
Abstract
Introduction
Necrotic cell death is characterized by a gain in cell volume, swelling of organelles, plasma membrane rupture, and subsequent release of intracellular components. Although initially described as an accidental type of cell death, it is now proposed that the execution of necrotic cell death can also be finely regulated by specific signal transduction pathways and catabolic processes (necroptosis). Necrosis has been reported to occur in inflammatory and neurodegenerative disorders, heart disease, neuronal ischemia and toxicity, muscular dystrophy, diabetes, infections, and in apoptotic cells that fail to be engulfed by phagocytic cells (secondary necrosis) (135, 170).
Autophagy is a major catabolic pathway by which eukaryotic cells degrade and recycle macromolecules and organelles. It has an essential role in differentiation, development, and cellular response to stress. Autophagy is initiated by the selective or nonselective engulfment of cytoplasmic constituents by a phagophore, which forms a closed double-membrane structure, the autophagosome. The autophagosome subsequently fuses with a lysosome to become an autolysosome whose content is degraded by acidic lysosomal hydrolases (105). Autophagy is a homeostatic mechanism involved in both survival and cell death. Autophagic cell death is morphologically defined by massive autophagic vacuolization of the cytoplasm in the absence of chromatin condensation. Although autophagy deregulation has been associated with distinct pathologies, it is primarily regarded as a pro-survival mechanism and there are only a limited number of cases where increased autophagy has been established as the cause of cell death (55, 136, 218).
Redox signaling events are important regulators of cell death pathways (40, 115, 181, 209). Although oxidative stress and ROS/RNS formation have long been thought to be major players regulating cell death, other redox-dependent signaling mechanisms have been identified as key players in the activation of the cell death machinery. GSH depletion is an early hallmark in the progression of distinct cell death mechanisms (36, 74, 107, 234). We and others have extensively reviewed the mechanisms by which alterations in GSH homeostasis regulate the activation of the cell death machinery (39, 74, 77, 130). Other excellent reviews address the role of compartmentalized GSH pools (mitochondria and endoplasmic reticulum) (6, 166), specific GSH-dependent antioxidant systems (119, 159), and GSH-based protein modifications in cell death (2, 50). Furthermore, several review manuscripts address the role of GSH in regulating cell death pathways in distinct pathologies (14, 71, 77) such as neurodegenerative disorders (167), cancer (63), hepatotoxicity (252), autoimmunity (196), and pulmonary diseases (22). This review article aims at highlighting the role of plasma membrane GSH efflux in GSH depletion during apoptosis and the mechanisms by which GSH depletion, by its extrusion, might contribute to alterations in the cellular redox balance and cell death progression. Furthermore, this works aims at summarizing the current evidence regarding the molecular identity of plasma membrane GSH transporters.
Overview of GSH Homeostasis
GSH (L-γ-glutamyl-L-cysteinyl-glycine) (Fig. 1) is the most abundant nonprotein thiol in mammalian cells acting as a major reducing agent and antioxidant defense by maintaining a tight control of the redox status. The peptidic γ-linkage between glutamate and cysteine protects GSH from hydrolysis by intracellular peptidases. The presence of the C-terminal glycine protects GSH against cleavage by intracellular γ-glutamylcyclotransferases. The cysteinyl moiety of GSH provides the reactive thiol group (-SH group) that mediates GSH biological functions, including oxidation-reduction (redox) and nucleophilic addition-type reactions (Fig. 1). GSH is also involved in the metabolism of xenobiotics, thiol disulfide exchange reactions, and acts as an important reservoir of cysteine. GSH synthesis is initiated by generation of γ-glutamylcysteine from glutamate and cysteine via the glutamate-cysteine ligase (GCL), and the subsequent addition of glycine by the activity of GSH synthetase (GS) (172, 219).
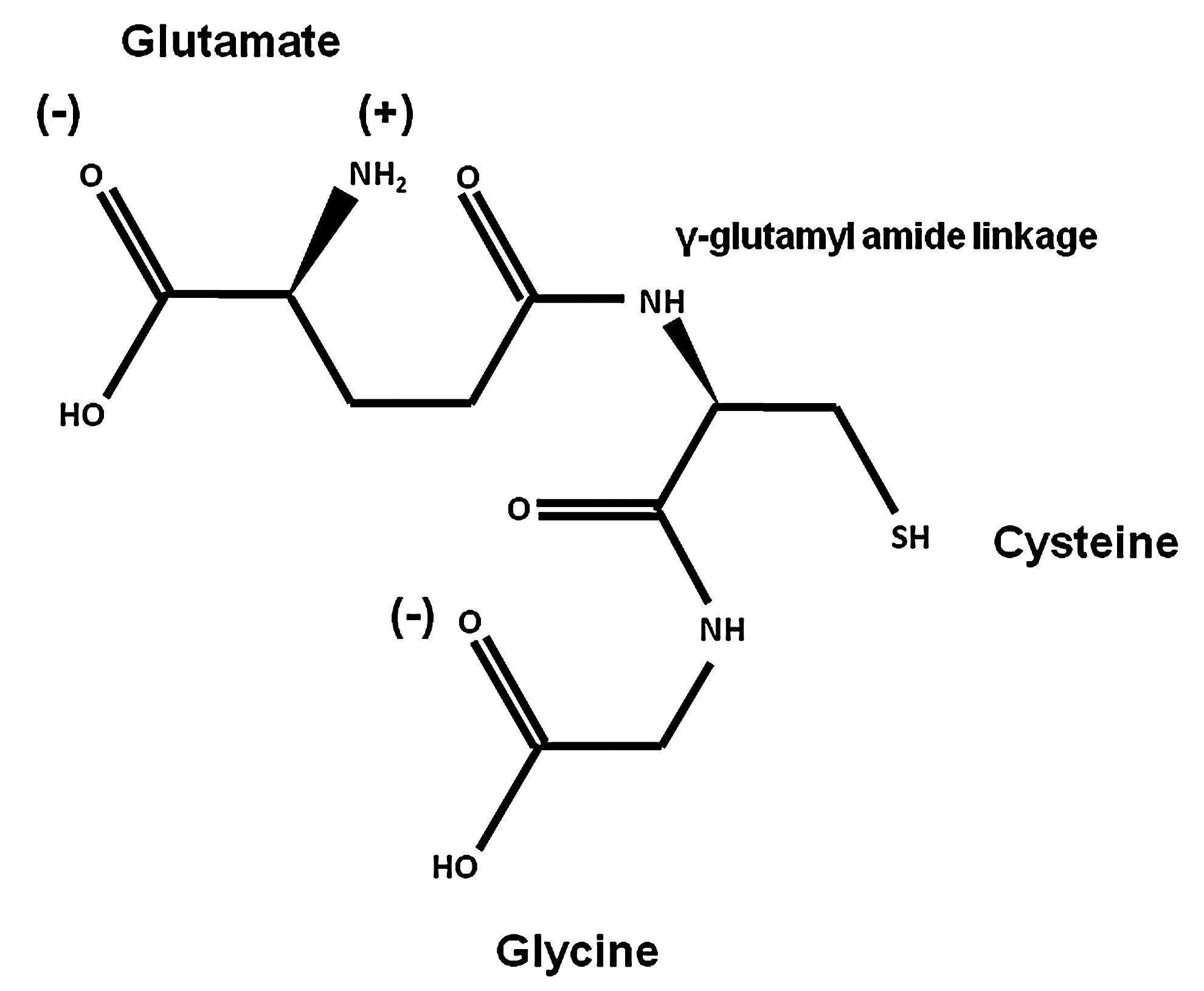
Changes in the intracellular thiol-disulfide (GSH/GSSG) balance are considered major determinants in the redox status/signaling of the cell (123, 212). Almost all physiological oxidants react with thiols, and GSH has the ability to directly scavenge ROS/RNS. A large variety of unique GSH oxidation species can be generated on ROS/RNS formation, and their chemical profile depends on the magnitude and identity of the ROS/RNS generated. Similar to protein thiols (cysteines), GSH can be subject to one-electron oxidation by ROS such as superoxide anion (O2•−), which mediates derivatives with an unpaired electron, including the thiyl radical (glutathionyl radical [GS•]) and the thiyl peroxyl radical (GSOO•). Two-electron oxidation of GSH by ROS/RNS such as hydrogen peroxide (H2O2) and peroxynitrite (ONOO−) mediates the formation of other distinct oxidized states of GSH, which include the homo-disulfide glutathione disulfide (GSSG), glutathione sulfenic (GSOH), sulfinic (GSO2H) and sulfonic acids (GSO3H), glutathione disulfide S-oxide (GS(O)SG), glutathione disulfide S-dioxide (GS(O)2SG), glutathione thiosulfenamide (GSNHSG), glutathione N-hydroxysulfenamide (GSNHOH), and S-nitrosoglutathione (GSNO) (200, 246). Except for GSNO and GSSG, the physiological relevance of other oxidized GSH derivatives has not been studied in detail primarily due to the lack of accessible and selective techniques to quantify them, and their high instability/reactivity (116, 222, 230).
GSH Depletion During Cell Death: Where Does It Go?
GSH is essential for cell survival as demonstrated by the observations that the GCL knockout mice die from massive apoptotic cell death (51), and that the knockdown of GCL in distinct cell types induces time-dependent apoptosis (58, 238). However, GSH itself is not required for survival, only the reducing equivalents provided by its reducing power (238). GSH depletion is a hallmark of the progression of cell death. More importantly, GSH depletion has been clearly shown to occur in apoptosis before the rupture of plasma membrane integrity (secondary necrosis) or cellular fragmentation, suggesting an active mechanism involved in its depletion (73). Distinct mechanisms have been reported to contribute to GSH depletion during cell death progression as summarized next (Table 1 and Fig. 2).

ABC, ATP-binding cassette; ABCC, ATP-binding cassette (ABC) transporter, subfamily C; CFTR, cystic fibrosis transmembrane conductance regulator; Cyt C, cytochrome C; G6PD, glucose-6-phosphate dehydrogenase; GCL, glutamate-cysteine ligase; GLAST, glutamate/aspartate transporter; GPx, glutathione peroxidase; GR, glutathione reductase; Grx, glutaredoxin; GS•, glutathionyl radical; GSH, glutathione; GSNO, S-nitrosoglutathione; GST, glutathione-S-transferases; MRP, multidrug resistance protein; N2O3, dinitrogen trioxide; NADPH, nicotinamide adenine dinucleotide phosphate; NO•, nitric oxide; NO2, nitrogen dioxide; OA−, organic anion; OATP, organic anion transporting polypeptides; PSSG, protein glutathionylated; PSOH, protein sulfenic acid; PSNO, protein nitros(yl)ation; RLIP76 (RALBP1), Ral-binding, Rho/Rac-GAP and Ral effector; ER, endoplasmic reticulum; VRAC/VSOAC, volume-regulated/volume-sensitive organic osmolyte-anion channels.
GSH depletion during cell death progression has been largely ascribed to its oxidation in response to ROS/RNS formation. Indeed, during the apoptosis induced by cytotoxic agents, which by themselves induce oxidative stress such as pro-oxidants, xenobiotics, mitochondrial toxins, chemotherapeutics, and metals, GSH depletion is mediated by its oxidation to GSSG by ROS/RNS (61, 104, 169, 199, 237) (Table 1 and Fig. 2). Glutathione reductase (GR) reduces GSSG back to GSH using reduced nicotinamide adenine dinucleotide phosphate (NADPH) as the electron donor reductant, and glucose-6-phosphate dehydrogenase (G6PD) is indispensable for the regeneration of NADPH from NADP+ (Table 1 and Fig. 2). The depletion/oxidation of NADPH and the inactivation of G6PD occur during apoptotic cell death, which might impair GSH recycling and contribute to GSH depletion (54, 85, 88, 198, 255). Besides G6PD, other NADP+-dependent dehydrogenases can also regenerate NADPH in the cytoplasm, including the 6-phosphogluconate dehydrogenase, the cytosolic NADP+-dependent isocitrate dehydrogenase (IDPc), and the cytosolic NADP+-dependent malic enzyme. Knockdown of IDPc increases GSSG levels and augments the sensitivity of cells to cell death induced by oxidative stress (149).
Previous findings have shown that GCL is a direct target of caspase 3 (78, 79), which during apoptosis should not only prevent GSH replenishment but also contribute to GSH depletion, as GSH's half-life has been estimated to be between 2 and 5 h (18, 113, 207). Furthermore, the impairment of cysteine uptake during cell death induced by parkinsonian neurotoxins has also been suggested as contributing to GSH depletion (5) (Table 1 and Fig. 2).
Protein (S-)glutathionylation (PSSG, also known as [S-]glutathiolation) refers to the formation of a protein-mixed disulfide between the thiol group of GSH and a cysteine moiety of a protein. During cell death, increased PSSG has also been reported, which might also contribute to GSH depletion (1, 38, 57, 137, 201) (Table 1 and Fig. 2). GSH can also form other GSH derivatives on reaction with distinct ROS/RNS (Fig. 2). GSNO regulates apoptosis (72, 155, 174, 228), and a recent report suggests that released cytochrome C (Cyt C) during apoptosis has the ability to catalyze GSNO formation (17). GSNO is metabolized via the GSH-dependent formaldehyde dehydrogenase class III alcohol dehydrogenase, also known as GSNO reductase (GSNOR) (20). In thymus, GSNOR deficiency increases apoptosis, reducing the number of CD4 single-positive thymocytes (250).
GSH efflux also participates as a major contributor in the alterations of the cellular redox balance associated with cell death (Table 1 and Fig. 2). In addition, the formation of GSH-adducts by xenobiotics and electrophiles, and their subsequent extrusion by specific plasma membrane transporters, has also been reported to contribute to GSH depletion during apoptosis (23, 171, 185, 205, 244, 247) (Table 1 and Fig. 2). It is important to mention that multiple mechanisms are likely to participate in GSH loss during apoptosis (56, 61, 67). In the next section, we will review the mechanisms involved in GSH efflux during apoptosis.
Transport Mechanisms Involved in Plasma Membrane GSH Efflux During Cell Death
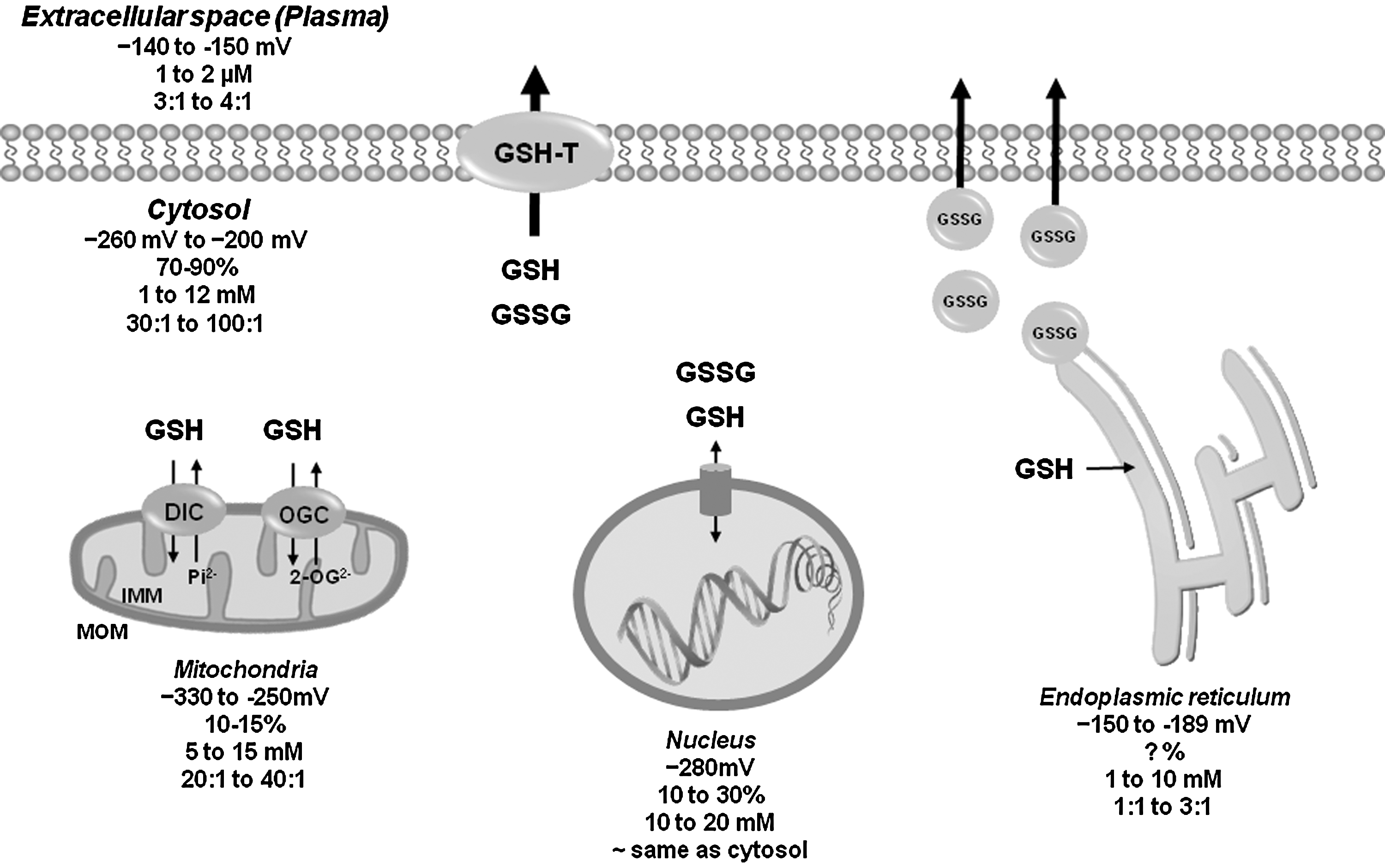
GSH is a ubiquitous tripeptide produced intracellularly that is not only 85%–90% freely distributed in the cytosol, but can also be found compartmentalized in mitochondria, peroxisomes, nuclear matrix, and endoplasmic reticulum (ER) (Table 1 and Fig. 3). Specific transport mechanisms have been evolved to maintain compartmentalized GSH/GSSG homeostasis. The concentration of mitochondrial GSH is similar to that of cytosol (10–14 mM). GSH can cross easily the outer mitochondrial membrane (OMM) through porin channels. A significant pool of GSH is compartmentalized in the mitochondria matrix by dicarboxylate carrier or 2-oxoglutarate transporters (OGC) [reviewed in this Forum and in Refs. (143, 166)] (Fig. 3). In contrast to mitochondria and cytosolic compartments, where GSH is predominantly found in its reduced form, in the ER, GSH exits mainly as GSSG acting as a source of oxidizing equivalents favoring disulfide bond formation for the proper folding of nascent proteins. Protein-dependent facilitated diffusion in the ER membrane is thought to mediate GSH permeation (Table 1 and Fig. 3). The ER is the initiating organelle of the secretory pathway, where secretory and membrane proteins are synthesized. In the cytosol, GSSG can be recycled back to GSH by GR or effluxed by specific transporters (discussed next). Since mitochondria lack a GSSG efflux mechanism, they rely on GR to counteract the pro-oxidant effects of GSSG. In contrast, the fate of the GSSG in the ER is unclear; it could be reduced within the ER by GR, transported to the cytosol for its reduction, or it could be secreted via the secretory pathway (6). Indeed, high levels of GSH have been found in secretory granules (112). GSSG extrusion through the secretory pathway can decrease GSH levels in the cell. However, if extracellular GSH is subject to recycle via the γ-glutamyl transpeptidase, its extrusion can promote cysteine recycling and de novo GSH synthesis. In this review, we focus only on the plasma membrane efflux mechanisms for GSH and GSSG and their role in cell death progression.
Apoptosis induced by distinct stimuli, particularly death receptors, has been reported to promote GSH depletion via the activation of a plasma membrane efflux transport (41, 67, 73, 90, 98, 106, 190, 220, 239). Inhibition of GSH depletion under these conditions rescues cells from apoptosis (73, 90, 96, 99). However, controversy still exists regarding the transport mechanism(s) involved in GSH depletion. A variety of protein transporters have been reported to act as GSH transporters (Table 1 and Fig. 4). Most studies to date have suggested that multidrug resistance proteins (MRPs) act as GSH efflux transporters during apoptosis (21, 67, 99, 106, 139). The human ATP-binding cassette (ABC) transporter, subfamily C (ABCC) subfamily of transporters contains 13 members from the ABC superfamily with sizes from 1325 to 1545 amino acids. The ABCC subfamily includes the cystic fibrosis transmembrane conductance regulator (CFTR, ABCC7), two sulfonylurea receptors SUR1 (ABCC8) and SUR2A/B (ABCC9), and nine MRPs. ABCC proteins are energy-dependent transporters, except for the CFTR that acts as a channel gated by ATP binding and hydrolysis, and SURs, which act as ATP-dependent potassium channel regulators. The MRP transporters have been demonstrated to act as cotransporters of organic anions (OA−) and GSH (12, 43). In addition, they also transport GSH-conjugated xenobiotics (GS-XN) and GSH-conjugated metabolites that must be exported to avoid deleterious effects. This efflux confers drug resistance to tumor cells and can protect normal cells from toxic insults. MRP1 functions as a GSH-conjugate transporter not only at the plasma membrane but also in intracellular secretory vesicles (240). The transport of organic anions, including drugs and conjugated OA−, by MRP, requires the hydrolysis of ATP (12, 43) (Table 1 and Fig. 4). Experimental conformation analysis has demonstrated that in solution, GSH is found as a mixture of different protonation states. Due to the presence of the two carboxylic acid groups, the thiol group, and the amino group, 16 different charged species of GSH with net charges ranging from +1 to −3 are found in solution. However, GSH has been found to possess a net negative charge of −1 at physiological pH (Fig. 1). Within GSH, the l-glutamic acid predominantly exists in its zwitterionic form, while the carboxyl group of the glycine fragment prefers to be deprotonated, and the cysteine moiety is in the neutral thiol form (142). Co-transport of two anions is an unusual mechanism. Thus, GSH transport by MRP1 transport might only target the GSH pool in a neutral or cationic state, which would explain the low affinity of MRP1 for GSH (see next). This pool could be slightly increased by acidification of the intracellular milieu during apoptosis (140). To date, there is no experimental evidence demonstrating that the co-transport of GSH and OA− molecules by MRP requires GSH to be in its anionic form. MRP1 can transport GSH alone, but this requires its stimulation by specific xenobiotics, for example, phenylalkylamines such as verapamil or bioflavonoids such as apigenin (43). Alterations in GSH levels reciprocally regulate MRP levels as shown by a recent report demonstrating that sustained GSH depletion prompts ubiquitin/proteosomal degradation of MRP2 (217).

Pharmacological activation of MRPs induces apoptosis by GSH depletion (139, 197, 232, 235). However, contradictory results have been reported regarding the role of MRP1 in GSH efflux during apoptosis. We previously demonstrated that pharmacological inhibition of MRP1 with MK571 (10–50 μM) and probenecid (250–1000 μM) stimulated rather than inhibited GSH depletion and apoptosis induced by Fas ligand (FasL) (73). Interestingly, some reports have demonstrated that in some cell types, inhibitors of MRP1-mediated drug transport stimulate GSH-efflux via MRP1 (45, 177). Similarly, the inhibition (6.5–50 μM MK571) and genetic knockdown of MRP1 stimulates anti-Fas- and tumor necrosis factor-alpha (TNF-α)-induced apoptosis in human epithelial cells (25). In contrast, Hammond et al. (99), using the same experimental model (Jurkat lymphoid cells), reported that the inhibition of MRP1 using high concentrations of MK571 (75 μM) and probenecid (7 mM) resulted in a significant reduction of GSH loss induced by either intrinsic or extrinsic pathways (99). Unfortunately, neither of these inhibitors are specific, especially at high concentrations. In their study, probenecid, an MRP1 blocker with poor selectivity, almost completely abolished GSH depletion and apoptosis, while MK571, a more selective MRP1 inhibitor (15, 60), only marginally reduced GSH loss (99). Furthermore, although the authors demonstrated that siRNA knockdown of MRP1 decreased GSH loss induced by Fas activation, the effect of MRP1 knockdown on apoptosis was not evaluated (99).
Several other factors likely also contribute to the contradictory results presented by Hammond et al. (99) and in our study (73). Although in both studies GSH depletion and its extracellular accumulation were determined using the GSH recycling assay, we also corroborated our results with flow cytometry analysis, which allows the discrimination between dead cells, cellular debris, and cells at distinct stages during the apoptotic program. These studies represent a more accurate discrimination between early GSH loss (before the loss of plasma membrane integrity) and passive GSH depletion after the plasma membrane integrity has been compromised. In addition, some differences might exist regarding the signaling pathway triggered by the Fas receptor. While we used the physiological ligand (FasL) (73), Hammond et al. triggered apoptosis using anti-Fas antibodies (99), which do not reliably mimic FasL (114, 211). Finally, in a follow-up study, the same group recently reported that overexpression of MRP1 protects rather than stimulates Fas-induced apoptosis, contradicting their own published results (164).
In addition to GSH, GSSG has also been shown to be detoxified by its efflux across the plasma membrane through MRP transporters (41, 62, 109, 110, 129, 151, 177, 182, 188), suggesting that MRPs might play a role in the cellular response to oxidative stress. In fact, MRP1 affinity for GSSG (Km ∼100 μM) is significantly higher than that for GSH (Km ∼5–10 mM), which explains its protective role during apoptosis, as the accumulation of GSSG has deleterious effects in cells (43). GSSG directly induces or sensitizes cells to apoptosis by activation of stress-activated protein kinases JNK (c-jun-n-terminal kinase) and p38 (68, 70). A recent study demonstrates that MRP1 activity in retinal pigment epithelial cells mediates both GSH and GSSG efflux upon oxidative stress and that its inhibition protects against oxidative damage by facilitating the intracellular reduction of GSSG and preventing GSH depletion (221). In sickle cell disease erythorcytes, an increase in GSSG efflux by MRP1 is linked to GSH depletion and oxidative stress (188). Other MRP proteins have also been reported to mediate GSH and GSSG efflux, including MRP2, 4, and 5 (13, 206), but their role in apoptosis has not been studied (Table 1).
Bi-directional GSH/OA− has been reported in different cell types, including human cell lines (86, 118, 150, 152, 153, 186, 229), and organic anion transporting polypeptides (OATP) have been proposed to mediate GSH efflux by a GSH/OA− exchange (Table 1 and Fig. 4). GSH efflux by OATPs is stimulated by the presence of a wide range of structurally unrelated OA− substrates (trans-stimulation), demonstrating the wide nonspecificity of the OA− binding site in the OATP proteins. GSH is present at high concentrations within the cells (>1 mM), whereas blood plasma concentrations are at least two orders of magnitude lower (<0.01 mM). Furthermore, since GSH is negatively charged at physiological pH, there is a large negative intracellular potential (−30 to −60 mV) that facilitates its extrusion from the cell (12, 97). Since GSH transport by OATPs is driven by the outwardly directed electrochemical gradient across the plasma membrane, it is reversed by increases in the extracellular GSH concentration, demonstrating its bidirectionality. OATPs were initially reported to mediate this exchange transport (97, 152, 153). However recent studies suggest that GSH/OA− exchange is not mediated by this family of transporters (12, 29, 160). We previously proposed a role for an OATP-like transporter in GSH depletion based on the observation that not only a variety of structurally unrelated OA− stimulate GSH depletion, but also that GSH loss was paralleled by an increased uptake of OA− in the absence of plasma membrane permeabilization. However, there remains a possibility that GSH efflux and OA− uptake are also mediated by different and still uncharacterized molecular entities (73).
The CFTR has been suggested to mediate the transport of GSH during apoptosis (125). Recently, staurosporine-induced apoptosis and GSH/GSSG depletion (138), as well as cigarette smoke-induced GSH efflux in the lung were associated with CFTR activity (94) (Table 1 and Fig. 4). More recently, another ABC transporter, the subfamily G member 2 (BCRP/ABCG2), was identified in human epithelial cells as a GSH efflux transporter, but its role in apoptosis remains to be studied (28). In addition, it has been recently demonstrated that, in Candida albicans, the ABC transporter Cdrp1 mediates GSH depletion and apoptosis. Cdrp1 protein sequence shows a higher similarity to human BCRP/ABCG2 than other ABC transporters [BCRP/ABCG2>p-glycoprotein (ABCB1)>MRP1 (ABCC1)]. However, whether GSH depletion mediated by Cdrp1 is via efflux of the reduced or conjugated form of GSH has not been determined (256) (Table 1 and Fig. 4).
Several other proteins are proposed to mediate GSH transport. The organic anion transporter 3 (OAT3) has been suggested to mediate renal GSH transport (144) (Table 1). RLIP76 (RALBP1) is a 76 kDa Ral-binding, Rho/Rac-GAP, and Ral effector protein that was proposed to be a multispecific transporter of xenobiotics as well as GSH-conjugates with inherent ATPase activity (10) (Table 1 and Fig. 4). Connexins and glutamate/aspartate transporters (GLAST) have also been suggested to mediate the efflux of GSH in excitable cells (87, 204, 224, 225) (Table 1 and Fig. 4). Finally, cell swelling is reported to induce GSH depletion (148). Since volume-regulated/volume-sensitive organic osmolyte-anion channels (VRAC/VSOAC) are activated during apoptosis (27) (Table 1), GSH depletion might be mediated by these efflux pathways driven by the electrochemical gradient of GSH across the plasma membrane. Accordingly, we have recently demonstrated that GSH depletion regulates cell shrinkage during apoptosis (apoptotic volume decrease) and activation of ion fluxes (75).
It is clear that further studies are required to elucidate the molecular identity(ies) of the transporter(s) mediating GSH efflux during apoptosis. However, the study of GSH depletion by its efflux is hampered by the lack of more sensitive and accessible approaches to determine accumulation of extracellular GSH, as the GSH recycling assay commonly used to measure GSH and GSSG levels might not be sensitive enough (low μM detection limit) to accurately determine their presence extracellularly. In vitro, the extracellular medium is infinitely bigger compared with the intracellular space, and this would result in profound dilution of GSH levels (202). More sensitive methods to detect GSH and GSSG based on high-performance liquid chromatography combined with mass spectrometry analysis could provide a better means for evaluating GSH accumulation in the extracellular milieu, but the application of these approaches is limited by their accessibility (180).
Redox Signaling, GSH Depletion, and Cell Death Progression
GSH content is a determinant of cell death progression. Several studies have demonstrated that high intracellular GSH levels are associated with apoptotic-resistant phenotypes in several models of apoptosis (33, 80), while by itself, GSH depletion either induces or stimulates apoptosis (4, 9, 165). Conversely, GSH supplementation prevents the apoptosis induced by distinct stimuli (32, 46, 73, 76, 133). GSH depletion induced by inhibition of the GCL potentiates death receptor-induced apoptosis in T-cells (9, 80), but by itself, it does not trigger cell death. However, this might be attributed to the observation that pharmacological inhibition of GCL depletes only the cytosolic GSH pool, having little effect on mitochondrial GSH (91, 241, 253). The precise contribution of cytosolic versus mitochondrial GSH pools in apoptosis is not fully understood, although some reports suggest that apoptosis correlates directly with cytosolic rather than with mitochondrial GSH depletion (245). In contrast, other studies have shown that mitochondrial GSH depletion is essential in triggering the cell death cascade [reviewed in this Forum and in Refs. (143, 166)]. Another explanation to why GSH depletion might not induce cell death in some cell types is given by reports demonstrating that prolonged GSH depletion up-regulates antiapoptotic proteins such as B-cell lymphoma 2 (Bcl-2), heat shock proteins, and nuclear factor-kappa B (NF-κB) (47, 69, 236), as well as other antioxidant systems, including the nuclear factor (erythroid-derived 2)-like 2 (Nrf2) and heme oxygenase-1 that might inhibit cell death progression (41, 81, 108, 147, 183). Interestingly, excessive GSH overload has also recently been shown to mediate mitochondrial toxicity and cell death by reductive stress (254).
The signaling pathways that regulate the progression of apoptosis have been extensively studied and characterized (Fig. 5). Induction of apoptosis via the extrinsic pathway is triggered by the activation of the death receptors Fas (CD95/Apo-1), TNF-related apoptosis-inducing ligand (TRAIL) receptors 1 and 2 (DR4/DR5), and TNF receptor 1 (TNFR1) by their respective ligands FasL, TRAIL, and TNF-α. Activation of death receptors leads to the formation of the death-inducing signaling complex, which includes the Fas-associated death domain (FADD), initiator caspase 8 or 10, and the cellular FADD-like interleukin-1 beta-converting enzyme (FLICE)-inhibitory protein (FLIP). In contrast, TNFR1 signaling results in the formation of two signaling complexes. TNF-induced complex I formation lacks FADD and pro-caspase 8, but induces the recruitment of the receptor-interacting protein (RIP), TNFR-associated death domain protein (TRADD), and TNFR-associated factor (TRAF)-1/2, which translocate to the cytosol where FADD, caspase 8/10, and FLIP are recruited to form the traddosome or complex II, leading to the activation of initiator caspases (145). Activation of NF-κB antagonizes programmed cell death induced by TNFR1, and GSH depletion has been shown to down-regulate TNF-induced NF-κB activation and sensitize hepatocytes to apoptotic cell death (157).
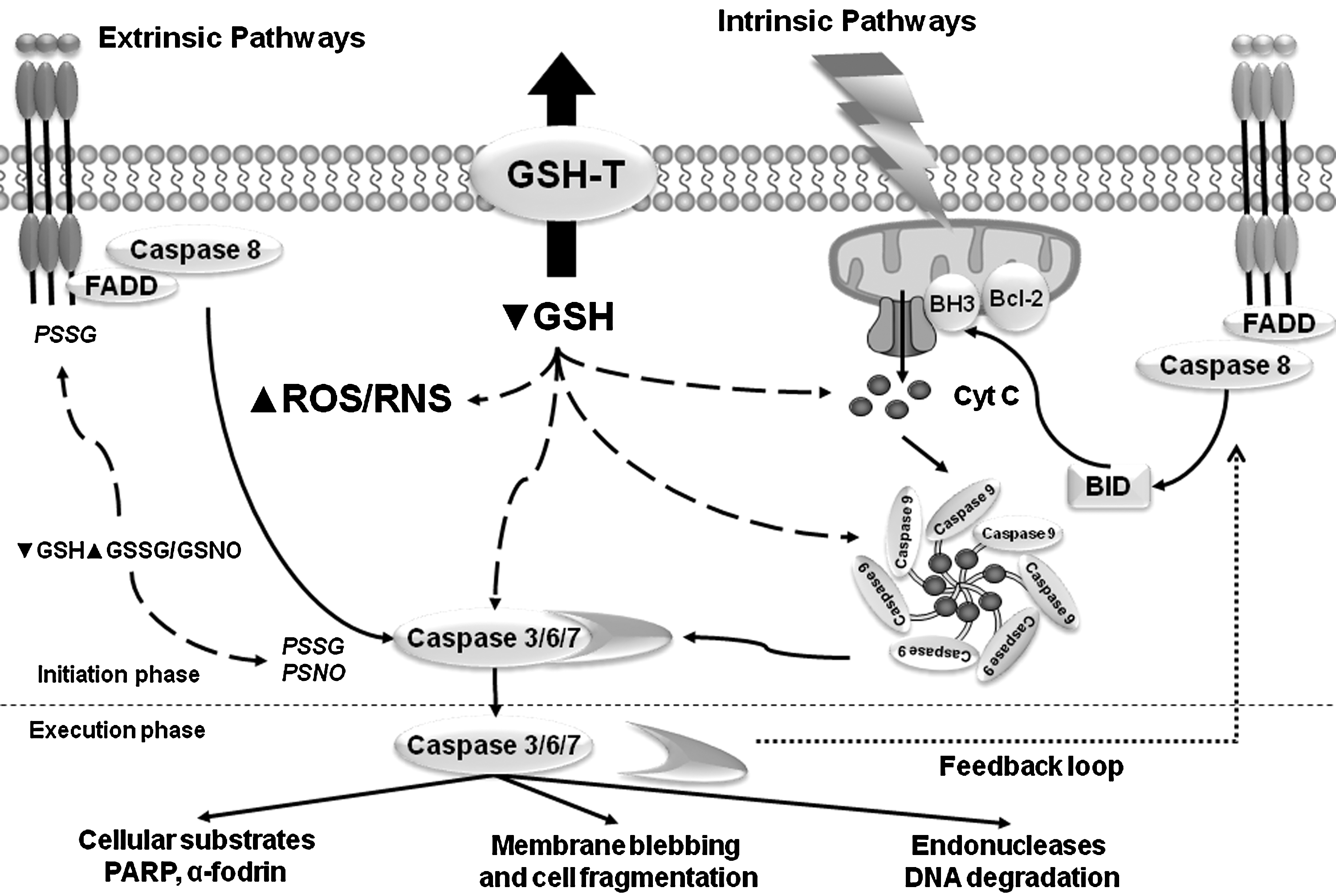
The extrinsic/death receptor pathway has the ability to crosstalk to the intrinsic pathway of apoptosis by an amplification loop induced by caspase-dependent cleavage of the Bcl-2-family protein BH3 (Bcl-2 homology 3) interacting-domain death agonist (Bid), which translocates to the mitochondria and promotes the release of Cyt C. The intrinsic pathway of apoptosis is activated by a wide variety of stimuli, including chemotherapeutic/cytotoxic agents (environmental pollutants, xenobiotics, and drugs), stress (radiation, hyperglycemia, hypoxia, oxidative and osmotic stress), and cytokine withdrawal. Activation of the mitochondria pathway mediates the release of Cyt C that is regulated by the Bcl-2 protein family. The BH3-only Bcl-2 family members Bcl-2-associated death promoter (Bad), Bid, Bcl-2-like protein 11 (Bim), NOXA, and p53 upregulated modulator of apoptosis (PUMA) regulate the antiapoptotic Bcl-2 proteins Bcl-2 and Bcl-xl (B-cell lymphoma-extra large) to promote apoptosis. Bcl-2 and Bcl-xl inhibit Bcl-2 associated X protein (Bax) and Bcl-2 homologous antagonist/killer (Bak), and activation of BH3-only proteins derepresses Bax and Bak by direct inhibition of Bcl-2 and Bcl-xl. Bax and Bak are crucial for inducing the permeabilization of the OMM and the release of Cyt C. Subsequently, released Cyt C leads to the recruitment of Apaf1 into an apoptosome and activates caspase 9 (37).
GSH depletion is necessary for the formation of the apoptosome (210) and also triggers cell death by modulation of the permeability transition pore of the mitochondria and the activation of executioner caspases (3, 8, 42, 189, 238, 241) (Fig. 5). In addition, GSH depletion activates the intrinsic apoptotic pathway initiator Bax and Cyt C release (49, 96) (Fig. 5). Released Cyt C requires cytosolic GSH levels to be depleted for its pro-apoptotic action (31, 89, 103, 194). Depletion of intracellular GSH also overcomes Bcl-2-mediated resistance to apoptosis (8, 208). The antiapoptotic role of Bcl-2 has been linked to GSH content by several studies, where it was reported that Bcl-2 regulates GSH content and distribution in different cellular compartments (121, 126, 242). Bcl-2 overexpression also reduces GSH efflux, but the mechanism involved remains unclear (191, 192). A recent study suggests that Bcl-2 regulates mitochondrial GSH content by a direct interaction of the BH3 groove with GSH (257), while the antiapoptotic effect of Bcl-xl has also been attributed to the regulation of GSH homeostasis by preventing GSH loss (26). However, these effects appear to be cell-type specific and context -dependent (175, 214, 231).
GSH depletion might also be a prerequisite for oxidative stress and the activation of cell death pathways. By itself, GSH depletion promotes nistrosative stress and cell death, suggesting an important role of basal GSH levels in the maintenance of a homeostatic reductive environment and the buffering of ROS/RNS (7). GSH depletion occurs at earlier stages of the cell death program and is followed by a delayed accumulation of ROS, which requires GSH depletion (48, 76, 139). GSH depletion by its efflux has been shown to be independent from oxidative stress and ROS generation (76, 96). We and others have recently shown that GSH depletion is necessary for the generation of ROS during FasL-induced apoptosis (76, 139, 156), and that GSH content, but not the excess in ROS formation and oxidative stress, regulates apoptosis induced by Fas activation (76) (Fig. 5). Other studies have also shown that apoptosis seems to be actively regulated by GSH content and not by excessive oxidative stress and ROS generation (53, 101, 198). The role of ROS/RNS in apoptosis has been extensively studied (40, 209), and several GSH-dependent antioxidant enzymes protect cells from undergoing programmed cell death. However, protective effects of thiol compounds on apoptosis in the absence of excessive ROS formation are also observed (53, 102). Ceramide accumulation is induced by different pro-apoptotic signals, including Fas ligation, irradiation, and anticancer drugs. A recent report shows that GSH depletion independent of ROS mediates ceramide generation and apoptosis by inhibition of sphingomyelin synthase, which converts ceramide to sphingomyelin (134).
GSH catalytically detoxifies cells from peroxides such as H2O2, OONO−, and lipid peroxides (LOO•) by the action of GSH peroxidases (GPx), leading to the accumulation of GSSG (Figs. 2 and 6). The accumulation of GSSG upon oxidative stress has been observed to be toxic to the cell (68, 70). GPx has been shown to protect against apoptosis induced by Fas activation (92). However, death receptor- (Fas and TNF) induced cell death was shown to be similar in animals deficient in GPx compared with WT (11). GPx also protects against apoptosis induced by oxidative stress (127), ischemia/reperfusion injury (44), and doxorubicin (93), and reduces pro-apoptotic Bax expression (65). Phospholipid hydroperoxide glutathione peroxidase (PHGPx or GPx4) directly reduces phospholipid hydroperoxides. GPx4 overexpression has also been reported to protect against oxidative-stress induced apoptosis by preventing cardiolipin oxidation and Cyt C oxidation (154, 203), while its down-regulation induces apoptosis-inducing factor (AIF)-mediated cell death (216). Overexpression of the mitochondrial GPx4 was also shown to protect against apoptosis induced by the intrinsic mitochondrial pathway by reducing mitochondrial hydroperoxide accumulation (187).
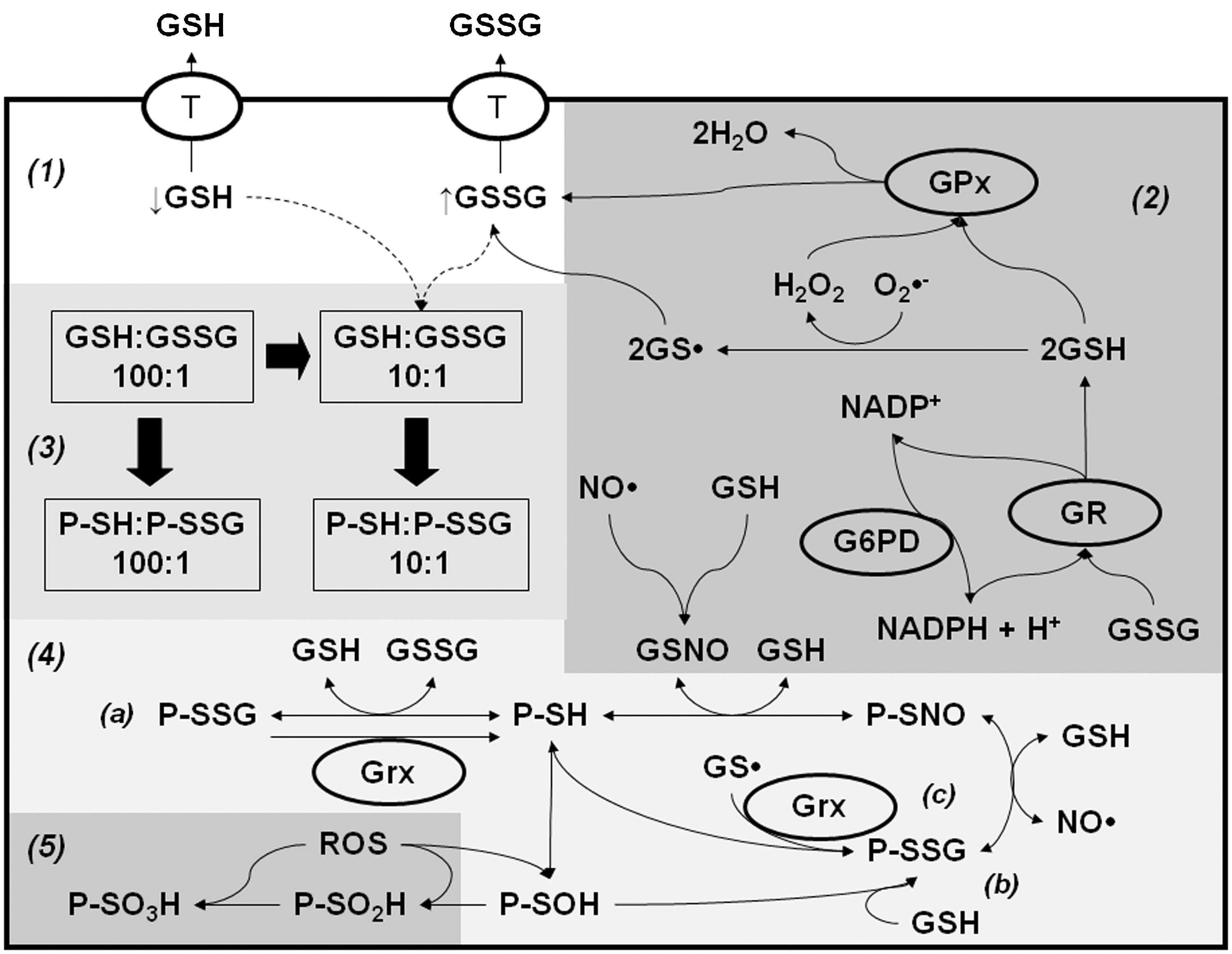
As indicated previously, another fate of GSH during oxidative stress and apoptosis is the formation of mixed disulfides with protein cysteines or PSSG. Since this subject is also reviewed in detail in this Forum, we will only briefly describe some major findings in this area. GSH depletion induced by oxidative stress, or by its active efflux across the plasma membrane, exerts prefunds alterations in the GSH/GSSG redox balance that might regulate PSSG levels (Fig. 6). Both GSSG and GSH can induce PSSG formation, depending on the oxidized/reduced status of the cysteine residue and the redox potential of the protein. Apoptosis is accompanied by increased PSSG formation (1, 57, 227). TNF-α-induced apoptosis is reported to be paralleled by increased PSSG formation, which is inhibited by overexpression of Bcl-2 (227). Loss or suppression of NF-κB enhances sensitivity to apoptosis. Glutathionylation of NF-κB inhibits its DNA-binding capacity and enhances apoptosis induced by hypoxic conditions (201). FasL-induced apoptosis has also been reported to increase PSSG, which amplifies the apoptotic signaling cascade by glutathionylation of the Fas receptor (1) (Fig. 5). In contrast, caspases can be glutathionylated under basal conditions and become de-glutathionylated upon the induction of apoptosis (193) (Fig. 5). GSSG is commonly viewed as a byproduct of GSH metabolism, which is either recycled to GSH o or exported out of the cell (Fig. 6). However, pathophysiological significance of GSSG per se remains poorly studied. An early and transient rise in intracellular GSSG has been shown to precede Cyt C release and caspase 3 activation (39, 199). Interestingly, GSSG-induced caspase 3 glutathionylation inhibits its enzyme activity (117). A recent report shows that GSSG-induced toxicity is mediated by 12-lipoxygenase (12-LOX) activation via its glutathionylation (195). GSH depletion and GPx4 down-regulation induce cell death by the activation of 12-LOX (35, 146, 216, 243).
PSSG reductases glutaredoxins (Grxs) have been demonstrated to protect against apoptosis by decreasing PSSG formation. In contrast, knockdown of Grx1 significantly inhibits TNF-α-induced cell death via increased glutathionylation of caspase 3 and impaired activation of the enzyme (111, 193). GSNO is a well-known inducer of protein nitros(yl)ation (PSNO) (Fig. 6), which regulates apoptosis (161, 162, 173). Caspases have been shown to be nitrosylated under basal conditions, and their de-nitros(yl)ation is required for their activation during apoptosis (132, 163, 178). In addition, several other proteins whose signal transduction cascades modulate apoptosis have been demonstrated to be regulated by nitros(yl)ation including Bcl-2 and FLIP (34, 59).
Most of the evidence regarding the role of GSH in the activation of cell death pathways refers to apoptotic signaling cascades. However, recent reports also suggest a protective role of GSH in cell death processes other than apoptosis. For example, N-acetyl-L-cysteine (NAC) has been shown to prevent ROS-induced formation of autophagosomes and the subsequent degradation of proteins during starvation-induced autophagy (213). Lipopolysaccharide-induced autophagy is paralleled by ROS formation and GSH depletion, which was also prevented by NAC (251). Treatment with γ-glutamylcysteinyl ethyl ester, a precursor of de novo GSH formation, decreases autophagy after traumatic brain injury (141). Excessive GSH depletion and oxidative stress have been reported to switch apoptosis to necrotic cell death (66, 158, 233, 234). GSH-depleting agents at doses that decrease mitochondrial GSH levels induce necrosis. However, modest doses of these agents resulting in selective cytoplasmic GSH depletion sensitize hepatocytes to TNF-α -induced apoptosis (100, 168, 184). Ceramide has been implicated as a secondary messenger for TNF-α-induced cell necrosis, and NAC or GSH-monoethylester can delay the onset of ceramide-induced necrosis (52). Recently, necrostatin-1, an inhibitor of programmed cell necrosis or necroptosis, was shown to inhibit cell death in mouse hippocampal cells induced by GSH depletion (248).
Conclusions and Perspectives
GSH depletion has been observed to occur at early stages during the cell death progression. Although GSH depletion was initially associated mainly to its oxidation by ROS/RNS generated during oxidative stress, it is now recognized that GSH depletion occurs by a variety of distinct mechanisms. GSH depletion by its efflux has been described as an active process that in many cases is independent from oxidative stress and precedes ROS accumulation. More importantly, GSH depletion has also been demonstrated to directly regulate the cell death machinery independently from ROS accumulation and oxidative damage. Several protein transport mechanisms have been proposed to mediate GSH efflux, but controversy still exists regarding its role in GSH depletion during apoptosis. The understanding and identification of GSH tranpsorters involved in GSH depletion is hampered by the lack of sensitive and accessible approaches to determine extracellular GSH accumulation. More research is necessary to accurately determine the transporter or transporter entities regulating GSH depletion during cell death, and the signaling mechanisms regulating/activating them.
Footnotes
Acknowledgments
This work was supported by the Intramural Research Program of the NIH/National Institute of Environmental Health Sciences 1Z01ES090079 (J.A.C.), P20RR17675 Centers of Biomedical Research Excellence (COBRE), and an Interdisciplinary Grant from the Research Council and the Life Sciences Grant Program of the University of Nebraska-Lincoln (R.F).