
Abstract
Introduction
Nitrosative stress is induced by a reaction of superoxide with nitric oxide (NO), which generates PN, and is implicated in diabetic conditions (38). Increased nitrosative stress could (i) induce protein nitration, (ii) damage membrane proteins and fatty acid, leading to changes of cellular signaling transduction, (iii) upregulate inflammatory response, and (iv) to an extreme extent, activate the apoptotic pathway (17, 27, 38). Hyperglycemic episodes are closely associated with increased oxidative and nitrosative stress, which can trigger the development of diabetic complications (6, 29). However, the exact role of nitrosative stress in DR remains largely unknown.
The Wnt family includes 19 secreted, cysteine-rich glycoproteins, which bind to a coreceptor complex composed of a frizzled (Fz) receptor and low-density lipoprotein receptor-related protein 5 or 6 (LRP5/6), and regulates expression of multiple target genes (10, 16, 20). In unstimulated cells, cytoplasmic β-catenin, a key effector of the canonical Wnt pathway, is phosphorylated by a kinase complex containing glycogen synthase kinase-3β, leading to its degradation. Upon binding of the canonical Wnt ligands to the LRP6-Fz receptor complex, the kinase complex is dissociated. As a result, β-catenin is unphosphorylated, stabilized, accumulates in the cytoplasm and ultimately translocates to the nucleus. Nuclear β-catenin dimerizes with transcription factors such as lymphoid enhancer-binding factor 1/T cell-specific transcription factor to regulate transcription of target genes such as vascular endothelial growth factor (VEGF), connective tissue growth factor, intracellular adherent molecule 1 (ICAM-1), and tumor necrosis factor-α (4, 11, 12, 21, 49).
The Wnt-signaling pathway regulates multiple physiological and pathological processes and plays a crucial role in embryogenesis and carcinogenesis (10, 39). Our previous studies have shown that activation of the Wnt pathway plays a pathogenic role in DR in humans and in animal models, and that blockade of the Wnt pathway has beneficial effects in DR models (8, 19, 30, 43). Moreover, we have recently reported that oxidative stress plays an important role in activation of the Wnt pathway in DR (50). However, the pathogenic role of nitrosative stress in DR and the relationship between nitrosative stress and the Wnt pathway in the development of DR remain unclear. In the present study, we investigated the role of nitrosative stress in retinal inflammation and vascular leakage in diabetes, and further determined whether nitrosative stress contributes to activation of the Wnt pathway in DR.
Results
Uric acid attenuates PN-induced tyrosine nitration and suppresses Wnt pathway activation
Previously, we showed that the Wnt-signaling pathway is activated in the retinas of DR models (8). To determine whether nitrosative stress contributes to Wnt pathway activation in DR, PN (ONOO−), a reactive nitrogen species (RNS), was employed as an inducer, which is formed (among other reactions) by combination of superoxide (O2 −) and NO radical (15). In ARPE19 cells, nitrosative stress was triggered 10 min after the addition of PN, as shown by the significant increases of 3-nitrotyrosine (3-NT), a biomarker of nitrosative stress, as shown by measurements from both Western blot analysis and enzyme-linked immunosorbent assay (ELISA) (Fig. 1A, B). Subsequently, elevated levels of phosphorylated LRP6 (pLRP6, Fig. 1C, D) and nonphosphorylated β-catenin (Np-β-catenin, Fig. 1E, F) were observed at 20 min of PN treatment, demonstrating activation of the Wnt pathway. The Wnt signaling induced by PN was significantly attenuated by uric acid (UA), a natural scavenger of PN (Fig. 1). These results suggested that nitrosative stress contributes to activation of the Wnt-signaling pathway.

UA attenuates 4-hydroxynonenal-induced tyrosine nitration and downregulates VEGF and ICAM-1 expression
4-Hydroxynonenal (HNE), a product of lipid peroxidation, is known to induce oxidative/nitrosative stress, which is associated with cellular signaling and cell damage (37, 40, 42). As shown by ELISA specific for 3-NT, treatment with 10 μM HNE resulted in a significant increase of 3-NT formation, which was inhibited by preincubation with UA for 90 min. The inhibitory effect of UA on HNE-induced reactive oxygen species (ROS)/RNS generation was further analyzed using dichlorofluorescein (DCF) fluorescence assay. As shown in Figure 2B, ROS/RNS were increased by 3-fold by HNE. Pretreatment with UA suppressed the ROS/RNS formation in a concentration-dependent manner.
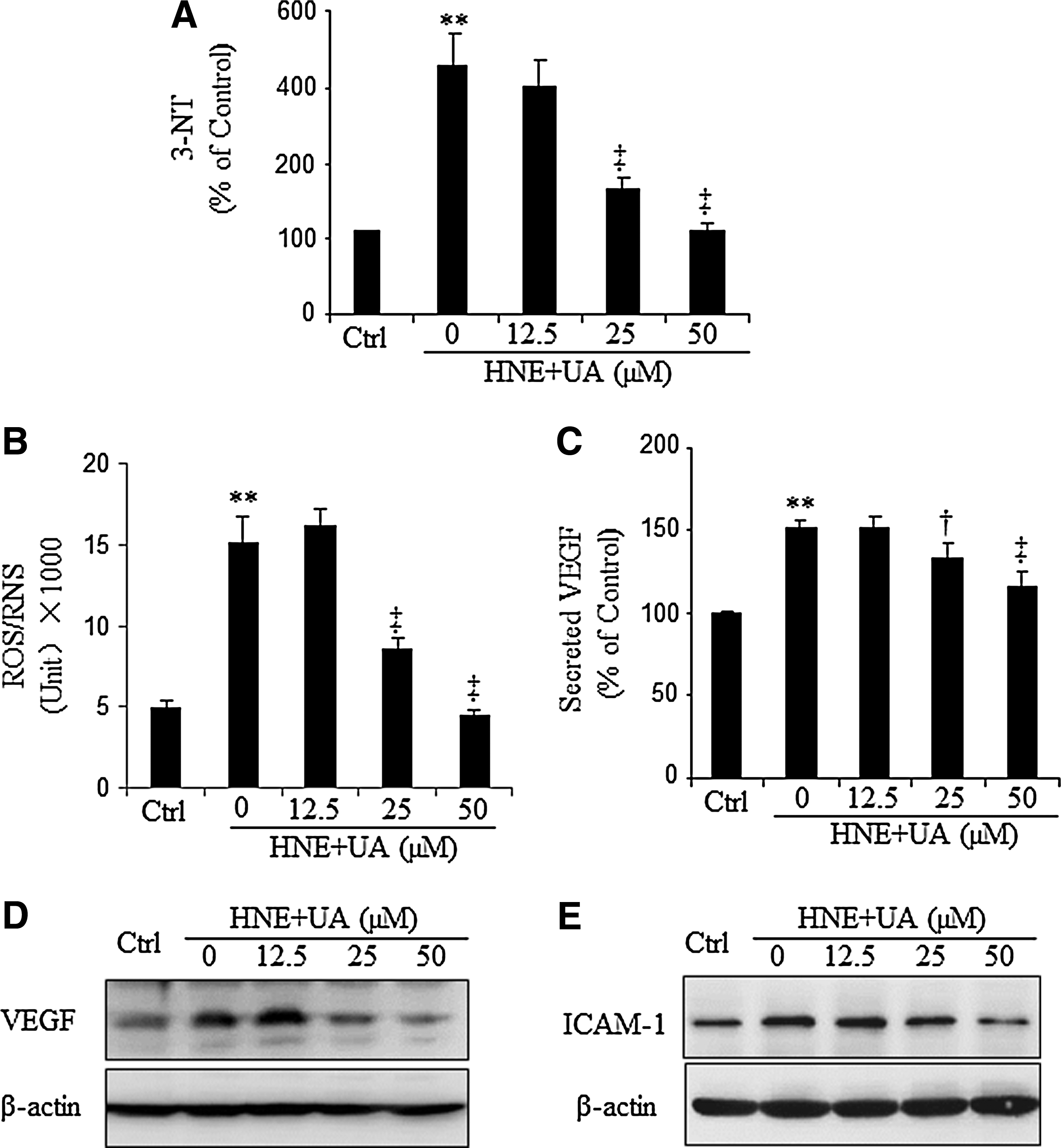
VEGF, an angiogenic and vascular permeability factor, and ICAM-1, a major proinflammatory adhesion molecule, are both target genes of Wnt signaling and are overexpressed in the diabetic retina (13, 26, 45, 46). To address the causative role of nitrosative stress on DR, ARPE19 cells were treated with HNE for 16 h with or without various concentrations of UA. Intracellular VEGF and ICAM-1 levels were determined by Western blot analysis. The results showed that HNE markedly upregulated VEGF (Fig. 2D) and ICAM-1 (Fig. 2E) expression, which was reversed by the UA treatment. As shown by ELISA, UA also decreased secreted VEGF levels in a concentration-dependent manner (Fig. 2C).
UA inhibits the Wnt-signaling pathway activation induced by HNE
To evaluate the effect of UA on Wnt signaling induced by HNE, ARPE19 cells were treated with 10 μM HNE with and without UA. As shown by Western blot analysis, treatment with HNE for 1 h significantly increased pLRP6, which was attenuated by UA in a concentration-dependent manner (Fig. 3A, B). A Wnt3a-conditioned medium (WCM) was used as a positive control for induction of LRP6 phosphorylation. To determine whether UA treatment affects β-catenin activation, the cytosolic and nuclear β-catenin levels were determined by Western blot analysis. After 6-h incubation with HNE, with or without the pretreatment of different concentrations of UA, ARPE19 cells were fractionated. As shown by Western blot analysis using subcellular fractions, HNE induced cytosolic β-catenin accumulation, which was prevented by UA (Fig. 3C, D). Measurement of nuclear β-catenin levels showed that the HNE-induced β-catenin nuclear translocation was significantly reduced by UA in a concentration-dependent manner (Fig. 3C, E).

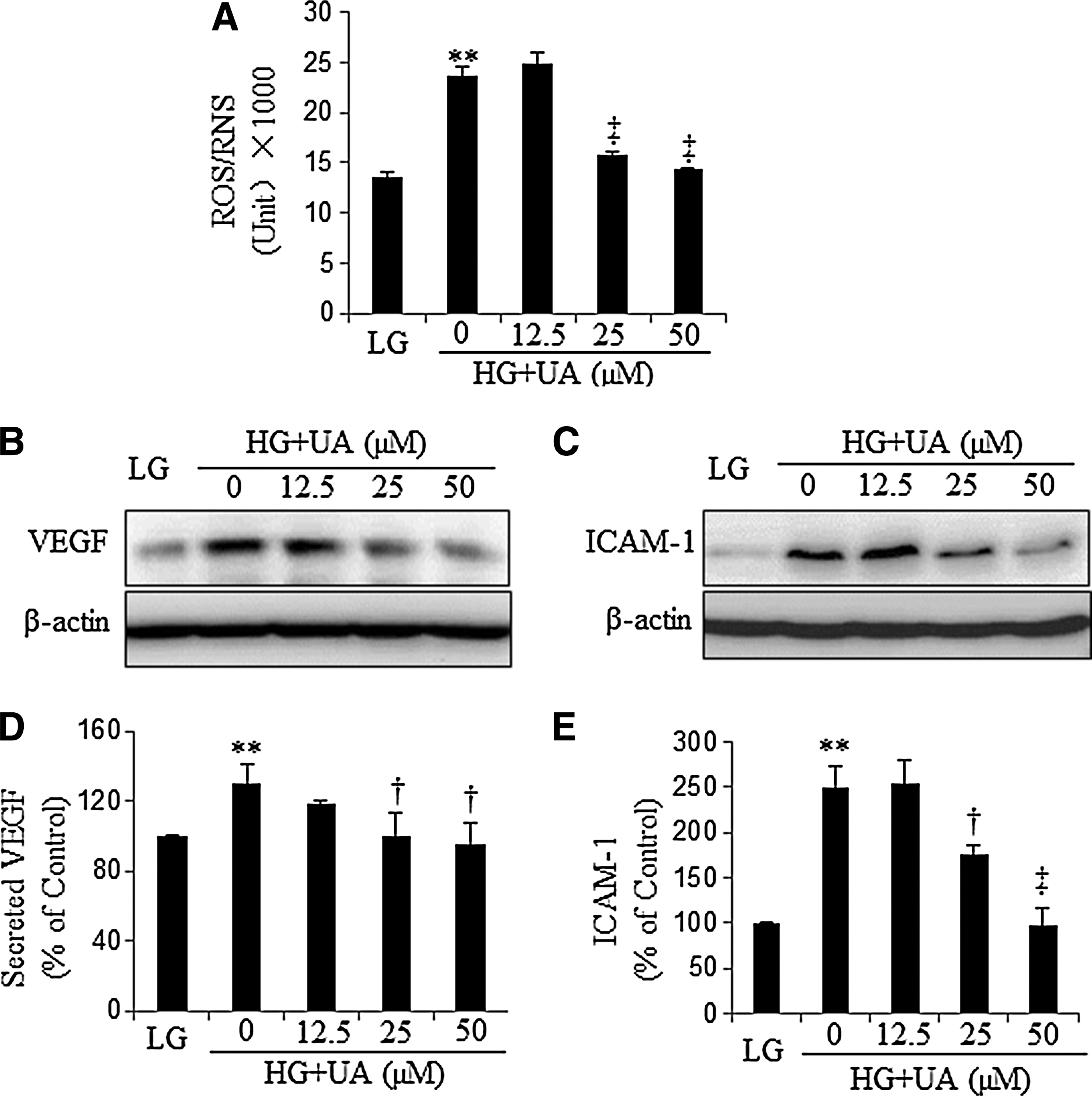
UA attenuates the overexpression of VEGF and ICAM-1 induced by high glucose
Hyperglycemia is known to cause mitochondrial dysfunction and increases of superoxide (O2
−) production (5). Meanwhile, hyperglycemia could also upregulate inducible nitric oxide synthase (iNOS) expression to produce NO (7). Reaction of O2
− and NO generates nitrosative stress anion PN. To determine UA's effect on high-glucose (HG)-induced nitrosative stress, ARPE19 cells and human retinal Müller cells (HMC) were incubated with 30 mM glucose (HG) for 48 h, and then treated with different concentrations of UA for another 16 h, with 5 mM glucose plus 25 mM
UA suppresses HG-induced activation of the Wnt signaling pathway
To further investigate whether UA inhibits HG-induced activation of Wnt signaling, ARPE19 cells and HMC were cultured with HG for 48 h and subsequently treated with different concentrations of UA for another 16 h. UA inhibited HG-induced LRP6 phosphorylation in a concentration-dependent manner in both ARPE19 cells (Fig. 5A and B) and HMC (Fig. S1). Phosphorylation of β-catenin is the initial step for its degradation. As shown in Figure 5C and D, phosphorylated β-catenin (p-β-catenin) was significantly decreased by HG, which resulted in subsequent increases of both cytosolic (Fig. 5E, F) and nuclear (Fig. 5G, H) β-catenin levels in ARPE19 cells. These changes of β-catenin induced by HG were reversed by UA (Fig. 5). Notably, UA at the concentration of 50 μM decreased the pLRP6 and cytosolic and nuclear β-catenin to levels similar to those in the

UA ameliorates the Wnt signaling activation induced by Wnt ligand, but does not inhibit Wnt signaling induced by a constitutively active β-catenin mutant
To further confirm that the suppression of the VEGF and ICAM-1 expression by UA is through blockade of the Wnt-signaling pathway, ARPE19 cells were exposed to 20% WCM for 16 h with or without UA. As shown in Figure 6A and B, WCM significantly elevated pLRP6 levels, which was attenuated by UA. Similarly, UA also inhibited WCM-induced phosphorylation of LRP6 in HMC (Fig. S1). A Luciferase activity assay was used to evaluate the transcriptional activity of β-catenin. Human telomerase reverse transcriptase-immortalized retinal pigment epithelial (HTERT-RPE) cells were transfected with the TOPFLASH construct and then treated with 20% WCM with or without different concentrations of UA for 16 h. The FOPFLASH vector was used as a nonspecific Luciferase control. Luciferase assay showed that WCM induced a 50-fold increase of β-catenin transcriptional activity, which was attenuated by UA in a concentration-dependent manner (Fig. 6C).

The constitutively active β-catenin mutant was generated by a point mutation at the Ser37 phosphorylation site, and is known to activate transcription of Wnt target genes in the absence of the Wnt ligand. Overexpression of S37A using an adenoviral vector (Ad-β-cat-S37A) induced higher TOPFLASH activities, which was not blocked by UA (Fig. 6D). These results suggest that UA does not inhibit Wnt signaling induced at an intracellular level, and that UA may regulate Wnt signaling at an extracellular level.
5,10,15,20-Tetrakis (4-sulfonatophenyl) porphyrinato Iron III Chloride inhibits Wnt ligand and HG-induced Wnt-signaling activation
To verify the role of nitrosative stress on Wnt-signaling activation, we utilized another PN decomposition catalyst, 5,10,15,20-Tetrakis (4-sulfonatophenyl) porphyrinato Iron III Chloride (FeTPPS), to scavenge nitrosative stress. As shown in Figure 7A, FeTPPS significantly suppressed HG-induced LRP6 phosphorylation and β-catenin accumulation in a concentration-dependent manner. Similar to UA, FeTPPS dose-dependently inhibited Wnt ligand-induced LRP6 phosphorylation and β-catenin accumulation (Fig. 7B). Moreover, FeTPPS attenuated WCM-induced β-catenin transcriptional activity in a concentration-dependent manner, as shown by Luciferase assay (Fig. 7C).

Clinical characteristics of diabetic animals
To determine the effect of nitrosative stress on Wnt signaling in the retina, we used streptozotocin (STZ)-induced diabetes rats, which exhibit Wnt pathway overactivation in the retina (8). Before the STZ injection, all of these rats had similar blood glucose levels (∼100 mg/dl) and body weights (∼140 g). Upon onset of STZ-induced diabetes, the diabetic rats were randomly assigned to two groups. One group was fed with UA (160 mg/kg/d) in drinking water for 6 weeks, and the other group was fed with regular drinking water as diabetic control. Six weeks after the treatment, no significant difference was observed in blood glucose levels and body weights between UA-fed diabetic and untreated diabetic control rats (Table 1) at any of the time points, suggesting that oral administration of UA had no effect on hyperglycemia and body weight loss in the diabetic rat model.
Values are means±SD. p-value 1: NDM versus DM; p-value 2: DM versus DM+UA. n=6.
DM, diabetic rats; NDM, nondiabetic rats; STZ, streptozotocin; UA, uric acid.
UA suppresses Wnt signaling in the retina of diabetic rats
To investigate whether UA inhibits the Wnt-signaling pathway in vivo, we fed the diabetic rats with UA. As shown by immunoblotting, 3-NT levels were significantly increased in the retinas of diabetic rats, compared to the nondiabetic control (Fig. 8A). Immunostaining using an antibody specific for 3-NT showed more intense 3-NT signals in the diabetic rat retinas, primarily in the inner retina (Fig. 8B). Treatment of diabetic rats with (160 mg/kg/d) UA for 6 weeks markedly reduced 3-NT levels in the retina as indicated by both Western blot analysis and immunostaining (Fig. 8A, B). Retinal levels of pLRP6, total LRP6, and β-catenin were significantly upregulated in diabetic rat retinas, consistent with our previous study (Fig. 8C–F). Nuclear translocation of β

UA downregulates VEGF and ICAM-1 expression and ameliorates vascular permeability and inflammation in the retina of diabetic rats
Our previous study has shown that Wnt pathway activation in the retina of diabetic animals is responsible for retinal vascular leakage and inflammation in DR models (8). Here, we evaluated the effect of UA on retinal inflammation using Western blot analysis of ICAM-1 and VEGF and examination of inflammatory cell infiltration. As shown by Figure 9A–C, ICAM-1 and VEGF were both overexpressed in the retina of diabetic rats, which was completely reversed by UA treatment, as demonstrated by Western blot analysis. We also evaluated the effect of UA on inflammatory cell infiltration by immunostaining of the monocyte marker CD11b in the retina after thorough perfusion. Consistent with the ICAM-1 changes, UA treatment decreased CD11b-positive cells in the perfused diabetic rat retina, suggesting decreased inflammation in the diabetic retina (Fig. 9D, E). Retinal vascular leakage was examined using Western blotting of extravascular albumin in the perfused retina. The results demonstrated that albumin leakage was significantly increased in the retina of diabetic rats. UA treatment significantly reduced albumin leakage in the retina of diabetic rats (Fig. 9F, G). Taken together, these results suggest that the nitrosative stress-activated Wnt signaling may play a causative role in retinal inflammation in DR.

Discussion
Accumulating evidence suggests that diabetes is associated with nitrosative stress and PN formation in multiple tissues in diabetic animal models and in diabetic patients (28, 38). Nitrosative stress has been suggested to induce diabetic complications (17, 29). Our previous studies showed that the Wnt pathway overactivation in the retina of diabetic patients and diabetic animal models plays a pathogenic role in DR (8, 50). However, association of the nitrosative stress with Wnt pathway activation has not been reported previously. The present study demonstrated for the first time that nitrosative stress plays an important role in retinal inflammation and vascular leakage in diabetes. Furthermore, our results showed that nitrosative stress is responsible, at least in part, for activation of the Wnt pathway in DR. Therefore, these observations have revealed a novel regulatory effect of nitrosative stress on Wnt signaling and a pathogenic mechanism by which nitrosative stress leads to retinal inflammation and retinal vascular leakage in DR.
Nitrosative stress is caused by overproduction of RNS. A number of studies have shown that nitrosative stress is increased in the diabetic retina (27, 28, 48). Diabetic stimuli could trigger generation of excess superoxide, which is rapidly converted to PN (reaction with NO), hydroxyl radicals (Fenton reaction or the iron-catalyzed Haber–Weiss reaction), and hydrogen peroxide (reaction catalyzed by superoxide dismutase). The reaction rate of superoxide with NO is at least 1-fold faster than other aforementioned reactions. Furthermore, under pathological conditions, even a modest increase of superoxide and NO simultaneously could greatly stimulate PN production (27, 31). PN can modify tyrosine residues in proteins to form nitrotyrosine. Nitrotyrosine is a well-accepted indicator of RNS generation, and this stable end product is involved in inactivation of mitochondrial and cytosolic proteins, resulting in damage of cellular constituents. Moreover, it can initiate lipid peroxidation, increase DNA damage, deplete intracellular GSH levels, and induce overexpression of proinflammatory factors and adhesion molecules. Therefore, tyrosine nitration is being increasingly proposed as a contributor to tissue injury in human diseases (3, 5, 27, 35). Thus, nitrosative stress is as important as oxidative stress in pathogenesis of DR in the clinical scenario, and antinitrosative stress should be a more attractive modality for treatment of diabetic complications.
HNE, a major lipid peroxidation product of ω-6 polyunsaturated fatty acids, is known to modulate different signaling pathways, dependent upon its concentration (33). HNE regulates cell proliferation and signal transduction at lower concentrations, but at higher concentrations, it can cause cell differentiation and apoptosis (34, 40). Previous studies have found that HNE can be upregulated in oxidative stress/nitrosative stress, and is considered a biomarker of oxidative stress/nitrosative stress (37, 42). Our previous study also showed that HNE increases oxidative stress in retinal cells (50). In the present study, we demonstrated that HNE also induced tyrosine nitration and ROS/RNS production. Meanwhile, the HNE-induced nitrosative stress is significantly decreased by UA (a scavenger of PN) and FeTPPS (a PN decomposition catalyst) in a concentration-dependent manner. Therefore, we chose PN and HNE as inducers of nitrosative stress in this study.
The Wnt pathway is involved in angiogenesis and chronic inflammation in multiple pathological conditions (22). It is well known that VEGF is a key angiogenic factor in DR and is regulated by the Wnt pathway (23, 47), and the Wnt pathway also regulates endothelial cell migration in angiogenesis (18, 32). Documented studies have shown that the Wnt pathway participates in many ocular diseases, such as vascular disorders in the retina and age-related macular degeneration (14, 23, 49). Our recent study showed that the Wnt pathway is activated in the retinas of diabetic patients and diabetic animal models, and blockade of the Wnt pathway ameliorates retinal inflammation, vascular leakage, and NV in DR models (8, 50). Consistently, the present study demonstrates activation of the Wnt pathway and overexpression of VEGF and ICAM-1, target genes of the Wnt pathway in retinal cells under diabetic stressors and in the retinas of type 1 diabetic rats. Moreover, we find that activation of the Wnt pathway correlates with increases of retinal vascular leakage and inflammatory cell infiltration in the diabetic retina. In the retina of diabetic animal models, our results indicate that the increasing 3-NT levels correlate with the phosphorylation of LRP6, a key coreceptor in the Wnt pathway, and accumulation and nuclear translocation of β-catenin, an essential effector of canonical Wnt signaling. Similarly, nitrosative stress inducers, such as PN, HG, and HNE, can activate Wnt signaling in cultured cells.
To further confirm the causative role of nitrosative stress in Wnt pathway activation and retinal inflammation and vascular leakage in DR, we suppressed nitrosative stress by UA, as UA is a commonly used inhibitor of nitrosative stress. As shown by LRP6 phosphorylation and β-catenin accumulation and nuclear translocation, Wnt signaling activation by PN and diabetic stressors can be attenuated by UA. UA attenuated Wnt pathway activation induced by the nitrosative stress inducers in cultured cells and in the retina of diabetic animals. UA also blocked the overexpression of VEGF and ICAM-1 induced by diabetic stressors and in the retina of diabetic rats. Furthermore, we demonstrated that FeTPPS, a PN decomposition catalyst, conferred a similar effect as UA in inhibition of the Wnt ligand and HG-induced Wnt pathway activation. Taken together, these in vitro and in vivo results suggest that nitrosative stress is responsible, at least in part, for the Wnt pathway activation in diabetes, and activation of Wnt signaling by nitrosative stress may represent a pathogenic mechanism by which nitrosative stress induces retinal inflammation and vascular leakage in DR.
To define the target by which nitrosative stress activates the Wnt pathway, we have determined the effect of UA on Wnt signaling induced by Wnt3a, a ligand of the canonical Wnt pathway. We also activated the Wnt target genes using a constitutively active mutant of β-catenin, which is known to activate the target genes in the absence of the Wnt ligand. The results showed that UA inhibited Wnt signaling induced by Wnt3a, but not that by the constitutively active β-catenin mutant. Collectively, these results indicated that UA inhibits the Wnt pathway at the extracellular or cell membrane levels. It is possible that PN could cause tyrosine nitration in some components of the Wnt pathway, which may affect protein kinase/phosphatase activity, and thus regulate LRP6 phosphorylation. Our results indicate that UA and FeTPPS prevent the tyrosine nitration by decreasing PN; however, the exact molecular mechanism by which UA and FeTPPS inhibit the Wnt pathway remains to be identified.
Hyperglycemia is a direct risk factor for DR (1, 41). Hyperglycemia can induce mitochondrial dysfunction, which leads to superoxide overproduction, subsequently upregulating iNOS and endothelial NO synthase expression, increasing NO production (5). Superoxide can react with NO to generate PN, which can directly, or through tyrosine nitration and DNA damage, lead to endothelial dysfunction (7). PN also upregulates adhesive molecules and proinflammatory cytokines, which actively contribute to DR (5, 36). Our in vitro results showed that HG increased ROS/RNS, and thereby induced nitrosative stress. In turn, nitrosative stress activated the Wnt pathway and induced expression of Wnt-signaling target genes, such as VEGF and ICAM-1. All of these events were significantly attenuated by UA and FeTPPS. Taken together, these results suggest that nitrosative stress is a causative factor for activation of the Wnt pathway in diabetic conditions. The results from diabetic animal models provide further evidence supporting this conclusion. 3-NT is significantly increased in the retina of diabetic rats compared to that of nondiabetic control, correlating with Wnt pathway activation. Attenuation of nitrosative stress by UA dramatically suppressed the Wnt pathway activation, downregulated VEGF and ICAM-1 expression, ameliorated the vascular leakage, and inhibited inflammatory cell infiltration. These findings provide further evidence, suggesting that nitrosative stress is responsible for the development of retinal inflammation and retinal vascular leakage in DR through a Wnt signaling-dependent mechanism.
In conclusion, our study for the first time demonstrated that nitrosative stress in diabetes contributes to Wnt pathway activation in the retina. Thus, Wnt pathway activation induced by nitrosative stress may represent a pathogenic mechanism for retinal inflammation and vascular leakage in DR.
Materials and Methods
Experimental animals
Care, use, and treatment of all of the animals in this study were in strict agreement with the Statement for the Use of Animals in Ophthalmic and Vision Research. The experimental diabetes was induced as described previously (44).
Cell culture
ARPE19 cells and HTERT-RPE cells, both derived from human RPE cells, were purchased from American Type Culture Collection (ATCC; Manassas, VA). L-cells and L-cells expressing Wnt3a were obtained from ATCC and maintained in the Dulbecco's Modified Eagle Medium (DMEM) with 10% FBS and 0.4 mg/ml G418 (Invitrogen, Carlsbad, CA). The L-cell-conditioned medium and WCM were collected following the ATCC guidelines.
Subcellular fractionation
Cells were fractionated using the FractionPREP™ Cell Fractionation Kit (BioVision, Mountain View, CA) following the manufacturer's instructions.
Measurement of intracellular ROS generation
Intracellular PN anion was measured by 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCF-DA, Molecular Probe, Invitrogen). The generated fluorescence intensity was determined by a fluorescence microplate reader (Perkin Elmer, Waltham, MA) with excitation wavelength at 485 nm and emission wavelength at 535 nm.
Plasmid transfection and TOPFLASH reporter assay
The TOPFLASH or FOPFLASH vectors were cotransfected with a Renilla luciferase pRL-TK vector into the HTERT-RPE cells. TOPFLASH activity was measured using a dual luciferase reporter system (Promega, Madison, MI) and normalized by Renilla luciferase activity.
Western blot analysis
Fifty micrograms of cellular proteins was subjected to 7.5% to 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred onto nitrocellular membranes (Bio-Rad, Hercules, CA) and blotted with primary antibodies as described previously (43). The primary antibodies were diluted as the following: anti-pLRP6 (Ser1490, 1:1000; Cell Signaling Technology, Danvers, MA), anti-LRP6 (1:2000; generated in our lab), anti-non-phospho-β-catenin (1:1000; Cell Signaling Technology), anti-β-catenin (1:3000; Santa-Cruz Biotechnology, Santa Cruz, CA), anti-3-Nitrotyrosine (3-NT, 1:1400; Abcam, Cambridge, MA), anti-VEGF (1:500; Santa-Cruz Biotechnology), anti-ICAM-1 (1:500; Santa-Cruz Biotechnology), and anti-β-actin (1:50,000; Sigma-Aldrich, St. Louis, MO).
ELISA for VEGF and 3-NT
VEGF secreted into the culture medium was determined using the VEGF ELISA Kit (R&D Systems, Minneapolis, MN) according to the manufacturer's instructions. Intracellular tyrosine nitration was quantified by the Nitrotyrosine ELISA kit (Millipore, Bellerica, MA) according to the manufacturer's instructions.
Immunohistochemistry
Immunohistochemistry was performed as described (9). The primary antibodies were used as following: anti
Statistical analysis
Data were presented as mean±S.D. Statistical analyses were performed using ANOVA with Bonferroni's post hoc test. A p-value<0.05 was considered as statistically significant.
Footnotes
Acknowledgments
This study was supported by the National Basic Research Program of China (Project 973) Grant 2011CB504606 (to LZ) and by the NIH grants EY018659, EY012231, EY019309, and P20RR024215 (to JXM).
Author Disclosure Statement
No conflicts in financial interests.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.