
Abstract
Introduction
Protein disulfide isomerase (PDI) is a chaperone member of the thioredoxin superfamily, located in the ER lumen, in the cytosol, and at the cell surface (9, 22). In addition to its chaperone activity, this multifunctional protein is an oxido-reductase that catalyzes the formation of disulfide bonds on newly formed proteins (22, 44). The presence of two cystein-rich thioredoxin-like catalytic domains present at the N- and C-terminal moiety (39), determines its redox potential (15). PDI is antiapoptotic and attenuates neuronal cell death resulting from ER stress induction (39). This protective effect may depend on its subcellular localization, but the precise function of the ER, cytosolic or cell-surface associated isoforms is only partly undestood (15). PDI is a target of S-nitrosylation and the resulting structural modifications are associated with a loss of its protective properties, with probable implication in neurodegenerative processes (39). In the cardiovascular field, PDI could protect cardiomyocytes of ischemic insult (34), but its possible role in atherosclerosis is not known.
OxLDL-induced modification of cellular proteins disturbs the expression and the regulation of pathways involved in cell homeostasis, such as the ubiquitin/proteasome system (40), and in cell signaling, such as the PDGFR pathway, which are progressively inhibited after prolonged contact with toxic oxLDL concentrations (5, 41). We recently reported that apoptosis of vascular cells induced by oxLDLs is associated with a sustained endoplasmic reticulum stress (ER stress) and unfolded protein response (UPR), characterized by a persistent phosphorylation of the ER stress sensor IRE1α involved in the c-Jun N-terminal kinase pathway, and an increased expression of the ER stress markers XBP1 (spliced form) and CHOP (25, 26). Recent advances indicate that ER stress and UPR are chronically activated in atherosclerotic lesions, which contributes to maintain inflammation, and lesional cell apoptosis, all of which promoting atherosclerosis progression (31). As a matter of fact, the proapoptotic factor CHOP is involved in ER stress-induced apoptosis (30), and its deletion in mice results in a reduction of atherosclerotic lesions and of necrotic areas, supporting the fact that prolonged ER stress within the vascular wall, is proapoptotic and proatherogenic (33).
As the accumulation of lipid oxidation products (26) and PDI modification (35, 39) are strong inducers of prolonged ER stress and apoptosis (24, 47), we hypothesized that PDI could be targeted and inhibited by carbonyl compounds, such as 4-HNE, present in oxLDLs and in atherosclerotic lesions (5, 18, 37), and that PDI inhibition may result in increased protein misfolding and sustained ER stress, thereby promoting apoptosis and progression of atherosclerotic lesions (31, 33).
The aim of this study was to investigate whether PDI is a target of oxLDLs and whether PDI modification plays a role in oxLDL-induced ER stress and apoptosis in vascular cells.
Results
OxLDLs inhibit PDI enzyme activity
In preliminary experiments, we observed that the activity of the antiapoptotic chaperone PDI was reduced in cells incubated with toxic concentration of oxLDLs. Since i) PDI is an abundant ER-resident enzyme/chaperone that is involved in the folding of proteins, ii) oxLDLs trigger both ER stress and apoptosis (25, 26) in vascular cells, and iii) nitrosative stress elicits PDI inactivation and increases neuronal apoptosis (39), we aimed to investigate whether PDI alteration by oxLDLs play a role in oxLDL-induced ER stress and apoptosis.
In HMEC-1 incubated with oxLDLs, PDI reductase activity determined with the fluorescent substrate Di-E-GSSG (di-eosin glutathione disulfide) (20) was altered in a time- and dose-dependent manner (Fig. 1A and Supplementary Fig. 1; supplementary data are available online at
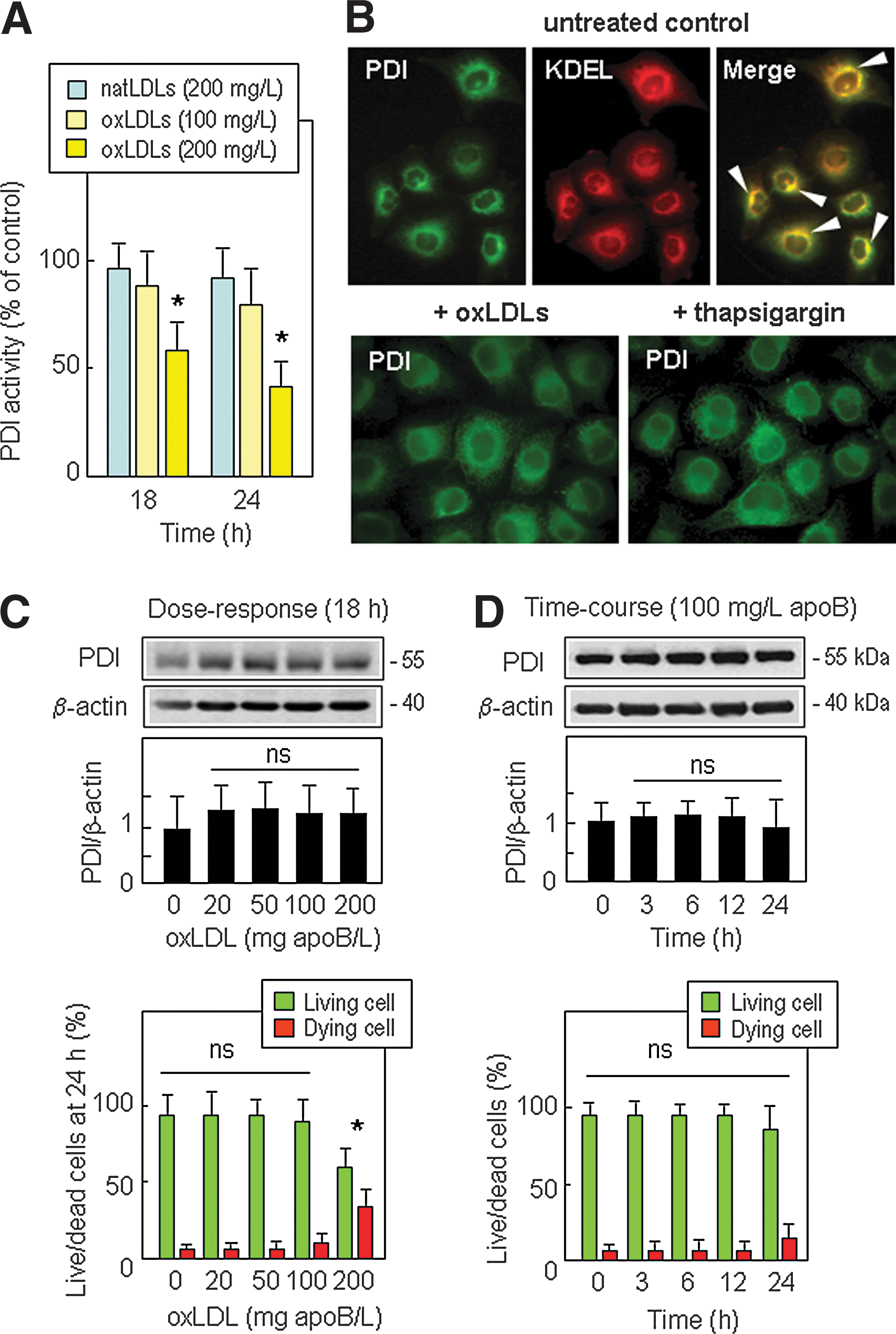
As PDI expression can be regulated by several stresses (27, 39), we checked whether the increase or loss of PDI activity in cells treated with oxLDLs was associated with changes in the PDI protein level or changes in subcellular distribution of PDI. In resting HMEC-1, immunofluorescence staining showed that PDI was mainly co-localized with KDEL motive-bearing proteins (Fig.1B, upper panel), in agreement with the main subcellular location of PDI in the ER. Noteworthy, incubation of cells with oxLDLs (200 mg/L) or by thapsigargin (1 μmol/L) for 18 h induced no obvious change in the subcellular distribution of PDI (Fig. 1B, lower panel). Dose-responses (0–200 mg apoB/L for 18 h incubation) and time-course experiments (with 100 mg/L apoB/L), showed no significant change of PDI expression over the time of the experiment, in HMEC-1 (Fig. 1C and 1D) and in the macrophage-like cell line U937 (Supplementary Fig. S2). This suggests that PDI inhibition in cells treated by 200 mg/L oxLDLs (Fig. 1A) does not result from a decrease of PDI protein level (Fig. 1C and 1D).
Interestingly, the comparison between the time-course of PDI activity (Fig. 1A) and oxLDL-induced toxicity (Fig. 1C and 1D) showed that PDI inactivation occurred before cell death. This led us to investigate whether PDI inactivation is involved (or not) in the susceptibility of cells to the apoptotic effect of oxLDLs.
PDI inhibition by bacitracin increases both ER stress and apoptosis induced by oxLDLs
PDI is generally thought to be protective, because its upregulation during brain ischemia protects against neuronal apoptosis (32) and, reversely, PDI inactivation abrogates the protection against neuronal cell death triggered by ER stress (2, 39). However, a recent report showed that PDI inhibition in rat brain cells suppressed the toxicity of mutant huntingtin and Aβ amyloid peptides (7). This led us to investigate whether PDI inhibition plays a role in oxLDL toxicity. This question was addressed by examining whether the toxic effect of oxLDLs was affected when the activity of PDI was modulated, either reduced by inhibiting PDI by the classical PDI inhibitor bacitracin (13), or increased by transfection of a myc-tagged active PDI cDNA.
Interestingly, under the used conditions, PDI inhibition (Fig. 2A) by bacitracin was not associated to changes of ER stress markers levels (CHOP mRNA and sXBP1) (Fig. 2A–2C) and was not toxic per se (up to 10 μmol/L) in the absence of oxLDLs (Fig. 2D–2G). In contrast, bacitracin potentiated dramatically the ER stress (increased CHOP and sXBP1 mRNAs) and the toxic effect of oxLDLs, since oxLDLs (100 mg apoB/L) used alone were not toxic but became toxic in the presence of bacitracin (Fig. 2D and 2E). This synergistic effect was obvious with 10 μmol/L bacitracin and 100 mg/L oxLDLs that are highly toxic (MTT<50%) when they are incubated simultaneously with HMEC-1, whereas they are not or only slightly toxic (<10%) when each is incubated alone with the HMEC-1 (Fig. 2D). In the same way, bacitracin potentiated the toxic and apoptotic effects oxLDLs (200 mg apoB/L) in HMEC-1 (Fig. 2E–2G) and in U937 cells (Supplementary Fig. S3).

This mutual potentiation of the toxicity of oxLDLs and bacitracin may suggest that PDI inhibition by bacitracin enhances the toxic effect of oxLDLs, and, conversely, that the active PDI participates in the defense against oxLDL-induced toxicity. In order to test the hypothesis that active PDI may reduce both ER stress and toxicity elicited by oxLDLs, we used another approach utilizing HMEC-1 transduced and expressing an active myc-tagged wild-type PDI (myc-PDI wt) and an inactive myc-tagged mutated PDI cDNA (myc-PDI mut) (Fig. 3).
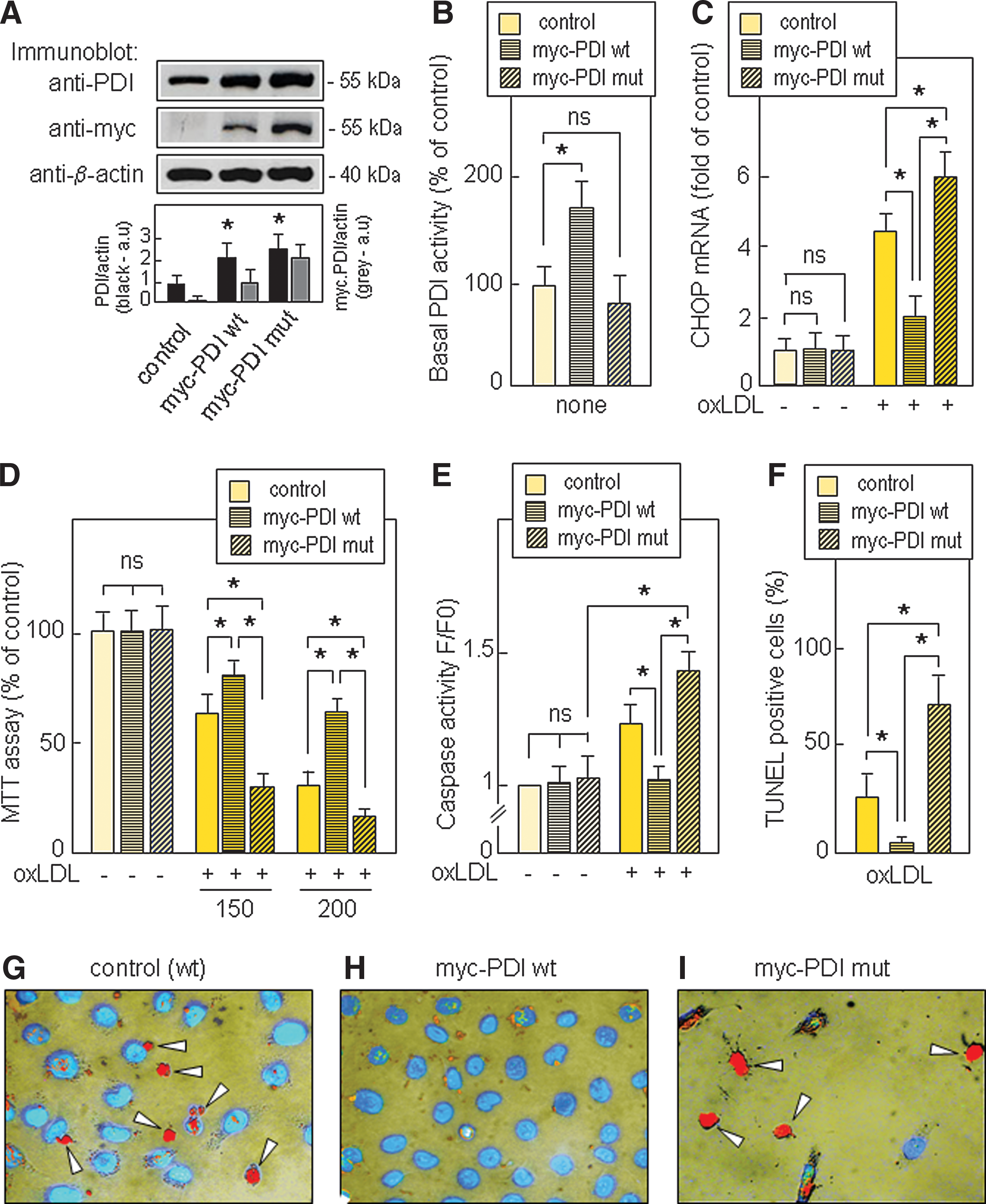
The expression of PDI in transfected cells was assessed by Western blot (Fig. 3A). The PDI reductase activity in myc-PDI wt HMEC-1 was about twice that of the parental HMEC-1, whereas in myc-PDI mut HMEC-1 it was in the same range as in control (untransfected) HMEC-1 (Fig.3B) and in mock-transfected HMEC-1 (data not shown). As expected, in myc-PDI mut HMEC-1, the expression of the inactive PDI does not prevent and even increases the toxicity of oxLDLs (Fig. 3D–3G), thus suggesting that the enzymatic activity of PDI is required for the protection. In contrast, the forced expression of the active PDI in myc-PDI wt HMEC-1 increased the PDI reductase activity, reduced CHOP expression and the toxicity of oxLDLs (Fig. 3D–3I and Supplementary Fig. S4). The data from transfection experiments are consistent with those obtained with bacitracin, and all these data contribute to support the hypothesis that active PDI participates in the cellular defenses against oxLDL-induced toxicity, in agreement with the protective role of PDI against neuronal apoptosis by nitrosative stress (39) and cardiomyocyte apoptosis during ischemia (34).
Interestingly, PDI overexpression prevented the toxic effect of two other ER stress inducers, thapsigargin (Supplementary Fig. S5) and tunicamycin (data not shown).
PDI inhibition by oxLDLs is associated with the formation of 4-HNE-PDI adducts
The data reported in Figure1 pointed out that toxic concentration of oxLDLs inhibited PDI activity, without altering protein expression. OxLDLs contain reactive carbonyl compounds able to react with thiol and free amino groups of proteins, thereby altering their conformation and/or their enzymatic function (18, 23, 38). Since the active sites of thioredoxin-like domains of PDI contain Cys-x-x-Cys motifs which are involved in the disulfide oxidoreductase activity (22, 39), we investigated whether oxLDLs might inhibit PDI via a post-translational modification by lipid peroxidation products.
In cells incubated with oxLDLs (200 mg apoB/L) or with 4-HNE (10 μmol/L), (one of the most abundant reactive carbonyl compound formed during the oxidation of LDLs or cellular lipids), we observed a modification of PDI protein, detected by immunoblot of PDI immunoprecipitates with an anti-4-HNE-Michael adduct antibody, and characterized by the formation of 4-HNE-adducts on high molecular weight PDI-reactive bands (Fig. 4A). Note that SDS-PAGE was performed with or without mercaptoethanol in order to avoid immunoglobulin interference to visualize the 55 kDa PDI and the modified/polymerized PDI. Western blots labeled with the anti-PDI antibody in untreated HMEC-1, showed only the presence of monomeric PDI, but revealed several additional bands with higher molecular weight in cells treated by 4-HNE or oxLDLs (Fig. 4A). PDI modification by 4-HNE was associated with a loss of enzymatic activity (Fig.4B) and a rise of toxicity (Supplementary Fig. S6) in a dose-dependent manner. Immunofluorescence and confocal microscopy showed that 4-HNE adducts co-localize with PDI in the ER (Fig.4C). These data confirm the formation of 4-HNE-PDI adducts observed on Western blots, and suggest that PDI modification by 4-HNE (and probably by other reactive carbonyl generated during lipid peroxidation) may lead to a progressive loss of PDI enzymatic activity.

Interestingly, antioxidants such as trolox and BHT, and carbonyl scavengers, such as pyridoxamine and N-acetyl cysteine (NAC), prevented both the modification and the inactivation of PDI by oxLDLs (Fig. 5A and Supplementary Fig. S7).

This strongly suggests that oxLDLs act mainly by triggering a cellular oxidative stress that induces in turn the peroxidation of cellular lipids and the formation of reactive aldehydes (among them 4-HNE). This is consistent with the fact that the major part of 4-HNE contained in oxLDLs, forms adducts with LDL components (e.g., apoB or phosphatidylethanolamine) and that only a minor part (10%–20 %) of 4-HNE remains free, dissolved in the lipid phase of oxLDLs (5). However, the free 4-HNE contained in oxLDLs is able to react with cell proteins (4), including PDI, as shown by incubation of cells with oxLDLs pre-loaded with radiolabeled [3H]4-HNE (Supplementary Fig. S8). The carbonyl scavengers NAC and pyridoxamine prevented PDI inactivation by 4-HNE (Fig. 5B) and inhibited CHOP mRNA increase and oxLDL toxicity (Fig. 5C and 5D). In contrast, the antioxidants trolox and BHT were almost completely inefficient to prevent PDI inactivation by 4-HNE (Fig. 5B). This suggests that 4-HNE-PDI adducts generated in cells treated with exogenous free 4-HNE may result from the direct reaction of the 4-HNE with PDI (without any additional oxidative stress), in contrast to oxLDLs that act mainly by triggering cellular oxidative stress. As expected, other aldehydes, issued from lipid peroxidation and glycoxidation, are able to inhibit PDI (Supplementary Table S1). Methylglyoxal (MGO) and glyoxal (GO), two alpha-ketoaldehydes formed under hyperglycemic conditions and precursors of advanced glycation end product (AGE) required higher concentrations (100 μmol/L) to inhibit PDI.
PDI modification by 4-HNE in atherosclerotic lesions
PDI was present both in human mammary arteries and in human carotid atherosclerotic plaques where it was relatively highly expressed in endothelium and subendothelial areas and in the central macrophagic core (Fig. 6A and Supplementary Fig. S9), consistent with the constitutive expression of PDI in cultured vascular cells and in human macrophage-like U937 cells (Supplementary Fig. S10). 4-HNE-adducts were detected in atherosclerotic lesions and were localized in the same areas as PDI, in the endothelium and in the central necrotic core of advanced plaques (Fig. 6A and Supplementary Fig. 9). Western blots of PDI immunoprecipitates showed the presence of 4-HNE-PDI adducts, particularly in high MW polymerized PDI from atherosclerotic carotid plaque, but not in control mammary artery (Fig. 6B). This was confirmed by immunofluorescence double staining, which showed the co-localization of PDI and 4-HNE in intact cells (Fig. 6C) and in cellular debris (some of them also stained by anti-CD68 specific for macrophages) in the central core area of carotid advanced plaques (Fig. 6D).
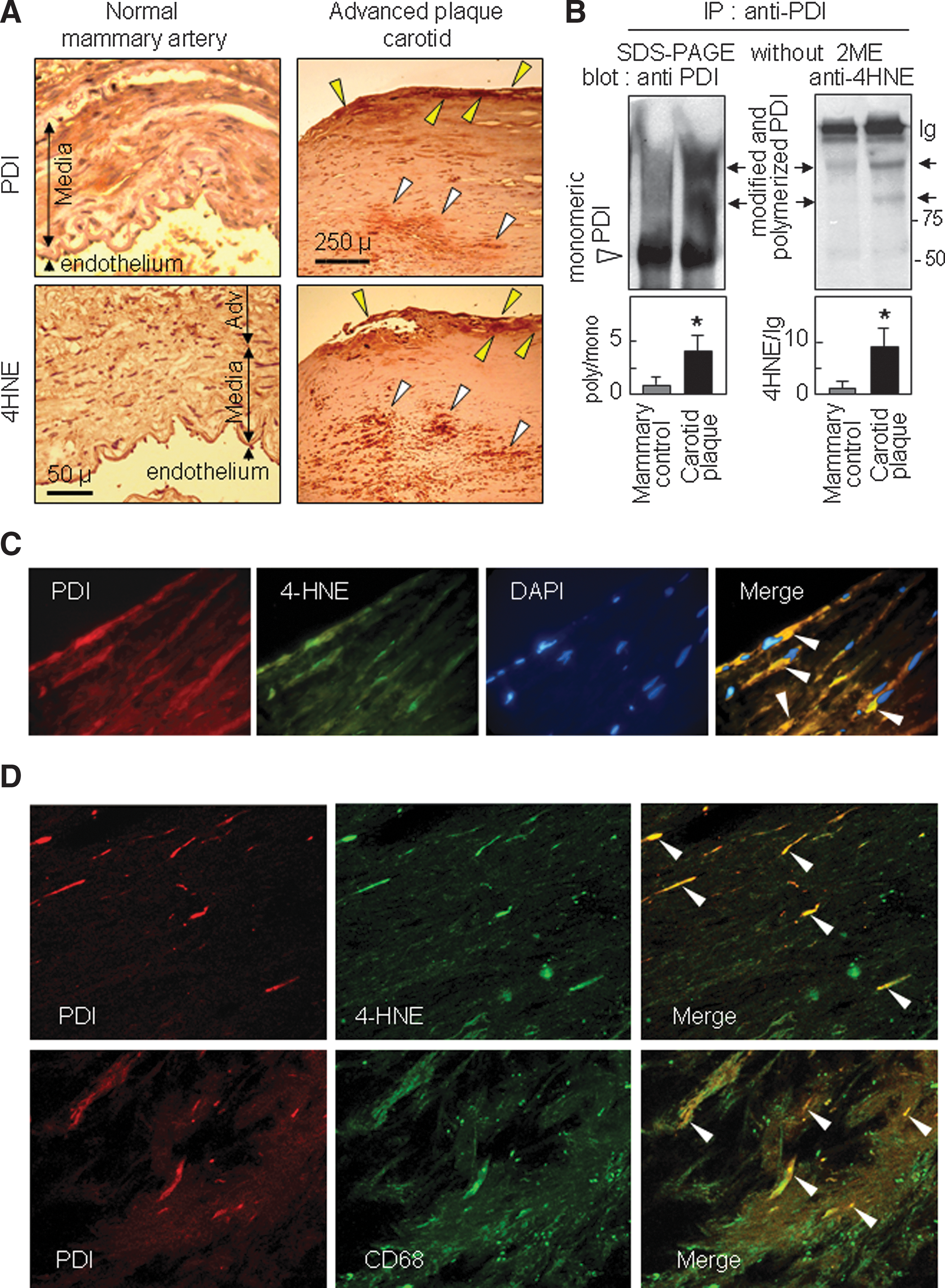
These data indicate that PDI modification occurs in vivo in atherosclerotic lesions, and suggest a possible loss of function (subsequent to 4-HNE-PDI adducts formation) in the endothelial/subendothelial areas and in the macrophagic core of atherosclerotic plaques.
Discussion
This study indicates that toxic concentrations of oxLDLs inhibit the activity of the ER-resident PDI. This was potentiated by bacitracin, and was associated to ER stress and apoptosis. PDI inhibition may result from its carbonylation by reactive lipid peroxidation derivatives, as supported i) by the presence of 4-HNE-PDI adducts in cells treated by oxLDLs; and ii) by the formation of similar adducts and PDI inhibition in cells incubated with 4-HNE. NAC and pyridoxamine prevented PDI inactivation by oxLDLs and 4-HNE, and reduced ER stress and toxicity. Interestingly, PDI modification by 4-HNE occurs in vivo in advanced atherosclerotic lesions.
Molecular mechanism of PDI inhibition by oxLDLs and 4-HNE
At the molecular level, PDI acts as a dithiol-disulfide oxidoreductase and a chaperone able to reduce, oxidize, and isomerize disulfide bonds, thereby contributing to the correct folding of proteins (9, 22, 44). The isomerase function can repair misformed disulfide bonds via a “scanning and escape” mechanism, in which the N-terminal thioredoxin active site forms a disulfide bond with a (to-be-corrected) protein (“scanning”), which is released by the C-terminal thioredoxin active site (“escape”), allowing in turn a regeneration of PDI (2, 42). This requires the integrity of the catalytic Cys-x-x-Cys motives and an important neighboring Arg (27, 42). Moreover, the cellular activity of PDI depends on GSH level, which is required to reduce the oxidized form of PDI, thus the redox state of the ER subcellular compartment, in which PDI is mainly located (2). Cys of the PDI active site and the neighboring Arg are targets for aldehydes, and Carbone et al. (2) report that the modification of a Cys of the PDI active site by 4-HNE or by the thiol reactive N-ethylmaleimide, is associated with a loss of the PDI isomerase activity. The integrity of the two SH groups of the N-terminal thioredoxin-like active site is required for the “scanning and escape” mechanism of PDI, and any mutation on Cys compromises the isomerase function (2, 42).
Since PDI activity depends on the ER redox state that allows disulfide formation (oxidation), while maintaining sufficient reducing power to break incorrect disulfide bonds, a ratio GSH/GSSG 5/1 is optimal for a rapid oxidative folding in vitro (45). As discussed by Raturi and Mutus (20), the ratio GSH/GSSG is around 2/1 in the ER, suggesting a half-reduced and half-oxidized state for PDI, potentially favorable to reduction/oxidation cycles required for the rearrangement of disulfide bonds of misformed proteins (42, 44). As oxLDLs induce a cellular oxidative stress (28), known to decrease the GSH/GSSG ratio, oxLDLs may alter the redox state of PDI and slow down its activity (45). However, in our conditions, PDI inactivation by oxLDLs began between 6 h and 12 h, (i.e., before the drop of GSH that starts during the late phase of cell apoptosis after 18–24 h incubation with oxLDLs) (28), thus suggesting that GSH depletion is not the primum movens of PDI inactivation. Likewise PDI inactivation, as assessed by the in vitro assay, is independent of the cellular GSH (since DTT, the reductive compound, is added in the assay mixture), thereby demonstrating that the inhibitory effect of reactive carbonyls results mainly from a direct action on PDI protein.
Finally, another molecular mechanism of PDI inactivation has been described in etoposide-induced apoptosis, in which the cytosolic PDI is cleaved by caspase-3, which results in the generation of 33 and 40 kDa fragments (15). In our system, caspases cannot explain the inhibition of PDI by oxLDLs, because i) we did not detect the formation of 33 or 40 kDa PDI fragments, even in apoptotic cells (data not shown); ii) while caspases are activated by oxLDLs (Fig. 3E and Ref. 41), their inhibition by the caspase inhibitors DVED-fmk and z-VAD-fmk did not prevent PDI inhibition induced by oxLDLs (data not shown).
PDI inhibition by oxLDLs may result from its modification by 4-HNE generated through intracellular oxidative stress and subsequent lipid peroxidation, as suggested by the protective effect of antioxidants trolox and BHT. A transfer of free 4-HNE (minor part of the whole 4-HNE content) (48) from oxLDLs to PDI may also occur, which is blocked by NAC and pyridoxamine. As previously reported (1), antioxidants such as trolox and BHT (which are devoid of carbonyl scavenger activity) were unable to protect PDI against the inhibitory effect of free 4-HNE, whereas the carbonyl scavengers, pyridoxamine and NAC, prevented the modification of PDI, and protected against ER stress and apoptosis induced by 4-HNE and oxLDLs. Altogether, these studies suggest that the main mechanism of PDI inactivation by oxLDLs or 4-HNE and other electrophilic aldehydes present in oxLDLs, results from the formation of carbonyl-PDI adducts, via oxidative and subsequent carbonyl stress, and via transfer of 4-HNE from oxLDLs to PDI.
Physiological role of PDI and consequences of its inhibition
PDI is one of the most abundant ER proteins, which catalyzes the folding of newly synthesized proteins and the rearrangement of incorrect disulfide bonds (9, 45). PDI activity tends to prevent the accumulation of misfolded proteins in the ER, and may thereby limit ER stress induction (22) and subsequent apoptosis, thus acting as a survival mechanism (12, 43). In our system, PDI inhibition (by bacitracin) sensitizes cells to ER stress and apoptosis induced by oxLDLs. This is in agreement with the concomitant inhibition of PDI and neuronal apoptosis induced by nitrosative stress (39), and cell death induced by chemotherapy drugs (12). Consistently, high PDI expression tends to limit or delay ER stress and apoptosis induced by oxLDLs and 4-HNE.
The data obtained in HMEC-1 expressing inactive ‘myc-PDI mut’ are apparently more puzzling, since under basal conditions (i.e., in the absence of oxLDLs), constitutive PDI activity is not inhibited by the expression of the inactive mutant PDI and CHOP expression and toxicity are not increased in the absence of oxLDLs. This suggests that the inactive mutant PDI has no inhibitory effect on the constitutive wt PDI. In HMEC-1 expressing inactive ‘myc-PDI mut’ treated by oxLDLs, both CHOP induction (ER stress) and the cytotoxicity are enhanced (in comparison to parental HMEC-1 not expressing the mutant PDI). The extra-potentiation of oxLDL toxicity in myc-PDI mut HMEC-1 requires the additional effect of lipid peroxidation compounds that may aggravate the misfolding of mutant PDI (possibly misfolded by mutations), which is relatively abundant (mut-PDI level is twice that of constitutive wt PDI) and could reinforce ER stress/UPR. Then the prolonged ER stress/UPR elicits the activation of caspase-dependent apoptosis, as assessed by caspase 3 activation and increase of TUNEL positive cells. It may be noted that this model is also consistent with the effect of thapsigargin that disturbs ER calcium homeostasis, induces ER stress and toxicity (but induces no major formation of 4-HNE-protein adducts), thus explaining that the toxic effect of thapsigargin was similar in control cells and in myc-PDI mut HMEC-1 (Supplementary Fig. 5).
The protective effect of PDI may result from its enzymatic (oxidase and isomerase) activities and/or from its chaperone activity (3, 32). However, this generally accepted protective role of PDI is challenged by a recent report showing that PDI may play a pro-apoptotic role in Huntingtin neuronal toxicity, which highlights a new mechanism linking protein misfolding and apoptotic cell death (7). Thus, PDI could participate in both phases of ER stress, the first one being protective when protein repair is effective, the second one being pro-apoptotic when the reparative phase is overwhelmed and misfolded proteins accumulate (24, 47).
PDI modification by 4-HNE, ER stress, and atherosclerosis
PDI modification by 4-HNE has been reported in the liver of aged mice (19), and in rat models for alcoholic liver disease (2, 29), and cysteine modification of PDI by S-nitrosylation occurs in neurodegenerative diseases (39).
In atherosclerosis, carbonyl stress is thought to be involved in foam cell formation, and cellular dysfunction in the vascular wall (18), but the precise molecular targets of reactive carbonyls are only partly identified (6), and to our knowledge, 4-HNE-PDI-adduct formation has never been reported. Our data show that PDI modification and inactivation by 4-HNE or oxLDLs contribute to ER stress and apoptosis, and that PDI modification by 4-HNE occurs in the endothelium and in the macrophage-rich core of advanced atherosclerotic lesions, thereby suggesting that this event may play a role in atherosclerosis. This hypothesis is consistent with the chronic activation of ER stress/UPR pathway at all stages of atherogenesis (49). The prolonged ER stress may be proatherogenic through several mechanisms, including (i) activation of inflammatory pathways in macrophages, (ii) endothelial cell apoptosis, which may contribute to lipoprotein influx into the plaque, thrombocyte aggregation, and plaque extension, and (iii) apoptosis of macrophages, which in turn leads to plaque necrosis if the apoptotic cells are not rapidly cleared (31). In addition, ER stress may link immune response (low chronic inflammation) with the metabolic disorders occurring in obesity, insulin resistance, and type 2 diabetes that are known to participate in the pathogenesis of atherosclerosis and its complications (8). Moreover, increased ER stress may induce apoptosis of smooth muscle cells and macrophages, thereby contributing to plaque vulnerability and athero-thrombotic events (14).
Materials and Methods
An expanded Methods section is available in Supplementary Data.
LDL isolation and oxidation
LDL from human pooled sera were prepared as reported (25). Mildly oxidized LDLs (oxLDLs) contained 71–104 nmol lipid hydroperoxide/mg apoB, 6–8 nmol TBARS/mg apoB, and 8–10 nmol 4-HNE/mg apoB under standard conditions.
Cell culture and transfection
Human Microvascular Endothelial Cell-1 (HMEC-1) (Dr. Candal, CDC, Atlanta), were grown in MCDB-131 culture medium supplemented with 10% fetal calf serum and 40 μmol/L of glutamine. HMEC-1 were transfected with 10 μg of Myc-tagged PDI wt cDNA (Myc-PDI wt) or Myc-PDI N-terminal and C-terminal mutant cDNA (Myc-PDI mutant) subcloned into pCR3.1 vector (a generous gift from Drs. Nakamura and Lipton, Boston, MA) using 20 μL of lipofectamine (Invitrogen) as reported (32). Transfected cells were cultured in the presence of G418 (1.5 mg/L) (PAA Laboratories). Cells were starved in serum-free medium for 12 h before the experiments.
Determination of PDI activity
Control, wild type, or mutant HMEC-1 were incubated with oxLDLs or 4-HNE, homogeneized in 250 μL of PDI buffer (0.1 mol/L potassium phosphate buffer pH 7.0 containing 2 mmol/L EDTA) and 20 μmol/L digitonin. The assay contained 150 nmol/L Di-E-GSSG, a fluorogenic PDI substrate (20), (a generous gift from Dr B. Mutus, Windsor, Canada), DTT (5 μmol/L), cell extract (50–100 μg cell protein), and PDI buffer in 500 μL final volume. The reduction of di-E-GSSG was measured fluorometrically at time 0 and after 30 min of incubation at 37°C (exc.525 nm em. 545 nm). The data are expressed as ratio of fluorescence/fluorescence of the unstimulated control.
Evaluation of cell viability, apoptosis, and necrosis
Cytotoxicity was estimated using the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay. Apoptosis and necrosis were evaluated using two vital fluorescent dyes, the permeant DNA intercalating green-colored DNA probe SYTO-13 (0.6 μmol/L), and the nonpermeant intercalating red DNA probe propidium iodide (15 μmol/L), (fluorescence microscope Fluovert FU, Leitz), (25). DEVDase activity and TUNEL experiments were done as indicated in the Expanded Method section.
Immunofluorescence
Cells grown on glass cover slides, were washed with PBS and fixed for 10 min in 30% paraformaldehyde containing 4% triton X100, then incubated with the anti-PDI antibody, revealed with Alexa Fluor-conjugated secondary antibody, and visualized using a Zeiss LSM 510 fluorescence confocal microscope.
Immunohistochemistry
Human advanced carotid plaques (5 patients 70–75 years old) were obtained after endarterectomy (Cardiovascular Surgery Department, CHU Toulouse), fixed in formalin and paraffin embedded. Serial 3 μm thin sections were incubated with anti-4-HNE-adduct, and anti-PDI antibodies, and revealed by using avidin-biotin horseradish peroxidase visualization system (Vectastain, ABC kit Elite, Vector Laboratories).
Western blot analysis
HMEC-1 and human tissue samples were homogenized in extraction buffer, and used for SDS-PAGE/immunoblotting either as total protein extracts or after immunoprecipitation (22). When indicated, beta-mercaptoethanol was omitted in the SDS-PAGE. Atheroscerotic plaque extract analysis was performed as described in Supplementary Data.
Quantitative and semi-quantitative RT-PCR analysis
Total RNA was used for Real-Time Quantitative PCR analysis to evaluate the expression of CHOP and spliced XBP1 (see detailed method in Supplementary Data).
Statistical analysis
Data are presented as mean±SD. Differences between means values were evaluated by unpaired t test (two groups) or by one-way ANOVA (more than two groups) followed by multiple comparisons versus a control by the Holm-Sidak Test. Estimates of statistical significance were performed using the SigmaStat 3.5 (Systat software). Values of p<0.05 were considered significant.
Footnotes
Acknowledgments
We thank Drs. T Nakamura and SA Lipton (Boston, MA) for giving the Myc-tagged wt and mutant cDNA PDI vectors, Dr. B. Mutus (Windsor, Canada) for providing the PDI substrate Di-E-GSSG, and Pr. S. Sasson (Jerusalem, Israel) for fruitful discussion. The authors wish to acknowledge Dr. R. d'Angelo (Cellular Imaging TRI Platform) for confocal microscopy, MH Grazide and C. Santiago for their excellent technical assistance.
Financial supports by INSERM, Université of Toulouse, Fondation pour la Recherche Médicale (DCV2007040927), and ANR Blanc SVSE1
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.