it is in fact a constitutively active enzyme that generates low levels of ROS under basal conditions (34, 53, 141). Another peculiar feature of Nox4 is its ability to generate predominantly H2O2 instead of superoxide (44, 141, 175, 214). The structural basis for this property of Nox4 has recently been suggested to be related to its E-loop (Fig. 3), which differs significantly from that of Nox2 and contains a highly conserved histidine moiety that may hinder superoxide egress and/or provide a source for protons, allowing dismutation to form H2O2 (190). Although H2O2 is more stable and membrane-permeable than O2 •− (163), it may still mediate spatially localized signaling because cells have very potent reducing mechanisms, such as peroxiredoxin (164). The propensity of Nox4 to produce predominantly H2O2 rather than O2 •− may have important implications for interaction with NO signaling (Fig. 4). Whereas O2 •− reacts with NO to generate peroxynitrite and disrupt NO signaling, H2O2 does not undergo this reaction and instead may even enhance NOS activity and signaling (29). Peroxynitrite may have its own distinct signaling effects too. Evidence in support of Nox4-dependent enhancement of NOS signaling is now emerging (36, 173) and is discussed further in later sections. It should be noted, however, that ROS concentration is also an important consideration; very high concentrations of H2O2 may have detrimental effects that outweigh any effect of enhanced NOS signaling. The lack of known regulatory subunits of Nox4 has led to the hypothesis that, in response to several pathological stresses, Nox4 activity is enhanced through an increase in its mRNA and/or protein levels (127, 175, 213). However, recent studies have also suggested that the interaction of Nox4 with two different proteins, Poldip2 and Tks5, could further increase Nox4 activity although the precise mechanisms remain unclear (42, 138). In addition, it has been suggested that increased levels of NADPH and/or FAD could fuel ROS production by Noxs (78, 211), although how such a mechanism may impact on different Nox isoforms remains to be established.


The differences between Nox2 and Nox4 in subcellular localization (previous section) and biochemical regulation seem to be important because both cellular studies and experiments in gene-modified mouse models indicate that changes in Nox2 or Nox4 activity generally result in distinct phenotypes and that one isoform does not usually “compensate” for lack of the other one (9, 34, 119, 161)—although there are exceptions. An important factor contributing to differences between the biological effects of the two isoforms is also likely to be the signaling targets for redox modification, related to the subcellular spatial colocalization of the Nox isoform and its target(s) within the cell (150, 197). For example, Nox2-dependent signaling has been shown to involve colocation of target proteins in the vicinity of the plasma membrane or endosomes (150, 197). In contrast, Nox4 was reported to modulate EGF-induced signaling by oxidizing an ER-resident protein tyrosine phosphatase in ECs (32). It was also reported that Nox4 might physically associate with toll-like receptor after stimulation of ECs with lipopolysaccharide (154). Far fewer such targets have been identified for Nox4 than Nox2 to date.
Role of Nox Enzymes in Cardiac Remodeling and Heart Failure
The failing heart has a complex phenotype that comprises several components, including cardiomyocyte hypertrophy, contractile dysfunction, arrhythmia, myocyte death, fibrosis, and chamber dilatation. Interestingly, many of these components are capable of being independently regulated or even dissociated from each other, suggesting that complex signaling pathways underlie the overall phenotype. A substantial body of evidence generated over the last few years implicates Nox proteins in modulating different components of the heart failure phenotype, generally through alterations in specific redox-regulated signaling pathways (Fig. 5). Nox expression and/or activity have been found to be increased in end-stage human CHF in several independent studies (21, 88, 139), supporting the potential clinical relevance of this pathway.

Cardiac hypertrophy
The role of different Nox isoforms in the development of left ventricular hypertrophy (LVH) has been best assessed in various gene-modified models (183). Increased activation of the renin-angiotensin-aldosterone system is of key importance in the development of heart failure, through multiple effects including the promotion of cardiac hypertrophy. Nox2 was found to be essential for cardiac hypertrophy that develops following short-term (2 weeks) subpressor infusion of AngII, with Nox2 knockout mice displaying less LVH and blunted increases in mRNA levels of molecular markers, such as atrial natriuretic factor (ANF) and β-myosin heavy chain (19). Similar results were found in mice with a cardiomyocyte-specific deletion of Rac1, which had defective Nox2 activation, indicating that it is Nox2 in cardiomyocytes that is stimulated in this setting (172). Consistent with this, AngII-induced hypertrophy of cultured neonatal rat ventricular myocytes (NRVM) involved the activation of Nox2 (149). AngII-induced Nox2-dependent hypertrophy appears to involve the enhanced activation of extracellular-signal-regulated kinase 1/2 (Erk1/2), Akt, and an apoptosis signal-regulating kinase 1 (ASK-1)/nuclear factor kappa-light-chain-enhancer of activated B cell (NF-κB) pathway (90, 91, 149, 172). In fact, in vivo LVH in response to AngII infusion is attenuated in ASK-1 deficient mice (97). Recently, it was reported that the signaling of AngII-induced cardiomyocyte hypertrophy involved a direct physical interaction of AT1 receptors with Nox2 and the induction of WNT1 inducible signaling pathway protein 1 (WISP1) (178). Cardiomyocyte hypertrophy mediated by other GPCRs, such as noradrenaline and endothelin-1, is also reported to involve Nox2 activation (80, 191, 208). In the case of α1-adrenoreceptor (AR)–stimulated hypertrophy, it was shown that Nox-mediated ROS induced Ras activation through the oxidation of specific thiols, thereby leading to activation of the MEK1/2-ERK1/2 pathway (115). Local AngII release and/or AT1 receptor activation may also contribute to the Nox2-mediated in vivo LVH induced by elevated aldosterone, as in DOCA-salt hypertension models (100, 209).
Early studies showed that Nox activity increased in parallel with progression of pressure overload-induced LVH in small animal models (124). However, Nox2 knockout mice developed a similar extent of hypertrophy in response to aortic constriction as wild-type controls (28, 144). This may suggest that the requirement for Nox2 is less important when hypertrophy occurs in response to primarily mechanical stimuli than to agonists such as AngII, in keeping with the knowledge that different signaling pathways are involved in GPCR-agonist-induced versus mechanically induced hypertrophy. Consistent with this notion, Nox2 deletion did not affect hypertrophy when renin-angiotensin system activation was accompanied by concomitant hypertension (and therefore a mechanical load) (100, 144). Nox2 activation nevertheless contributes to the development of contractile dysfunction and interstitial cardiac fibrosis during chronic pressure overload (19, 73), suggesting that these abnormalities involve distinct signaling perturbations than those that drive hypertrophy per se (172).
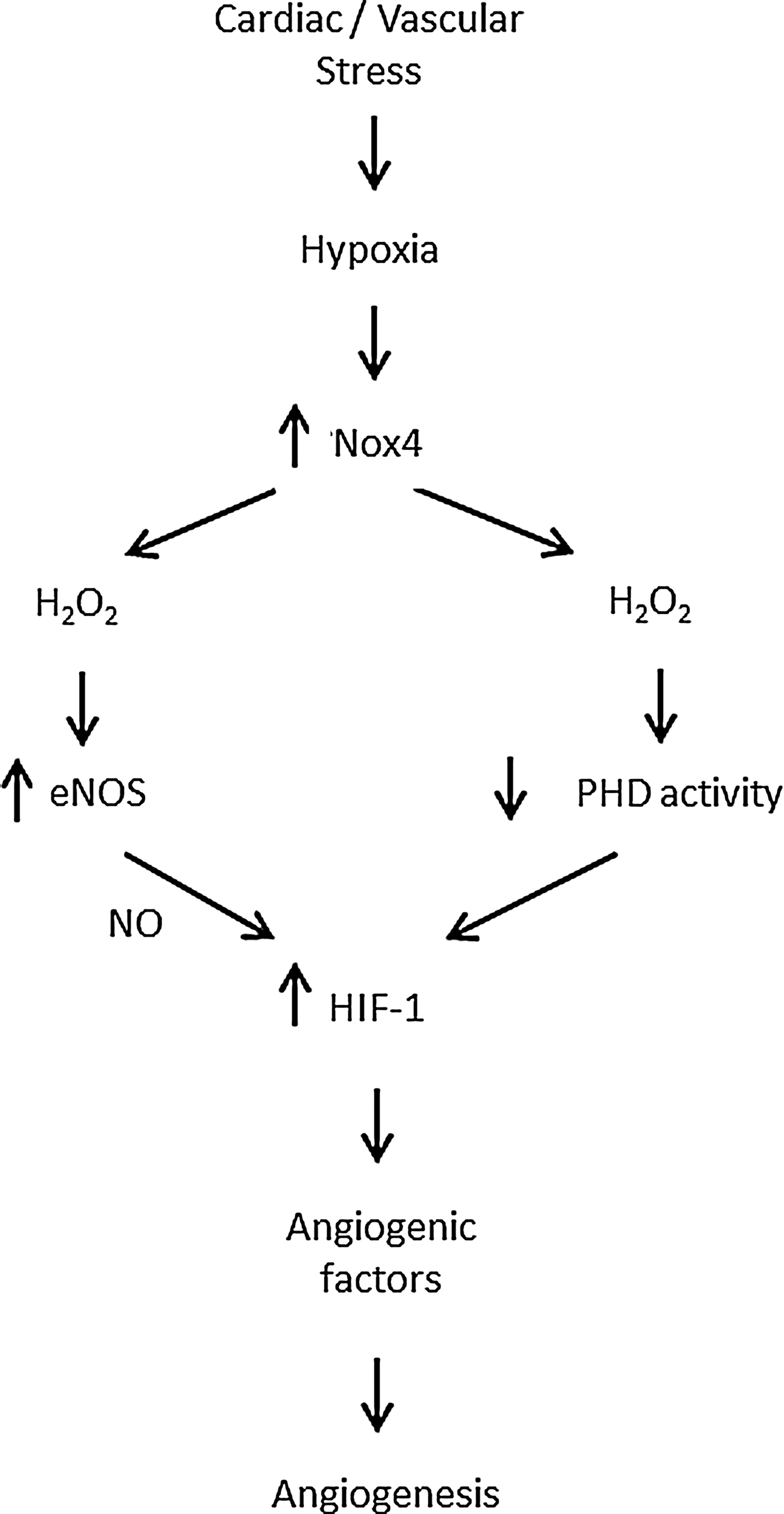
The lack of inhibition of pressure overload LVH in Nox2 knockout mice could be due to compensatory changes in other Nox isoforms or related signals. Indeed, we found that Nox4 levels were significantly increased by pressure overload (28). To investigate the role of Nox4 in pressure overload LVH, we studied mouse models with a cardiomyocyte-targeted Nox4 overexpression or a knockout of Nox4. These studies unexpectedly demonstrated that Nox4 was actually protective against chronic-load-induced cardiac stress, in that Nox4 knockout mice developed worse LVH, contractile dysfunction, and dilatation whereas Nox4-overexpressing mice showed the opposite phenotype (214). No differences in Nox2 levels upon perturbation of Nox4 were found in these studies. Investigation of underlying mechanisms revealed that an increase in Nox4 levels promoted a better preservation of myocardial capillary density after pressure overload through enhanced activation of hypoxia-inducible factor 1 (Hif1) and the release of vascular endothelial growth factor from cardiomyocytes to exert paracrine angiogenic activity (214). This mechanism is in line with studies in other cellular systems that support an important role of Nox4 in enhancing angiogenesis (41, 198, 207). These findings reveal Nox4 to be a novel inducible regulator of myocardial angiogenesis, a key determinant of cardiac structural and functional compensation to chronic pressure overload (171, 180). In addition to the enhancement of Hif1, cardiac-specific overexpression of Nox4 was found to activate the transcription factor Nrf2 and increase the expression of many genes involved in phase 2 defense (25, 173). Since Nrf2 has been reported to protect against load-induced cardiac hypertrophy (122), this could also contribute to the beneficial effects of Nox4.
The precise mechanism(s) by which Nox4 increases Hif1 activation remains to be fully defined. The major mechanism of regulation of Hif1α levels is via its hydroxylation at specific proline residues by prolyl hydroxylases (PHD) and subsequent targeting for proteosomal degradation (70). In the studies described just now, we found evidence of reduced Hif1α hydroxylation, suggesting that Nox4 may inhibit PHD activity. In fact, a previous study in tumor cells reported that Nox4-derived H2O2 inhibited Hif1 hydroxylation, with evidence that this involved redox modification of the iron center within PHD (66). In the vasculature, however, recent studies showed that Nox4-induced angiogenesis was associated with an increase in eNOS expression and activity (36, 173). NO can augment Hif1α levels through its S-nitrosylation and inhibition of hydroxylation (131), suggesting that an alternative or complementary mechanism for the effects of Nox4 on Hif1 may involve increased eNOS-derived NO (Fig. 6). The contribution of such an NO-dependent mechanism to the protective effects of Nox4 in the heart remains unclear although no changes in eNOS protein levels were found (214). Nevertheless, the potential for Nox4-derived H2O2 to increase eNOS levels and activity is a significant difference from Nox2, the activation of which is generally associated with a decrease in NO actions (26). Interestingly, beneficial effects of endothelial Nox4-derived H2O2 on vasodilatation were recently reported (161), which contrast to the detrimental effects of endothelial Nox2 on vascular function (20, 147).
In contrast to the work discussed just now, it should be noted that studies from the Sadoshima laboratory using independently generated mouse models of Nox4 overexpression and deletion reported a detrimental role of Nox4 in aortic constriction-induced cardiac dysfunction (113). The reasons for the marked differences between results remain to be elucidated and could possibly be related to methodological differences, such as mouse background strain, strategy of gene targeting, and severity of aortic constriction. Of note, this work found that an important mechanism for cardiac impairment was mitochondrial ROS generation and dysfunction (4, 113). Enhanced generation of mitochondrial ROS primarily from the electron transport chain is considered to be an important pathogenic mechanism in heart failure, especially in its advanced stages, acting via damage to mitochondrial DNA and energetic processes (195). Ultimately, this may result in cell death by causing the mitochondrial permeability transition, a process that results in mitochondrial membrane depolarization and the leak of pro-apoptotic and pro-necrotic proteins into the cytosol (79). Increased mitochondrial ROS result both from an increase in ROS generation and an impairment of mitochondrial antioxidants (10). Importantly, ROS production by other sources (including Noxs and monoamine oxidases) can induce further mitochondrial ROS production, which may act as an amplifying mechanism (48, 101, 108, 220). Future studies need to establish the precise inter-relationship between the generation of ROS by classical mitochondrial enzymes and those derived from different Nox isoforms and other sources in the setting of heart failure, as this data will be germane to the selection of potential therapeutic targets and may also help to decipher the conflicting results referred to previously.
Excitation-contraction coupling and contractile dysfunction
Abnormalities of cardiomyocyte excitation-contraction coupling (ECC) are a fundamental feature of CHF (177). The redox environment influences several components of ECC, typically through oxidative modification of critical reactive cysteine residues in ion channels and transporters (219), but also through modulation of expression levels (194, 218). Increasing evidence supports a role of Nox proteins in these effects but the relevance to heart failure is still poorly explored (Fig. 7).

The open-state probability of L-type calcium channels may be modulated by Nox2 in response to agonists, such as endothelin-1 (212), while AngII reportedly increases expression levels of L-type channel subunits through a Nox-dependent transcriptional pathway (194). Redox regulation of the ryanodine receptor (RyR), which regulates release of calcium from the sarcoplasmic reticulum (SR), has been extensively studied and involves S-glutathionylation and S-nitrosylation (47). An “NADH oxidase” activity associated with the SR has long been recognized (33), and Nox2 was reported to be present in canine cardiac SR (170). Both tachycardia and exercise could enhance Nox activity and increase RyR S-glutathionylation and calcium release rates (169, 170). On the other hand, Nox4 is also reported to localize to the SR (214). Interestingly, Sun et al. recently reported that Nox4 in the SR of skeletal muscle may act as a physiological sensor of O2 tension and generate H2O2 that modulates RyR function (186). Whether Nox4 may play a similar role in the heart is unknown. The SR calcium ATPase pump (SERCA) that mediates re-uptake of calcium into the SR is also susceptible to redox regulation. Increased SERCA oxidation and calcium re-uptake related to increased Nox2 activity have been reported in mouse models of diabetes (128). Contractile dysfunction may also result from abnormalities at the level of the myofilaments and oxidative modification of contractile proteins has been implicated in this process (86). However, whether Nox-derived ROS contribute to this abnormality remains to be addressed.
Recently, Prosser et al. reported a novel role of Nox2 in modulating mechanosensitive stretch-induced calcium release in normal cardiomyocytes (158). Increased calcium release in response to stretch is an important contributor to length-dependent regulation of cardiac function or the Frank–Starling relationship. These authors found that physiological stretch activated Nox2 in the sarcolemma and T-tubules, and that the ROS that were generated sensitized RyR to enhance calcium-induced calcium release—evident as calcium “sparks” on confocal microscopy. This is perhaps the first data to suggest an important physiological role for Nox2 in the cardiac myocyte and addresses a long-standing question as to why these proteins are present even in the normal healthy heart. While this mechanism was beneficial in the physiological setting, Prosser et al. found that excessive activation of Nox2 induced either by AngII stimulation or in a mouse model of muscular dystrophy resulted in abnormal calcium waves instead of sparks (158). Such a mechanism could have relevance to heart failure by contributing to calcium dysregulation, impaired contraction, and arrhythmia. This finding suggests that the level of Nox2 activation and the amount of ROS generated may be a crucial factor in determining the effects on cardiomyocyte function.
Arrhythmia
Atrial fibrillation (AF) is a common arrhythmia complicating CHF, especially in older patients, and increases morbidity. It is associated with electrophysiological and structural remodeling of the atrial myocardium that favors perpetuation of the arrhythmia but the mechanisms underlying these changes remain incompletely understood. Current evidence shows that increased atrial oxidative stress may play an important role in initiating and sustaining AF in animal models and humans (30, 146), and Noxs have emerged as important in this regard (50, 106, 107, 162). Induction of AF with rapid atrial pacing in pigs was associated with increased Nox activation and ROS production in the left atrium (50). Studies from the group of Casadei showed that Nox2 was the main source of ROS in human atrial myocytes and that Nox-derived ROS production was increased in right atrial appendages of patients undergoing cardiac bypass surgery who subsequently developed postoperative AF (106). Further, atrial NADPH-stimulated ROS production was independently associated with an increased risk of postoperative AF (107). Recent work from the same group reported that Nox2 appears to be important mainly in the initiation of AF, either in a goat rapid-pacing model or in humans who develop postoperative AF, whereas uncoupled NOSs and mitochondrial oxidases may be more important ROS sources in the setting of longstanding AF (162). Interestingly, AF develops spontaneously in mice with cardiac-specific overexpression of a constitutively active Rac1, which activates Nox2 (2).
Therapeutic agents that target Nox2 activation may have some potential in reducing the risk of developing AF. Statins, which downregulate Rac1 activity by suppressing its isoprenylation, are reported to have a favorable effect on risk of developing AF although they may be ineffective in persistent AF (2, 7, 162, 182, 210). Angiotensin-converting enzyme inhibitors and AngII receptor blockers both reduce risk of AF (52, 85), and it is possible that part of the mechanism may involve a reduction in Nox2 activation. Indeed, AngII promotes early afterdepolarizations in rabbit cardiomyocytes through activation of the Nox-ROS-Ca(2+)/calmodulin–dependent protein kinase II (CaMKII) pathway (217). An important role of oxidized CaMKII downstream of Nox2 activation is also supported by work from the Anderson laboratory showing that Nox2-dependent oxidation of CaMKII promotes cardiac sinus node dysfunction in a mouse model of AngII-induced dysrhythmia (189). However, whether any of the above mechanisms are relevant to AF in CHF remains to be established.
Myocyte death
A low level of cardiomyocyte death occurs during chronic cardiac remodeling and contributes to CHF (61). ROS and redox signaling potentially impact on apoptosis at several levels, including upstream signaling pathways that are pro-apoptotic and at the level of the mitochondria (60, 143). The former include pro-apoptotic pathways, such as the activation of ASK-1, JNK, p38MAPK, and CaMKII, as well as anti-apoptotic pathways, such as Akt, Bcl2, and heat shock proteins (143).
There is some evidence to support an involvement of Nox-derived ROS in cardiomyocyte apoptosis. AngII promotes apoptosis of cultured NRVM at least in part via Nox, most likely Nox2 (74, 134, 159). The activation of CaMKII might be a critical convergence point of pro-apoptotic signaling since it is activated both by Ca2+ and by Nox2-derived ROS, downstream of AngII (57). Cardiomyocytes from p47phox−/− mice or from mice expressing an inhibitory peptide against CaMKII were protected against apoptosis induced by chronic AngII infusion (57). The importance of this pathway in other species was further demonstrated by Palomeque et al. (153). Norepinephrine, aldosterone, and doxorubicin are also reported to promote cardiomyocyte apoptosis through the activation of Nox2 (67, 82, 129). Interestingly, apoptosis of rat cardiomyocyte-like H9c2 cells induced by metabolic inhibition was suggested to involve nuclear Nox2 (145). The exposure of rat cardiomyocytes to high glucose upregulates Nox2 activity and increases apoptosis (179), which may be relevant to diabetic heart disease (see the section below entitled Other forms of heart failure).
The possible role of Nox4 in myocyte apoptosis remains controversial. It was reported that adenoviral-mediated overexpression of Nox4 in cardiomyocytes enhanced myocyte apoptosis (4). However, studies in many other cell types reported anti-apoptotic effects of Nox4, for example, through enhancement of Akt activation (14, 41, 51, 120). We did not find any evidence of increased apoptosis in cardiomyocyte-targeted Nox4 transgenic mice up to the age of 12 months (214). It is interesting that Nox4 enhances Akt activation whereas Nox2 was shown to increase CaMKII activation; a likely reason for this could be the different subcellular localization of the two isoforms and presumably different coupling to downstream targets.
Myocardial fibrosis
Myocardial interstitial fibrosis is an important component of the hypertrophied and failing heart and impacts on cardiac passive filling characteristics, tissue hypoxia, and susceptibility to arrhythmia. Increased activation of the renin-angiotensin-aldosterone system, increased mechanical load, and increased cytokine activation are all known to be important in the development of fibrosis. Nox proteins appear to have a major role in the development of fibrosis in different settings. Interstitial fibrosis induced either by subpressor AngII infusion, pressor AngII infusion, or chronic activation of the renin-angiotensin system was inhibited in Nox2 knockout mice (19, 28, 100, 203) or in cardiomyocyte-specific Rac1 knockout mice (172). Nox2 was also implicated in mineralocorticoid receptor agonist aldosterone-induced cardiac fibrosis in mice and rats (100, 188). Moreover, interstitial cardiac fibrosis was abolished in Nox2 knockout mice subjected to aortic constriction or permanent coronary artery ligation (73, 136). Of note, deletion of Nox2 attenuated fibrosis even when the extent of myocyte hypertrophy was unaltered in some of the above studies, indicating a dissociation in the contribution of Nox2 to these phenotypes (100, 188). These data indicate a crucial role of Nox2 in fibrotic remodeling in vivo in response to diverse stimuli. The dissociation between effects of Nox2 on fibrosis and hypertrophy may indicate that the effects on fibrosis involve nonmyocytes or inflammatory cells (148). Some in vitro studies have suggested that Nox4 is involved in the proliferation of cultured human cardiac fibroblasts and their transformation into myofibroblasts in response to transforming growth factor β treatment (39). However, the relevance of this in vivo remains to be established. We did not find a role of Nox4 in promoting fibrosis in gene-modified Nox4 mice subjected to aortic constriction (214).
A number of molecular mechanisms through which Nox2 may promote fibrosis have been suggested. Nox2 may enhance the expression and/or activation of MMPs, which are involved in matrix turnover (11, 168). MMPs not only degrade normal matrix components but also modulate collagen synthesis, for example, by modulating the formation of matrikines and the release of pro-fibrotic growth factors from the extracellular matrix (130). As such, increased MMP activity is commonly associated with increased fibrosis in the failing heart, with the fibrotic tissue comprising altered collagen type, organization, and cross-linking (77, 87, 98). However, matricellular proteins, such as thrombospondin-1, that are activated by MMPs actually inhibit inflammation and fibrosis (63) so that the precise relationship between MMP activation and fibrosis may depend upon context. It was reported that TNF-α-induced induction of MMPs in cardiomyocytes involves Nox and PI3Kγ (11). Nox-mediated MMP activation has also been shown in rabbit hearts in response to combined volume and pressure overload (135), and in age-related cardiac fibrosis in rats (199). Aldosterone-induced in vivo myocardial fibrosis in mice involves the Nox2-dependent activation of NF-κB; upregulation of the profibrotic cytokine connective tissue growth factor; increased expression of procollagen-1, 3, and fibronectin; and increased MMP2 activation (100).
Infiltration by circulating inflammatory cells and the local release of cytokines constitute key pro-fibrotic stimuli within the myocardial tissue in response to pathological stress such as MI and hemodynamic overload (116). Inflammation contributes not only to fibrosis but may also affect cardiomyocyte hypertrophy and viability. Recent studies have highlighted the role of leukocyte PI3Kγ in regulating inflammatory cell recruitment and consequent fibrosis, which may also involve Nox2 activation (40, 92, 155). Mice with catalytically inactive PI3Kγ (PI3Kγ KD) have reduced cardiac fibrosis after transverse aortic constriction, accompanied by attenuated infiltration of inflammatory cells (155). Using a bone marrow chimera approach, it was conclusively demonstrated that the reduction in leukocyte recruitment and fibrosis in the myocardium after aortic constriction was attributable to bone marrow cells (40). Importantly, PI3K has a central role in regulating neutrophil Nox2 activation (81), suggesting that pharmacological targeting of PI3Kγ could impact on Nox2 activation and may at least in part be involved in the effects on fibrotic remodeling.
Post-MI remodeling
Post-MI remodeling includes cell death and inflammation in the early phase of infarct healing, followed by infarct expansion, progressive hypertrophy and fibrosis remote from the infarct site, LV chamber enlargement, and functional deterioration in the late phase. Increased oxidative stress has long been recognized to be involved in this process (109, 181, 187). The specific involvement of Nox proteins was suggested by the finding that Nox2 is upregulated in cardiomyocytes in the infarcted myocardium after acute MI in patients (111) as well as in experimental models (64, 142). Nonspecific inhibitors of Nox2, such as apocynin and statins, were found to have beneficial effects on adverse post-MI remodeling (15, 83, 160). More compelling evidence for the involvement of Nox2 in post-MI remodeling comes from studies in gene-modified mice. Mice deficient in p47phox had no reduction in infarct size after myocardial ischemia/reperfusion or permanent coronary ligation (46, 93). However, these animals were found to have attenuated ventricular dilatation and contractile dysfunction after permanent ligation, as well as higher survival than wild-type controls (46). Likewise, Nox2 knockout mice also displayed less LV dilatation and contractile dysfunction after MI, together with reduced cardiomyocyte hypertrophy, apoptosis, and interstitial fibrosis (136). It has been suggested that adverse effects following myocardial ischemia-reperfusion may involve the ROS-induced induction of lectin-like ox-LDL receptor-1, which in turn mediates the activation of MAPKs and inducible NOS (94).
Increased activation of the sympathetic and renin-angiotensin-aldosterone systems is known to be important in promoting heart failure after MI. It was recently shown that the β1-selective AR antagonist nebivolol exerted beneficial effects on post-MI hypertrophy and contractile dysfunction through the inhibition of Nox (185). However, this agent also has other effects, in particular, an enhancement of NO production (140), raising the question whether the effects on Nox and NO are related. Several studies have shown that increased ROS generation in the central nervous system is implicated in mediating sympathoexcitation post-MI (121, 132). It was reported that Nox4 is upregulated in the paraventricular nucleus after MI and that shRNA-mediated knockdown reduced sympathetic outflow from the brain and improved cardiac function in this setting (96). High plasma levels of aldosterone increase the risk of early death after MI, at least in part by increasing cardiac rupture. Recently, a novel Nox2-mediated mechanism contributing to this lethal complication has been identified (84). He et al. (84) found that Nox2-dependent oxidation of CaMKII mediated these cardiotoxic effects of aldosterone, through an increase in MMP9 expression by cardiomyocytes that promoted cardiac rupture post-MI (Fig. 8). Either the inhibition of CaMKII or a deficiency of Nox2 attenuated the increase in MMP9 activity and protected the heart against rupture after MI. Cardiac-specific deletion of the mineralocorticoid receptor also ameliorates post-MI remodeling, at least in part by reducing Nox2 activation (62).

Other forms of heart failure
Patients with diabetes may develop a greater extent of cardiac dysfunction and failure that can be attributed simply to myocardial ischemia or changes in cardiac workload (22). While the underlying causes are multifactorial and include prominent metabolic dysregulation, an increase in myocardial ROS production is a contributing factor (22). Nox-derived ROS production may be involved in this process. High glucose increases Nox2 activation in isolated cardiomyocytes and contributes to contractile dysfunction (13, 157) and apoptosis (179). Hyperglycemia-induced Nox2 activation has been shown to contribute to the formation of increased glycated proteins, NF-κB activation, and the upregulation of ANF mRNA in cardiomyocytes (215). Several studies have demonstrated that myocardial Nox activity is increased in models of type 1 and type 2 diabetes (128, 142, 174, 179, 202, 205) and treatment with apocynin was reported to protect against diabetes-induced myocardial fibrosis, hypertrophy, and dysfunction (123, 128, 165). Li et al. used mice with cardiac-specific Rac1 KO to demonstrate that activation of Rac1/Nox2 oxidase is involved in myocardial remodeling during the development of type 1 diabetic cardiomyopathy (123). However, definitive in vivo data to confirm the roles of specific Nox proteins in the development of diabetic heart disease are still lacking.
Anthracycline chemotherapeutic agents, such as doxorubicin, cause dose-dependent cardiotoxicity and heart failure that involves increased ROS generation and oxidative stress (58, 103, 133, 151). Emerging evidence indicates that Nox2-derived ROS are important in the development of doxorubicin-induced cardiotoxicity. Doxorubicin induces rapid Nox-mediated ROS generation in H9c2 cardiomyoblasts (67) and in the mouse heart in vivo (151). Conversely, Nox2 deletion attenuated doxorubicin-stimulated cardiac ROS production and cardiac dysfunction (204). Importantly, the latter group found that genetic polymorphisms of the Nox2 oxidase complex may influence the individual risk of doxorubicin cardiotoxicity in cancer patients (204). Recently, Zhao et al. showed that Nox2-derived ROS contributed to contractile dysfunction, myocardial atrophy, increased cardiomyocyte apoptosis, interstitial fibrosis, and inflammatory cell infiltration in an in vivo model of doxorubicin cardiotoxicity in mice (216).
Therapeutic Interventions Targeting Nox
The studies discussed previously suggest that targeted inhibition of Nox proteins, in particular, Nox2, may be a useful therapeutic strategy in heart failure and its antecedent conditions. The complex regulation and activation of Nox2 and the specific downstream signaling pathways that it modulates offer the potential for multiple complementary approaches to inhibition of detrimental signaling. A diverse range of compounds are potential Nox inhibitors and these have been comprehensively reviewed recently (49, 99). Commonly used agents such as apocynin and diphenylene iodonium are not specific but newer agents in development appear promising (49, 99). Here, we consider a few general issues that may be relevant in developing Nox-targeted therapies for heart failure. Whereas many existing agents or compounds in early development are inhibitors of multiple Nox isoforms, therapies of heart failure may require isoform-specific agents because Nox4 has beneficial effects, such as the preservation of appropriate myocardial vascularization during chronic pressure overload (214). Nox2 may also have some beneficial effects, for example, in ischemic cardiac preconditioning (18) or in ECC (158). Another issue to consider with respect to Nox2 inhibitors is the potential to compromise anti-microbial and/or immune functions of white cells, although it may be feasible to selectively target Nox2 in different cell types due to cell-specific differences in its regulation (99). Further, recent work that severe chronic granulomatous disease is only evident in individuals whose phagocytic ROS production is more than two orders of magnitude lower than healthy controls suggests that safe therapeutic targeting of cardiovascular Nox2 could be feasible (112). Targeting specific molecules upstream or downstream of Nox2 may also be worthwhile. For example, the inhibition of PI3Kγ could be a useful approach to Nox inhibition (167). Likewise, targeting molecules downstream of Nox2 that act as signaling hubs, for example, CaMKII, appears promising (56). Finally, better elucidation of the interactions among Noxs, NOS signaling, and mitochondrial ROS may also offer new targets. In conclusion, the evidence reviewed in this article indicates that Nox-dependent signaling plays important roles in the development of heart failure and may offer novel therapeutic approaches for this condition.
Footnotes
Acknowledgments
This work is supported by a Foundation Leducq Transatlantic Network of Excellence Award (E.H. and A.M.S.), the British Heart Foundation (RG/08/011/25922 and RE/08/003 to A.M.S.), and the Department of Health via a National Institute for Health Research (NIHR) Biomedical Research Centre award to Guy's & St. Thomas' NHS Foundation Trust in partnership with King's College London and King's College Hospital NHS Foundation Trust.
Author Disclosure Statement
E.H. is a cofounder of Kither Biotech. None of the other authors have any conflicts to disclose.