
Abstract
Introduction
Pathologic characteristics of pulmonary hypertension include vasoconstriction within the pulmonary vasculature as well as histological abnormalities of the vascular wall, with the latter including medial hypertrophy, intimal proliferation, fibrosis, adventitial thickening, thrombotic lesions, and inflammatory infiltrates. In advanced stages of pulmonary arterial hypertension, all of these characteristics are present in the plexiform lesions. The pathogenic mechanism of pulmonary hypertension involves various pathways that lead to vasoconstriction of pulmonary arteries and increase in pulmonary vascular resistance due in part to the abnormal production of vasoactive compounds caused by endothelial dysfunction. Increased vasoconstriction is also related to the abnormal expression and activity of potassium channels in pulmonary vascular smooth muscle cells. Endothelial dysfunction increases vasoconstrictive and proliferative substances such as endothelin-1 and thromboxane A2. Also, levels of factors with vasodilatory and antiproliferative properties, such as nitric oxide and prostacyclin, are decreased. These abnormalities increase the vascular muscle tone and promote vascular thickening. The increased pulmonary arterial pressure and resistance strains the right ventricle, leading to right ventricular hypertrophy and ultimately to heart failure and death.
Evidence for the Occurrence of Oxidative Stress in Human Pulmonary Hypertension
Studies providing evidence of increased oxidative status in patients with pulmonary hypertension are summarized in Figure 1. Cracowski et al. (6) reported that urine F2 isoprostane levels were 2.3 times higher in patients with pulmonary hypertension compared to healthy control subjects, measured using gas chromatography/mass spectrometry. Of patients in this study, 48% had idiopathic pulmonary arterial hypertension and 52% had secondary pulmonary hypertension. Similarly, Irodova et al. (18) reported higher concentrations of plasma malondialdehyde in patients with idiopathic pulmonary arterial hypertension than in healthy volunteers. In addition, a mass spectrometry study revealed that, compared to individuals without pulmonary hypertension, plasmas from patients with idiopathic pulmonary arterial hypertension and patients with sickle cell anemia who had pulmonary hypertension contained more pronounced malondialdehyde-modified albumin at the amino acid residue lysine 159 (31). These studies collectively demonstrate that pulmonary hypertension is associated with increased lipid peroxidation. In addition to lipid peroxidation, oxidation states of other biological molecules are reportedly higher in patients with pulmonary hypertension. Immunohistochemical studies by Bowers et al. (3) demonstrate that lungs from patients with idiopathic pulmonary arterial hypertension had intense 8-hydroxyguanosine staining in the plexiform lesions and in the luminal endothelial cells of concentric intima fibrosis lesions, indicating the occurrence of DNA oxidation. These authors also reported that lungs from patients with pulmonary hypertension had positive staining for nitrotyrosine. The occurrence of protein oxidation was also demonstrated in our laboratory by measuring the plasma protein carbonyl content using an immunological technique. We find that patients with idiopathic pulmonary arterial hypertension have a higher plasma protein carbonyl content compared to healthy subjects (51). Using high-performance liquid chromatography, our study also shows that, compared to control subjects, patients with idiopathic pulmonary arterial hypertension have reduced levels of lipophilic antioxidants in plasma, including α-tocopherol and β-carotene (33).
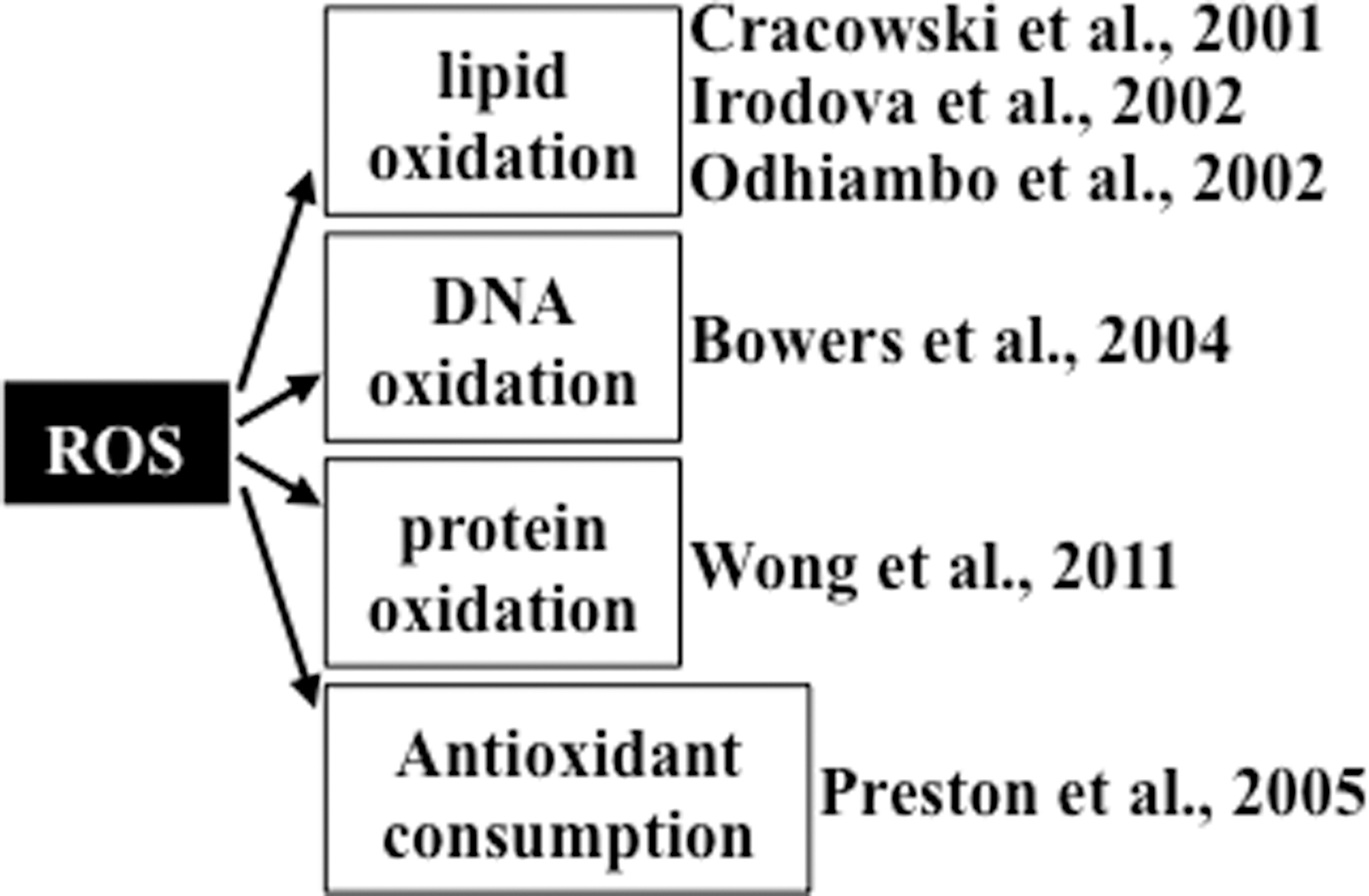
Evidence for Beneficial Effects of Antioxidants in Animal Models of Pulmonary Hypertension
In animal models, various compounds with antioxidant properties have been shown to suppress the progression of pulmonary hypertension (Fig. 2). In rats, compounds with antioxidant activities have been found to inhibit pulmonary hypertension and/or right ventricular dysfunction. Such compounds include probucol (19), N-acetylcysteine (16, 23), tempol (9, 20), erdosteine (46), allicin (43), pyrrolidine dithiocarbamate (17, 39), superoxide dismutase (22), allopurinol (20), sulfur dioxide (21), resveratrol (7, 52), and EUK-134 (35). In neonatal lambs with persistent pulmonary hypertension, superoxide dismutase was found to improve oxygenation, reduce oxidation (24), restore endothelial nitric oxide synthase expression and function (10), and normalize phosphodiesterase 5 (11).

Role of Reactive Oxygen Species in Cell Signaling for the Pathogenesis of Pulmonary Hypertension
Accumulating evidence suggests that reactive oxygen species (ROS) can regulate cell signaling, with NAD(P)H oxidase serving as the major source of ROS in such events (44). The role of NAD(P)H oxidase in the pathogenesis of pulmonary hypertension has been documented in animal models of pulmonary hypertension, including fetal lambs (4, 13), newborn piglets (8), and mice (30). These results suggest that NAD(P)H oxidase and components of downstream ROS signaling are possible therapeutic targets for the treatment of pulmonary hypertension.
In pulmonary artery smooth muscle cells, serotonin (26) and endothelin-1 (47) activate the production of ROS via NAD(P)H oxidase. The GATA4 transcription factor plays an important role in the regulation of growth of pulmonary artery smooth muscle cells, and antioxidants inhibit serotonin-induced GATA4 phosphorylation and activation (45). The serotonin signal for the nuclear translocation of extracellular signal-regulated kinase (ERK) and subsequent GATA4 phosphorylation is dependent on the activation of RhoA and Rho kinase (27). In response to serotonin, ERK has also been shown to activate GATA4 via monoamine oxidase-A-dependent production of hydrogen peroxide (H2O2), which promotes the translocation of phosphorylated ERK to the nucleus (25). This could operate through RhoA signaling, as our laboratory recently found that Rho guanine nucleotide dissociation inhibitor (RhoGDI), which functions to prevent the exchange of guanosine-5′-triphosphate (GTP) for guanosine-5′-diphosphate on the Rho-GTPases by guanine nucleotide exchange factors, is carbonylated in response to serotonin (Wong & Suzuki, unpublished results). Thus, carbonylation of RhoGDI may trigger RhoA/Rho kinase activation, leading to the nuclear translocation of ERK and GATA4 phosphorylation. Carbonylation of RhoGDI may also be a mechanism for ROS-dependent stimulation of RhoA as described by Resta and coworkers (5). Based on these studies, Figure 3 summarizes proposed ROS-dependent cell-signaling pathways for GATA4 activation by serotonin in pulmonary artery smooth muscle cells. Endothelin-1 also produces ROS in pulmonary artery smooth muscle cells via NAD(P)H oxidase, and antioxidants block endothelin-1-induced cell proliferation (47).

Mechanisms of ROS Signaling
While, as described above, the roles of NAD(P)H oxidase and ROS in the development of pulmonary hypertension have been supported by a number of published studies, the exact mechanisms of ROS actions are not well understood. Our laboratory provided a series of evidence that iron-dependent hydroxyl radical (HO
Despite the major and active role played by HO
Protein carbonylation is a process in which reactive aldehydes or ketones are introduced into proteins by iron-dependent biological oxidation; the resultant product can react with 2,4-dinitrophenylhydrazine or other hydrazine derivatives to form hydrazones. Protein carbonylation has been described as occurring largely on four susceptible amino-acid residues: arginine, proline, lysine, and threonine (2). The oxidation of proline or arginine side chains produces glutamic semialdehyde, and lysine oxidation forms aminoadipic semialdehyde, introducing carbonyl groups into the protein structure (36). Oxidation also converts the hydroxyl group of threonine side chains to a carbonyl group, 2-amino-3-ketobutyric acid (Fig. 4). Reportedly, these amino acids are susceptible to metal-catalyzed oxidation based on the characterizations of isolated proteins and of amino acid homopolymers that are subjected to oxidative stress (1). In addition, other amino acid residues, such as serine, leucine, valine, isoleucine, and alanine, are also capable of being carbonylated as shown in Figure 4 (14, 42).

We find that ligand-receptor stimulation in response to the treatment of pulmonary artery smooth muscle cells with endothelin-1 or serotonin, which are important mediators of pulmonary hypertension and pulmonary vascular remodeling, promotes carbonylation of various proteins (49). Two-dimensional gel electrophoresis and mass spectrometry identified that the carbonylated proteins include annexin A1, which functions as an anti-inflammatory, antiproliferative, and proapoptotic factor. We also find that annexin A1 is degraded by proteasomes subsequent to being carbonylated. Interestingly, in contrast to the situation of strenuous oxidative stress, in which most or all of carbonylated proteins are degraded, under the oxidant signaling condition, only small portions of carbonylated proteins are degraded (49). Thus, we propose that oxidant signaling triggers specific proteolysis of carbonylated proteins and that this defines the specificity and selectivity of signal transduction. Annexin A1 is one such protein that is specifically degraded under oxidant signaling conditions, so the degradation of annexin A1 should promote proinflammatory, proliferative, and antiapoptotic signaling. This “carbonylation-degradation pathway of signal transduction” may underlie the molecular mechanism of ROS signaling for the growth of pulmonary artery smooth muscle cells and pulmonary vascular remodeling (49). Thus, the inhibition of protein carbonylation may be a novel therapeutic strategy for preventing the development and progression of pulmonary hypertension.
ROS Signaling for Right Ventricular Hypertrophy
Pulmonary hypertension strains the right ventricle, initially causing compensatory concentric right ventricular hypertrophy, which in turn leads to right-sided heart failure and death. The transcription factor GATA4 plays an important role in the development of cardiac hypertrophy (32). Pressure overload exerted on the right ventricle by pulmonary artery banding or hypoxic pulmonary hypertension can increase GATA4 gene expression. Our laboratory identified the transcriptional start site of the Gata4 gene, finding that the 250-bp region that is proximal to the transcriptional start site is important for regulating the gene transcription of Gata4. This region contains the functionally important CCAAT box, where the CCAAT-binding factor/nuclear factor-Y (CBF/NF-Y) transcription factor binds (32). In the left ventricle, pressure overload by aortic banding activates GATA4 via post-translational modifications, while pulmonary artery banding increases GATA4 gene expression in the right ventricle, suggesting that the right ventricle specifically possesses a transcriptional mechanism for GATA4 activation (32). Using the hypoxic pulmonary hypertension model of right ventricular hypertrophy, we find that CBF/NF-Y activation occurs through the liberation from a complex with the annexin A1 protein via ROS-mediated protein carbonylation and proteasomal degradation of annexin A1, as summarized in Figure 5 (32). We find that the right ventricle has a higher ratio of CBF/NF-Y to annexin A1, compared to the left ventricle, making for more efficient CBF/NF-Y activation upon annexin A1 degradation (32).
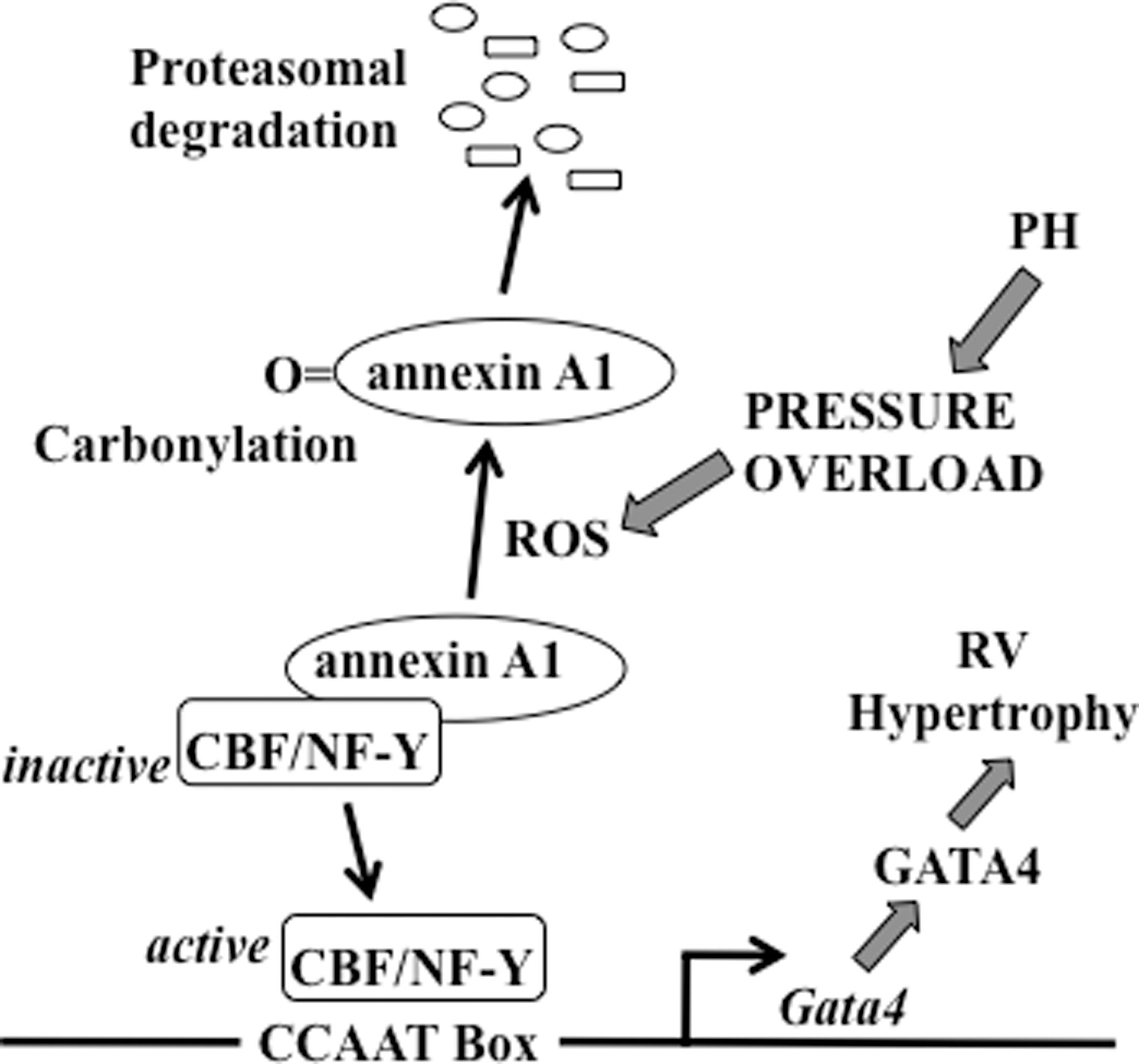
We also reported that treating perfused isolated rat hearts with serotonin promoted protein carbonylation, specifically in the right ventricle, but not in the left ventricle (28). We propose that these differential responses to serotonin between the right and left ventricles are defined by the low expression of monoamine oxidase A in the right ventricle compared to the left ventricle, which preserves the cytosolic level of serotonin and the ability of serotonin to produce superoxide via NAD(P)H oxidase, as described previously by Fanburg and coworkers (26). Since patients with pulmonary arterial hypertension have high levels of plasma serotonin (15), the ROS signaling mechanism triggered by serotonin may also play a role in the development of right ventricular hypertrophy.
These results reveal that ROS signaling regulates pulmonary hypertension-induced right ventricular hypertrophy and right heart failure. Consistent with this idea, right-sided heart failure due to monocrotaline-induced pulmonary hypertension was found to have increased ROS production (34), and the treatment of rats with the antioxidant EUK-134 attenuated pulmonary hypertension-induced right heart failure (35). Thus, antioxidant-based therapies to prevent and/or treat pulmonary hypertension-induced heart failure may be possible. A more precise understanding of the mechanisms of ROS actions should help to optimize such therapeutic strategies.
Role of Iron in Pulmonary Hypertension
As described above, accumulating evidence suggests that ROS plays a role in the progression of pulmonary hypertension. Since iron plays a vital role in ROS biology, iron chelation may inhibit pulmonary hypertension. This concept is supported by experiments from our laboratory demonstrating that deferoxamine (an iron chelator) can inhibit pulmonary vascular remodeling induced by chronic hypoxia (50). However, recent human studies reported that 43%–63% of patients with idiopathic pulmonary arterial hypertension are iron-deficient (37, 38, 41). Soon et al. (41) reported that, in their sample, 50% of premenopausal women with idiopathic pulmonary arterial hypertension were iron-deficient, while only 8% of premonopausal women with chronic thromboembolic pulmonary hypertension, 14% of postmenopausal women with idiopathic pulmonary arterial hypertension, and 28% of men with idiopathic pulmonary arterial hypertension were iron-deficient. Since these patients already had developed pulmonary hypertension, iron deficiency may not be related to the proposed roles played by iron and ROS in the mechanisms of the development and progression of the disease. However, it is unclear how lipid peroxidation could be increased (as described above) if these patients have reduced iron levels. Further work is needed to clarify this issue, which makes the design of antioxidant-based therapies less than straightforward.
Conclusions
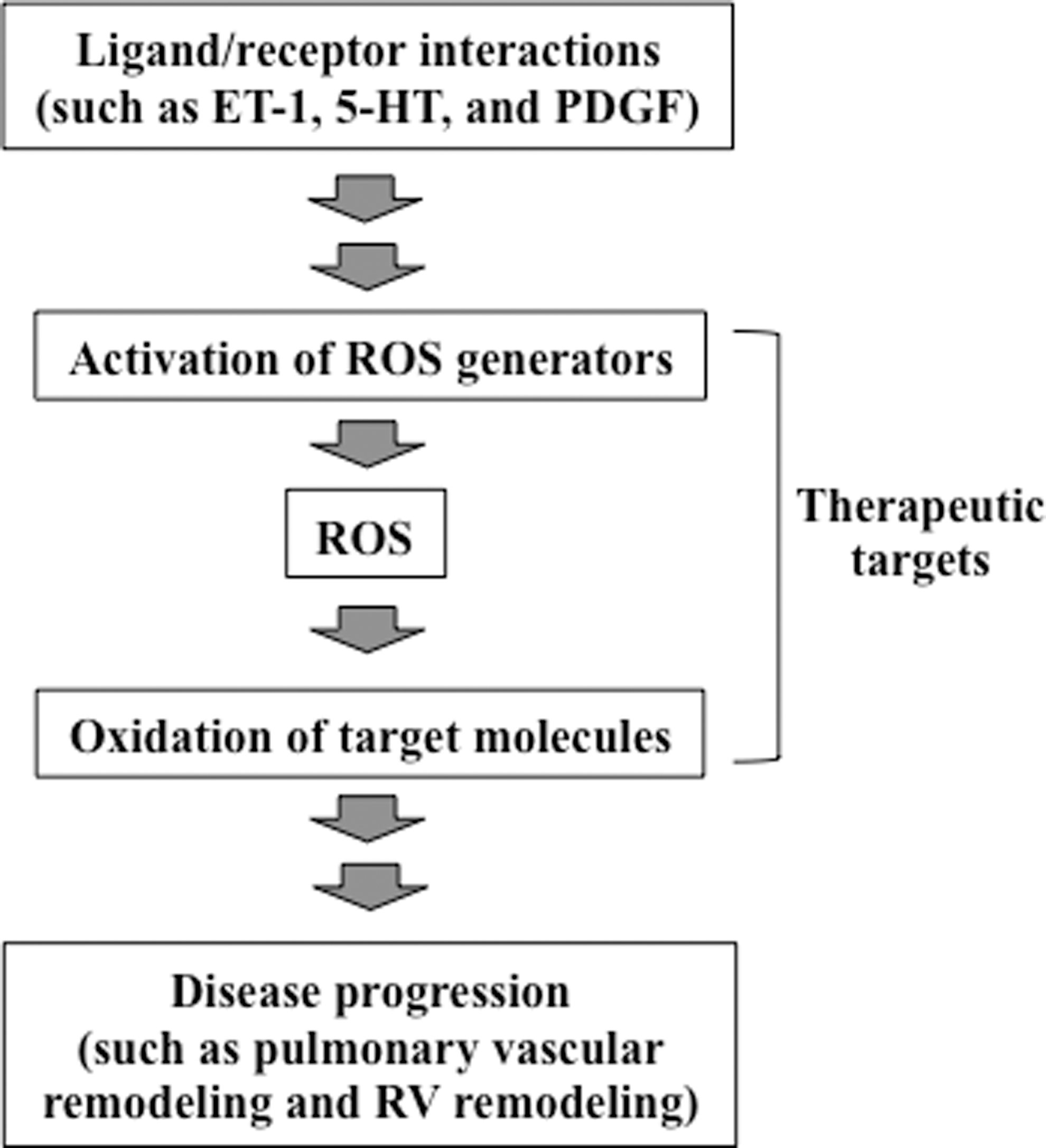
Using various experimental systems, including animal models of chronic hypoxia-induced pulmonary hypertension, many studies have shown that ROS are involved in the development and progression of pulmonary hypertension. ROS have also been implicated in the promotion of right ventricular hypertrophy and right heart failure in response to pulmonary hypertension. Human studies demonstrate that patients with idiopathic pulmonary arterial hypertension have increased oxidation status. Therefore, at least Group 1 (pulmonary arterial hypertension) and Group 3 (pulmonary hypertension due to lung diseases and/or hypoxemia) patients (12) may benefit from antioxidant-based therapies. However, it is unclear whether patients with pulmonary hypertension should receive antioxidants. For many diseases, clinical studies do not support the effectiveness of either pharmacological or dietary antioxidants in humans, despite the existence of ample evidence in experimental models for the importance of ROS. It is likely that we still do not understand the precise mechanisms of ROS actions; thus, we are not utilizing specific therapeutic strategies involving antioxidants that would eliminate unwanted ROS actions, while preserving the necessary functions of ROS. Understanding the molecular mechanisms of oxidant signaling should enable the specific, targeted use of antioxidants to effectively prevent and/or treat diseases, as depicted in Figure 6. In pulmonary hypertension, evidence suggests that ROS are second messengers for the growth of pulmonary artery smooth muscle cells during cell-signaling events that are triggered by mediators of pulmonary vascular remodeling and also for the development of right ventricular hypertrophy. NAD(P)H oxidase and monoamine oxidase-A have been shown to be generators of ROS during these cell-signaling processes. ROS likely react with protein thiols to elicit subsequent events. In addition, ROS may promote iron-dependent protein carbonylation as a means to send intracellular signals. The consequences of protein carbonylation may include the direct activation or inhibition of the activities of target molecules as well as the selective degradation by proteasomes. These studies of cell signaling also suggest that conditions of oxidative stress and ROS signaling may differ in terms of how biological processes operate and function. However, recent reports of some patients with idiopathic pulmonary arterial hypertension, particularly in premenopausal women (37, 38, 41), complicate the interpretation of the role of iron and ROS in pulmonary hypertension. Further study of the molecular mechanisms of ROS actions should contribute to the development of therapeutic strategies to treat pulmonary hypertension and right heart failure.
Footnotes
Acknowledgment
This work was supported in part by grants from National Institutes of Health (R01HL72844 and R01HL97514) to Y.J.S.