
Abstract
Introduction
AngII leads to the activation of signaling molecules in multiple downstream pathways, including kinases, transcription factors, cytokines, and growth factors, and much of this activity results from its ability to modulate the generation of unstable oxygen derivatives, known as reactive oxygen species (ROS), and oxidative stress. ROS continuously arise as a result of leakage from the mitochondrial electron transport chain, or may be generated as an enzymatic byproduct of metabolism or the unfolded protein response during endoplasmic reticular stress. In addition, nitric oxide synthase (NOS) uncoupling can lead to production of ROS instead of nitric oxide (NO•), and nicotinamide adenine dinucleotide phosphate (NAD[P]H) oxidases produce ROS as their primary product. Endogenous antioxidants, including superoxide dismutase (SOD), glutathione peroxidase (GPx) and catalase, as well as nonenzymatic scavengers, serve to counteract these sources of ROS, maintaining equilibrium and preventing oxidative stress. Although ROS have been shown to play a role in normal physiological cell signaling, excessive ROS production can be extremely detrimental, through direct damage to cellular components as well as through reduction–oxidation (redox) modification of signaling molecules. Like AngII, ROS and oxidative stress have been shown to play an important role in the progression of CVD and HF. Thus, there is considerable interest in clarifying the relative importance of the various mechanisms of ROS production in the heart, their downstream effects, and the effectiveness of therapeutically targeting them.
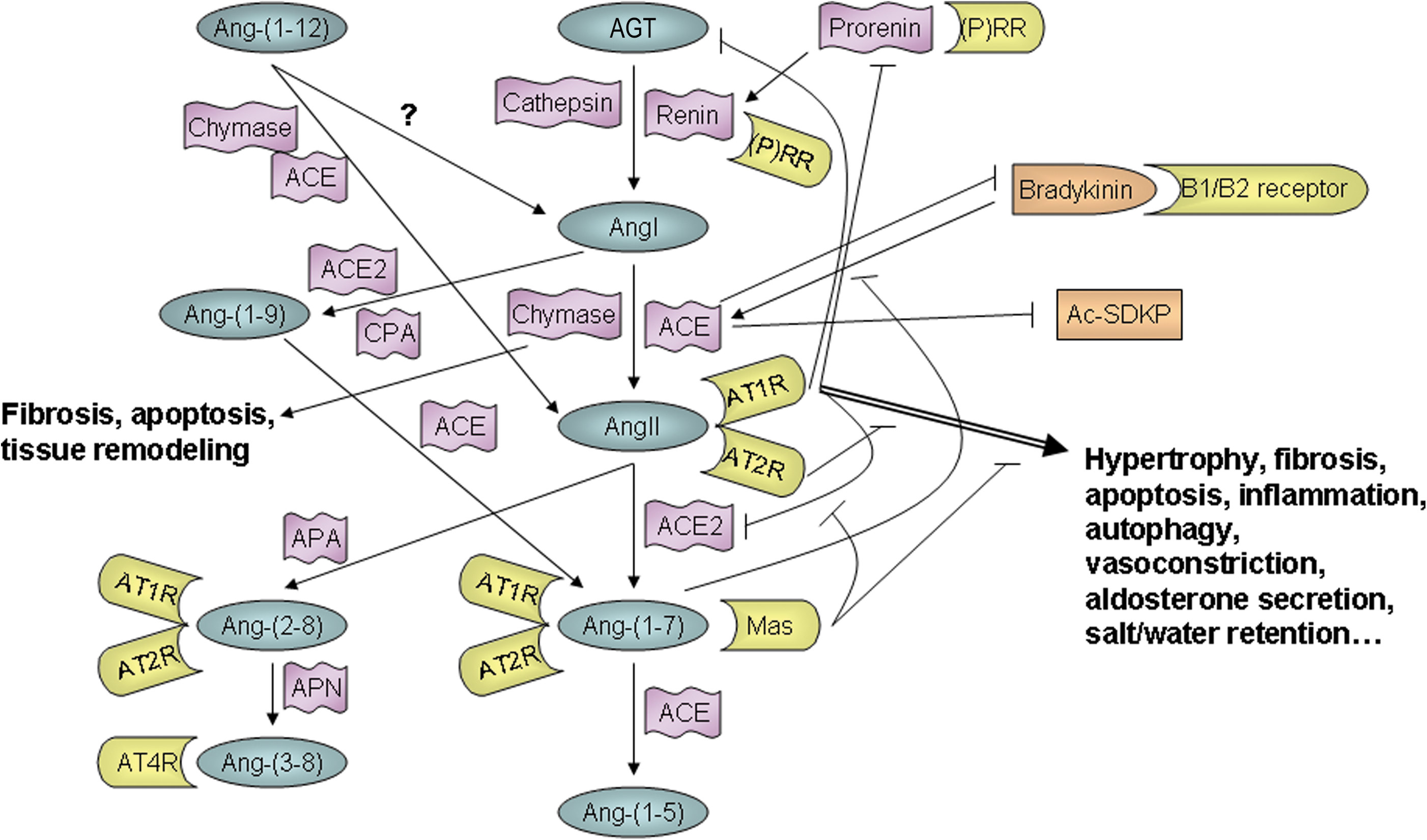
Overview of the RAS
In the classical RAS, angiotensinogen (AGT) produced in the liver and renin produced in the kidneys enter the bloodstream, where the renin cleaves the AGT to form the decapeptide, AngI. Angiotensin-converting enzyme (ACE), which is mainly produced in the lung vascular bed, and which is bound to the lumenal side of endothelial cells in the vasculature, then cleaves AngI to produce the biologically active octapeptide, AngII. Finally, AngII binds to either the AngII type 1 receptor (AT1R), a G protein–coupled receptor through which it exerts most of its known functions, or alternatively, to the AngII type 2 receptor (AT2R), another G protein–coupled receptor, whose function was until recently largely unknown, but is thought mainly to oppose AT1R signaling (52, 107, 117). However, it is now known that this linear systemically acting pathway is a greatly oversimplified version of the actual RAS.
Research conducted over the last few decades has shown that AngII is generated locally in some tissues, including the heart (51, 52), as well as systemically, and that there are actually many more components in the RAS than was initially thought. Several other Ang peptides have been discovered, including Ang(1 –5), Ang(1 –7), Ang(1 –9), Ang(2 –8) (also known as AngIII), Ang(3 –8) (also known as AngIV), and Ang(1 –12) (24, 36, 52). Although the functions of these additional Ang peptides have not all been well characterized and some may simply be intermediary or degradation products, there is increasing evidence that most, if not all, of these peptides are biologically active and may be important in modulating RAS function. In particular, Ang(1 –7), which is formed by cleavage of either AngII or Ang(1 –9), can bind to both the AT1R and AT2R, as well as to the Mas receptor, and plays an important role in opposing the effects of AngII signaling, including cardiac hypertrophy and fibrosis (25, 36, 75, 135). There is some evidence that Ang(1 –9) may be an important cardioprotective factor as well (90), and Ang(1 –12) serves as an alternate precursor for AngI and AngII formation (100, 130), bypassing the need for AGT in the RAS cascade. It is also now known that renin and ACE are not the only enzymes that take part in the RAS. AGT can be cleaved to form AngI by cathepsin as well as renin, and chymase, rather than ACE, is responsible for cleavage of AngI (18, 51, 79, 136) and possibly Ang(1 –12) to AngII (100) under certain circumstances. In addition, an ACE homolog, ACE2, has been identified and shown to be involved in cleaving AngI and AngII to form Ang(1 –7) (52). The relative importance of the various RAS components appears to be context- and species-dependent, and continues to be the subject of much research.
In addition, it is now known that the RAS is not the simple linear cascade it was once thought to be. Several endogenous negative regulators and feedback mechanisms exist that prevent excessive AngII/AT1R signaling under normal circumstances. Although its physiological importance requires further clarification, AT1R-associated protein has been shown to bind to the AT1R and promote receptor internalization, thus acting as an endogenous AT1R inhibitor (124, 133). Investigation into AT2R signaling has shown that many, though not all, of its effects oppose those of the AT1R, such that some of the beneficial effects of AT1R blockers (ARBs) in treating CVD may be due to increased AngII signaling through the AT2R (52, 84, 97, 107, 117, 145). However, it appears that the AT2R may also have some functions similar to those of the AT1R, since mice lacking the AT2R do not develop hypertrophy in response to pressure-overload (108), nor do they develop hypertrophy and fibrosis in response to AngII infusion (33). AngII/AT1R signaling can also be self-limiting in that it suppresses AGT and renin expression. Perhaps the most extensively studied negative feedback mechanism, however, is that of the ACE2/Ang(1 –7)/Mas axis, which has been shown to play a major role in opposing AngII signaling, both through AngII degradation and the positive and negative actions of Ang(1 –7) itself (19, 25, 45, 75, 129, 135, 151). Furthermore, some of the RAS enzymes have additional functions that do not involve Ang peptides. Binding of renin or its precursor, prorenin, to the (pro)renin receptor can activate extracellular signal-regulated kinase in an AngII-independent manner (23). In addition to Ang peptides, ACE also hydrolyzes a variety of other substrates, including the cardioprotective factors bradykinin, providing a link between the RAS and kallikrein/kinin system, and N-acetyl-seryl-aspartyl-lysyl-proline tetrapeptide (52, 95). Likewise, chymase function is not limited to Ang peptide cleavage, playing an important role in tissue remodeling through activation of transforming growth factor-β (TGF-β) and matrix metalloproteases (MMPs), as well as catalyzing degradation of thrombin and plasmin (18, 121).
Thus, the RAS is actually a complex network of intertwining pathways (Fig. 1), and much of the complexity stems from differences between systemic and local tissue AngII generation and signaling. While the question of whether AngII is generated intracellularly as well as extracellularly remains the subject of some debate, there is little doubt that the tissue RAS plays an important role in determining cardiac function. The effects of locally generated AngII may be intracrine or autocrine/paracrine, depending on the cell type and conditions (51, 52, 103).
AngII and HF
The first clinical evidence of the benefits of RAS inhibition in treating HF came from the results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS) in 1987, in which treatment with the ACE inhibitor, enalapril, significantly improved mortality and cardiac symptoms in patients with severe congestive HF (1). In the more than two decades since, multiple clinical studies have demonstrated the effectiveness of blocking AngII generation and/or signaling with ACE inhibitors or ARBs in treating hypertension, as well as congestive HF, acute myocardial infarction (MI) and coronary artery disease (47, 114). As a result, current guidelines recommend the use of these drugs as first-line therapy for the treatment of patients at risk for or suffering from HF (38). More recently, there has been some evidence that the use of renin inhibitors may be cardioprotective as well, and additional studies to further clarify their potential benefits are ongoing (34). Nevertheless, therapeutic approaches that inhibit AngII, while beneficial, have not proven to be entirely effective in eradicating CVD, and dual therapies combining different types of RAS inhibitors remain controversial. Two large trials conducted in patients with HF, the Valsartan- HF Trial (ValHeFT) and the Candesartan in HF: Assessment of Reduction in Mortality and Morbidity-Added (CHARM-Added) Trial, showed that combined treatment with ACE inhibitors and ARBs significantly improved morbidity and mortality due to CVD or congestive HF, but not overall mortality (50, 72). In the Ongoing Telmisartan Alone and in Combination with Ramipril Global Endpoint Trial (ONTARGET), dual therapy with ACE inhibitors and ARBs led to worse renal outcomes than with either ACE inhibitors or ARBs alone, suggesting that there may be significant risks associated with this combined therapy (68). On the other hand, the Aliskiren Observation of HF Treatment (ALOFT) study showed that treatment with the direct renin inhibitor, aliskiren, ameliorated the neurohormonal derangements and reduced left ventricle (LV) filling pressures in patients with congestive HF already under treatment with ACE inhibitors or ARBs (73). Thus, current evidence suggests that more efficient RAS system inhibition may lead to better clinical outcomes in patients with HF, and investigation into AngII function and its inhibition, including endogenous inhibitory mechanisms, continues to be an active area of research (146).
Among the most well-known effects of AngII are salt/water retention and vasoconstriction, and AngII-induced hypertension plays an important role in the progression of CVD and HF. However, AngII also has blood pressure-dependent and blood pressure-independent effects in the heart that play a critical role in causing cardiac remodeling and dysfunction as well. AngII is an important regulator of cardiac cell growth, causing both cardiac hypertrophy and fibrosis (17, 35, 39, 40, 43, 79, 85, 86, 89, 93, 107, 113, 134, 141, 142, 149). On the other hand, AngII is also involved in cell death, playing a role in cardiomyocyte apoptosis (12, 22, 41, 42, 60, 94, 101, 113, 142, 153) and, as demonstrated in recent studies, in autophagy as well (16, 17, 97, 98). Although generally considered a homeostatic mechanism, autophagy has been shown to be detrimental during reperfusion injury (29, 71), and autophagic cell death has been detected in the cardiac tissue of HF patients (28). AngII signaling activates tumor necrosis factor α (TNFα) and nuclear factor κB (NFκB) (40, 77, 109, 120, 122, 153), and upregulates endothelial chemokines and chemoattractant molecules, thus triggering an inflammatory response in models of diastolic HF and MI, as well as in aging-induced cardiomyopathy (45, 77, 89, 107, 120, 142, 143). In addition, intracellular AngII has been shown to have a negative effect on myocyte junctional conductance and ion channel function (109, 112). However, because it is a more immediate consequence of AngII signaling and acts upstream to promote most of the other effects mentioned above, perhaps the most important effect of AngII is increased production of ROS and oxidative stress.
Overview of ROS
ROS are unstable, reactive oxygen derivatives formed by the reduction of one or more electrons from molecular oxygen, including superoxide (O2 •−), hydrogen peroxide (H2O2), and hydroxyl radical (OH•), among others. O2 •− can be generated by a number of mechanisms in the cell, including leakage from the electron transport chain and as a metabolic byproduct of enzymes such as xanthine oxidase (XO) and cyclooxygenase. ROS can also be produced by oxidoreductases of the endoplasmic reticulum during the unfolded protein response or as a result of NOS uncoupling. In addition, NAD(P)H oxidases (Noxs) produce O2 •− as their primary product, using either NADH or NADPH as an electron donor. O2 •− is highly reactive but poorly lipid-soluble, such that its direct effects are largely confined to the subcellular compartment in which it is generated except in cases of active transport. However, O2 •− is rapidly converted to the less reactive but more highly membrane-permeable H2O2, either spontaneously or by SODs, allowing for a more-widespread effect throughout the cell. Additional highly reactive species are generated when H2O2 is reduced further through the iron-catalyzed Fenton reaction to produce OH•, and when O2 •− combines with NO• to form peroxynitrite (ONOO−).
This ongoing ROS production is normally counteracted and an equilibrium oxidative state is maintained by SODs and the reduction of H2O2 to H2O by catalase and peroxidases, as well as the actions of nonenzymatic antioxidants and radical scavengers. Many cellular proteins undergo changes in conformation and activity due to reduction and oxidation of their thiol groups, and ROS have been shown to play a key role in many important physiological processes, including host defense and inflammation, cellular signaling, gene expression and regulation of cell growth and death, and oxygen sensing (7). A recent study demonstrated that ROS play an essential role in starvation-induced autophagy by inhibiting the Atg4 cysteine protease through a redox-dependent mechanism (106). Despite a decrease in oxidative stress, antioxidant treatment exacerbated ischemia/reperfusion (I/R) injury in Dahl salt-sensitive hypertensive rats (69), and antioxidant treatment in conjunction with losartan treatment abolished the protective effect of the ARB against LV remodeling and in cardiomyopathic hamsters, apparently due to the inhibition of redox-dependent activation of NOS (70). Nox4 overexpression in cardiomyocytes and the resultant increase in H2O2 production protected mouse hearts against the effects of chronic pressure overload by increasing angiogenesis, possibly due to enhancement of hypoxia-inducible factor-1 activation and vascular endothelial growth factor release (147). Other examples include the regulation of calcium/calmodulin-dependent protein kinase II (CaMKII), class II histone deacetylases (HDACs), and the Na+-K+ pump in the heart, all of which were recently demonstrated to undergo reversible oxidative modification (3, 22, 26).
Oxidative Stress and HF
Under pathological conditions, however, the equilibrium oxidative state may be disrupted due to either an increase in ROS production or a decrease in antioxidant function, leading to the development of oxidative stress. Excess ROS can cause direct damage to proteins, lipids, and DNA, or can contribute to pathophysiological processes through inappropriate reversible or irreversible redox modification of cellular proteins.
ROS and oxidative stress have been implicated in a number of pathological processes that contribute to HF, including vasoconstriction, cardiac hypertrophy, myocyte apoptosis, fibrosis, inflammation, I/R injury, and myocardial stunning (48, 116). In addition, there is growing evidence that ROS are directly involved in the development of HF. In rats, the progression from hypertension-induced compensated left ventricular hypertrophy to decompensated HF was associated with an increase in myocardial ROS (132). Similarly, a number of studies have shown that O2 •− production and oxidative stress are significantly increased in the hearts of patients with dilated cardiomyopathy and HF (11, 30, 37, 66, 87, 104, 131). Thus far, clinical studies have not demonstrated a clear beneficial effect of antioxidant therapy in improving cardiovascular outcomes (46, 62, 128), however, possibly due to inhibition of beneficial ROS signaling mechanisms together with the detrimental ones.
AngII, Oxidative Stress, and HF
Numerous studies have shown that AngII signaling in the failing heart is associated with increased ROS generation and oxidative stress. AngII infusion for 2 weeks, which causes cardiac hypertrophy, doubled OH• production in mouse hearts in an AT1R-dependent manner (43). In rats, increased oxidative stress after MI coincided with an increase in RAS components in infiltrating macrophages, suggesting the presence of local AngII production, and was blocked by treatment with an ARB (65). Similarly, the protective effects of ARB treatment against myocarditis-induced HF and hypertensive diastolic HF in rats and pacing-induced HF in dogs were associated with decreases in ROS production and oxidative stress (27, 80, 119). In addition, oxidative stress markers were shown to be increased and correlated with circulating AngII levels in human HF patients, and were highest in patients with increased AT1R responsiveness to AngII due to homozygocity for the AT1R A1166C gene polymorphism (14).
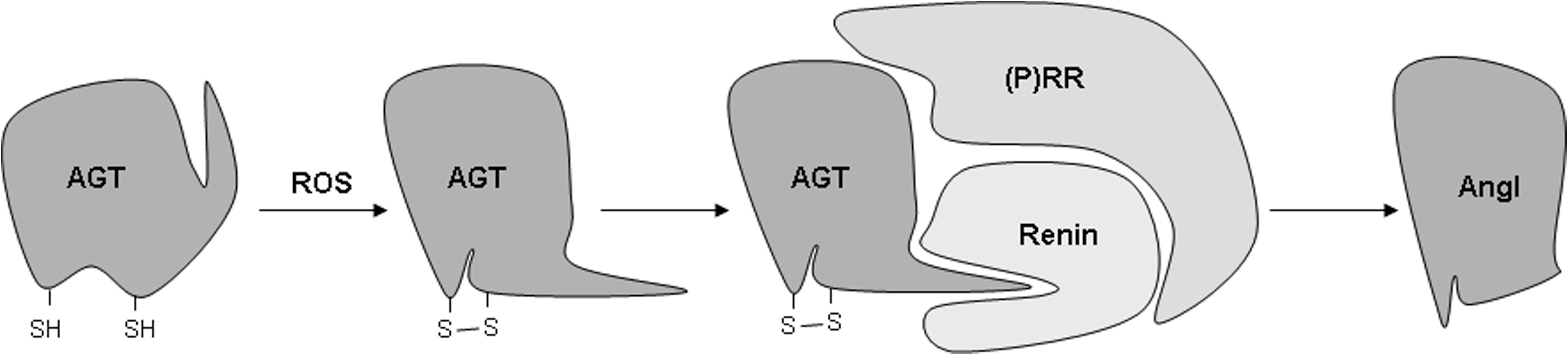
As mentioned above, many of the downstream effects of AngII signaling are mediated through ROS generation. AngII-induced ROS modulate gene expression through transcription factor activation, upregulate growth factors and cytokines, and activate kinases. Furthermore, AngII-induced oxidative stress can cause mitochondrial damage and dysfunction. Oxidative stress has been shown to be important in mediating AngII-induced cardiac hypertrophy, apoptosis, and fibrosis, as well as playing a role in AngII-dependent inflammation and cardiomyocyte contractile dysfunction. Importantly, cleavage of AGT by renin was recently shown to require a conformational change brought about by formation of a disulfide bridge between two cysteines as a result of oxidation (152), suggesting the presence of a positive feedback mechanism by which AngII-induced ROS further increase AngII formation (Fig. 2). How AngII increases cardiac oxidative stress is still not entirely understood. However, recent studies have pointed to the involvement of several different mechanisms.
NAD(P)H Oxidase
As mentioned above, Noxs are a family of membrane-bound enzymes that specifically produce O2 •− by transferring an electron from NAD(P)H to O2 as their primary function, rather than as a byproduct. The prototype Nox catalytic subunit, gp91phox or Nox2, was first identified as being responsible for the respiratory burst that plays an important part of the killing response in phagocytes. There are now seven known members of the Nox family, Nox1–Nox5 and Duox1–Duox2. Of these, Nox1–Nox4 have highly conserved structures, comprising six transmembrane domains, two hemes, and NAD(P)H- and flavin adenine dinucleotide–binding domains in the C-terminus, and require heterodimerization with a second membrane protein, p22phox, for stabilization and activation (7). In the heart, Nox activity is primarily due to Nox2 and Nox4 (2, 8, 13) (Fig. 3), which, despite their structural similarities, differ in cellular localization and functionality. Nox2 is located primarily on the plasma membrane, and, in addition to binding p22phox, its activation involves binding of cytosolic subunits p47phox and p67phox (and, in some cases, p40phox), and Rac1 GTPase, allowing for post-translational modulation of Nox2 activity through regulation of cytosolic subunit translocation (7). Nox4, on the other hand, is primarily located on perinuclear intracellular membranes, such as those of the mitochondria (2), and does not require cytosolic subunit binding, but rather is regulated primarily at the transcriptional level (7).

There is abundant evidence that Nox activity plays an important role in cardiac remodeling and HF (53, 67). Expression of Nox2 and its cytosolic cofactors is increased during the progression of cardiac hypertrophy to HF in guinea pigs subjected to pressure overload and in hypertensive Dahl salt-sensitive rats (56, 122), and Nox activity is increased in end-stage failing human hearts (30, 66, 87). Nox activation plays a critical role in AngII-induced apoptosis. ONOO− formed as a result of AngII-induced Nox activity causes DNA damage and p53 activation, leading to an increased Bax/Bcl-2 ratio, caspase-3 cleavage, and apoptotic cell death in H9c2 cells (60). Since p53 also binds to and activates the promoters of AGT and AT1R (96), this may represent a positive feedback mechanism by which AngII signaling self-amplifies. AngII-induced apoptosis also involves phosphorylation of p38 after activation of CaMKII by ROS generated by Nox (22, 94, 101). ROS lower the CaMKII activation threshold to subdiastolic Ca2+ concentrations (94), and activate the kinase through parallel mechanisms of autophosphorylation at threonine 287, possibly as a result of phosphatase oxidation and inhibition, and oxidation of two redox-sensitive methionine residues (22, 31) (Fig. 4). This ROS-dependent activation of CaMKII may also play a role in the development of cardiac hypertrophy through phosphorylation of class II HDACs. Nox2 and Nox4 have been implicated in AngII- and pressure overload-induced cardiac hypertrophy, respectively (8, 13), and phosphorylation of class II HDACs such as HDAC4 by CaMKII and other HDAC kinases leads to their nuclear export and de-repression of prohypertrophic transcription factors (4, 92). In addition, HDAC4 is subject to reversible redox regulation, with oxidation of conserved cysteine residues and disulfide bond formation leading to nuclear export regardless of its phosphorylation state (3). While a role for Noxs in this mechanism has yet to be established, Nox4 is expressed in the nucleus and is upregulated by hypertrophic stimuli such as AngII and phenylephrine (2), making such a role plausible. Increased Nox-dependent ROS also activate NFκB, TGF-β, MMPs, and growth factors, leading to increased fibrosis, extracellular matrix degradation, and tissue remodeling, and contribute to AngII-dependent cardiac contractile dysfunction (10, 40, 45, 58, 63, 105, 122, 149, 150). The latter may be due in part to a role in the regulation of cardiac ion channels. Nox-dependent O2 •− production and oxidative stress due to increased cellular AngII cause a decrease in the K+ currents of ventricular myocytes in diabetic rats (110 –112), and AngII inhibits transcription of the cardiac Na+ channel, leading to a decrease in the Na+ current, through activation of NFκB by Nox-dependent ROS (109). Glutathionylation by ONOO− produced as a result of protein kinase C-dependent Nox activity is also responsible for AngII-induced inhibition of the cardiac sarcolemmal Na+-K+ pump, and may contribute to myocardial damage and contractile dysfunction as a result of high Na+ and Ca+ levels (26, 102, 139). The form-specific functions of Nox2 and Nox4 in mediating these effects require further clarification, however. Many of the above studies examined p47phox expression and translocation as an indicator of Nox activity, suggesting the involvement of Nox2, but systemic knockout of p47phox in mice inhibited increases in both Nox2 and Nox4 expression after aortic constriction, and only Nox4 was inhibited in ACE2/p47phox double knockout mice (10). On the other hand, cardiomyocyte-specific deletion of Rac1, which attenuates association of Nox2 with its cytosolic cofactors, also inhibits AngII-induced cardiac hypertrophy and dysfunction (105), and systemic Nox2 knockout protects the heart against AngII-induced hypertrophy, and MI- and doxorubicin-induced adverse remodeling and dysfunction (8, 63, 150). Doxorubicin treatment increases both Nox2 and Nox4 mRNA levels, but neither one is increased in Nox2 knockout mice, suggesting that Nox2 may play a role in regulating Nox4 expression (150). Zhang and colleagues found that Nox4 expression in cardiomyocytes was protective against pressure overload-induced cardiac dysfunction, hypertrophy, and fibrosis due to paracrine activation of angiogenesis (147). However, studies in our laboratory demonstrated that Nox4 expression in cardiomyocytes is detrimental, leading to increased apoptosis and mitochondrial dysfunction in aged mice, and increased cardiac dysfunction, fibrosis, and apoptosis after aortic constriction (2, 54). In both aging and aortic constriction, the cardiomyocyte cross-sectional area was increased, but there was no organ-level hypertrophy (2, 54), suggesting that Nox4-associated hypertrophy may be secondary to its role in apoptosis and dysfunction. Neither cardiac-specific overexpression nor systemic or cardiac-specific knockout of Nox4 resulted in any baseline abnormality in young mice (2, 54, 147), and the discrepancies between these studies may be due to differences in genetic background or experimental conditions.

The vast majority of currently available data point to Nox as the source of AngII-induced ROS. In vitro and in vivo studies in rats and mice have shown that AngII treatment stimulates an increase in ROS production in the heart that is significantly inhibited by diphenylene iodonium (DPI, an inhibitor of flavoproteins such as Nox), apocynin (a Nox inhibitor), or dominant negative Rac, but not by N G-nitro-L-arginine methyl ester (L-NAME, a NOS inhibitor), rotenone (a complex-I mitochondrial electron chain inhibitor), or XO inhibitors, suggesting that Nox is the major source of AngII-induced ROS in cardiomyocytes (13, 27, 88, 89, 101, 118, 134, 138) (This increase in Nox activity and oxidative stress was not observed in female mice, however [21]). Similarly, rats known to have increased AngII levels, such as Ren2 and hypertensive Dahl salt-sensitive rats, displayed an AT1R-dependent increase in ROS generation, which was inhibited by DPI (27, 89, 138). AngII has also been shown to increase translocation of p47phox to the membrane (17, 58, 153), as well as membrane-associated Rac1 GTP-binding activity (86), both of which are involved in Nox2 activation, as mentioned above. In addition, RAS inhibition by ACE inhibitors or ARBs appears to inhibit the increased expression of Nox subunits p22phox, Nox2, p47phox, and p67phox observed in rats and mice with pathological cardiovascular conditions such as spontaneous hypertension, diastolic HF, MI, or cardiomyopathy due to experimental autoimmune myocarditis, doxorubicin treatment, or hyperglycemia (27, 35, 77, 99, 119, 138, 150).
The relative importance of Nox2 and Nox4 in mediating the effects of AngII in the heart remains a subject of some debate, however. Several in vitro and in vivo studies have shown that AngII stimulation increases expression of Nox2, p47phox, and membrane-associated Rac1 (17, 101, 134, 141, 149), and most of the studies investigating the role of AngII-induced Nox activity and expression in pathological cardiovascular conditions have focused on Nox2 and its cytosolic subunits, as noted above. Furthermore, studies with Nox2 knockout mice found that Nox2 deficiency abolished the AngII-induced Nox activity observed in wild-type mice (13, 40). However, these studies do not preclude the possibility that ROS produced by one Nox form might be involved in upregulating the other, with both playing a role in mediating the downstream effects of AngII. Furthermore, these studies used relatively short periods of AngII treatment, leaving untested the possibility that longer chronic exposure to AngII might directly upregulate Nox4. Indeed, our laboratory and others have recently demonstrated that AngII treatment increases Nox4 expression in mouse hearts (2, 17). In addition, Dai and colleagues found that, while AngII increased both Nox2 and Nox4 expression, the increase in Nox2 was inhibited by catalase while the Nox4 increase was not (17), suggesting that the two forms are regulated by different, though possibly interdependent, mechanisms.
Nitric Oxide Synthase
The three NOS isoforms, neuronal NOS, inducible NOS, and endothelial NOS (eNOS), are a family of homodimeric enzymes whose primary function is to produce NO• by transfer of electrons from NADPH to O2 and oxidation of L-arginine to L-citrulline via the stable intermediary, Nω-hydroxy-L-arginine. This process involves shuttling electrons from the C-terminal reductase domain of one monomer to the N-terminal oxidase domain of the other monomer, and requires the presence of several cofactors, including tetrahydrobiopterin (BH4) (57, 81). NO• is a powerful vasodilator, and it and sufficient NOS activity are generally considered to be protective factors in the cardiovascular system. However, NOS can also contribute to ROS production since, as noted above, in the presence of a nitroso-redox imbalance, O2 •− can combine with NO• to form ONOO−. Furthermore, oxidation of NOS by ONOO− or a deficiency in either L-arginine or BH4 causes uncoupling of the enzyme, resulting in generation of O2 •− instead of NO• (57). BH4 is itself readily oxidized by ROS to the inactive BH2, leading to a self-perpetuating cycle of BH4 deficiency, NOS uncoupling, and ROS generation (81).
ROS generation due to eNOS uncoupling was shown to play a major role in cardiac remodeling brought about by chronic pressure overload in mice (123). However, the role of NOS uncoupling in HF and AngII-induced ROS generation is not well understood. On the one hand, eNOS expression was significantly downregulated in the hearts of spontaneously hypertensive stroke-prone rats, but upregulated to normal levels in the presence of RAS inhibition (125), and NOS inhibition abrogated the protective effect of treatment with the ARB, losartan, against left ventricular remodeling in hamsters with cardiomyopathy, and I/R injury in Dahl salt-sensitive rats (69, 70), suggesting that NOS activity is protective against the effects of AngII. Similarly, adenoviral gene transfer of the human eNOS gene into rat hearts after MI resulted in increased cardiac NO• production and reduced cardiac ROS generation and remodeling (115). More specifically, as mentioned above, AngII-induced ROS generation in the hearts of rats was not inhibited by L-NAME (134). On the other hand, AngII treatment increased eNOS expression while decreasing NO• production, indicating the presence of eNOS uncoupling, in rat aortas (83), and treatment with the ACE inhibitor, captopril, normalized the increased eNOS expression observed in the LVs of hamsters with chronic congestive HF (82). There is some evidence that BH4 supplementation may be effective in counteracting some of the effects of AngII and treating CVD and HF (44, 81). However, BH4 can function as an antioxidant in addition to stabilizing NOS, so its importance in clarifying the role of NOS uncoupling remains to be determined. Losartan treatment increased eNOS expression and NO• production in the cardiomyocytes of obese rats, but had the opposite effect in control animals (32), suggesting that the relationship between AngII signaling and eNOS function may be context-dependent. eNOS expression may be increased by ROS via activation of redox-sensitive transcription factors or mRNA transcript stabilization, but cytokines such as TNFα, which are activated by AngII, may reduce eNOS expression (64). Furthermore, because ROS induce NOS uncoupling, generation of ROS by NOS may be secondary to production of ROS from other sources, so that considerable additional investigation may be required to elucidate its role in mediating the effects of AngII.
Xanthine Oxidase
XO is one of two forms of the metalloflavoprotein, xanthine oxidoreductase, both of which catalyze oxidation of hydroxanthine to xanthine and xanthine to urate, playing a role in purine catabolism. However, whereas xanthine dehydrogenase (XD) uses NAD+ as an electron acceptor and produces urate and NADH as end products, XO transfers electrons to O2, resulting in formation of urate and O2 •− instead. XO is derived post-translationally from XD, either by irreversible proteolytic cleavage or by reversible oxidation of sulfhydryl residues (57).
There is a small but growing body of evidence that suggests that XO activity and the resulting generation of ROS play an important role in HF. Protein expression of both XD and XO has been shown to be elevated in failing human myocardium (15), and long-term high-dose treatment with the XO inhibitor, allopurinol, reduced mortality in patients with HF (137). Similarly, chronic allopurinol treatment improved cardiac function in rats with chronic HF, in part through a reduction in oxidative stress (74). In spontaneously hypertensive/HF rats with established dilated cardiomyopathy, although expression of both XO and the Nox subunits, Nox2 and p67phox, was increased, only the XO activity was elevated above normal, while Nox activity remained unchanged (78). Treatment with an XO inhibitor reduced ROS levels and oxidative stress by inhibiting XO activity (but not Nox activity), and improved cardiac function by reversing the existing adverse remodeling in these rats. Similarly, an XO inhibitor reduced left ventricular O2 •− levels, prevented the progression of adverse cardiac remodeling, and improved survival in Dahl salt-sensitive rats with established diastolic HF (39, 143).
However, the role of XO in mediating the effects of AngII is less clear. TNFα, which is activated by AngII, can induce proteolytic cleavage of XD, thus contributing to increased expression of XO (57). Alternatively, AngII-induced XO activation could be secondary to ROS generation from other sources through thiol oxidation of the XD sulfhydryl residues. Indeed, increased ROS levels in cultured neonatal rat ventricular myocytes after AngII stimulation were slightly, though not significantly, inhibited by allopurinol, suggesting that XO may contribute to some extent to AngII-induced ROS generation (118). Perhaps more convincing is the fact that allopurinol reduced oxidative stress and improved cardiac fibrosis and function in a blood pressure-independent manner in mice with AngII-induced diastolic dysfunction (39). In addition, although both Nox and XO activities were increased in the hearts of rats with diastolic HF and both were reduced by treatment with the ARB, candesartan, treatment with an XO inhibitor reduced cardiac ROS and closely mimicked the broadly protective effects of candesartan, while apocynin did not (143), suggesting that AngII-induced O2 •− formation is XO-dependent rather than Nox-dependent in this model. Accordingly, ACE inhibition significantly reduced XO substrates in hypertensive subjects, suggesting that AngII may regulate the activity of XO by controlling the availability of its substrates (91). In addition, AngII was shown to directly increase endothelial XO activity (55). Nevertheless, studies addressing the relationship between AngII and XO remain scarce, and XO inhibitors can act as direct ROS scavengers in addition to inhibiting XO function, thus complicating interpretation of data obtained from studies using these compounds and making further investigation necessary.
Antioxidant Downregulation
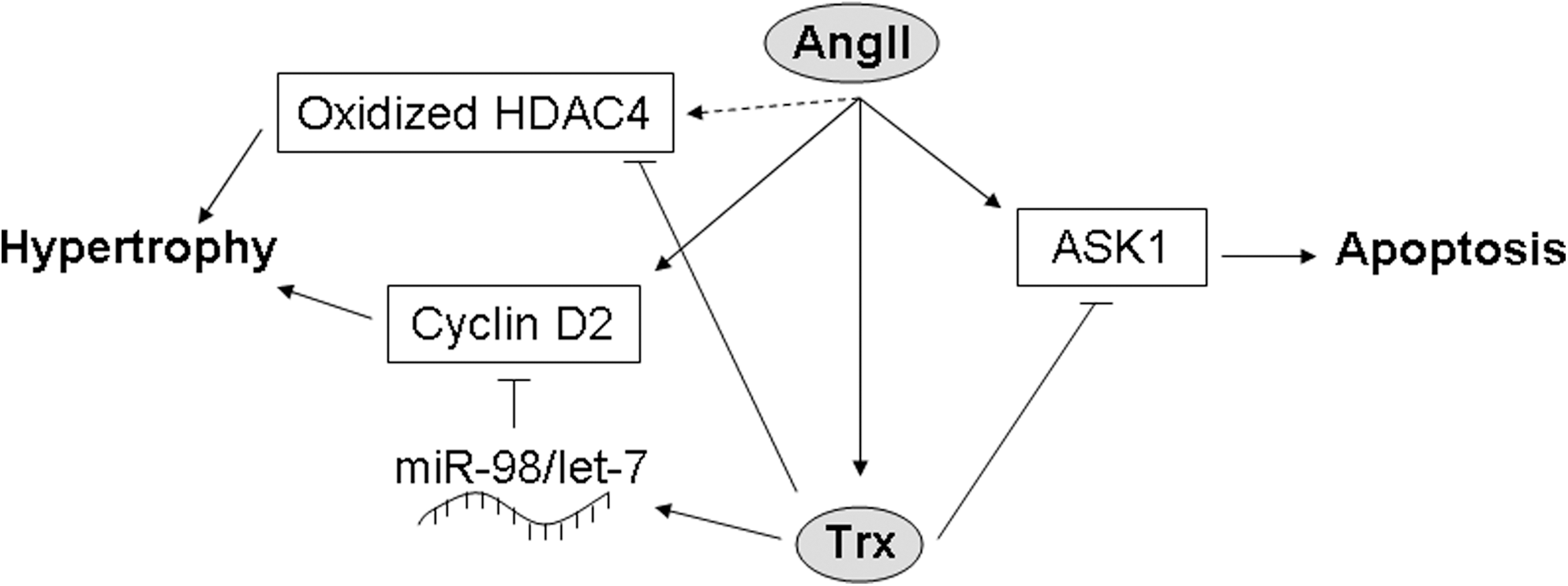
In addition to increasing generation of ROS, AngII could theoretically increase cardiac oxidative stress by downregulating antioxidant enzymes. While there is some evidence to support a role for such a mechanism in mediating the effects of AngII stimulation and in HF, the data are extremely mixed. Several studies have indicated that SOD activity is not significantly affected in the hearts of human HF patients (6, 11, 20). However, one study found that manganese SOD (MnSOD) activity (which comprised 90% of the cardiac SOD activity) and protein expression were significantly decreased in failing human hearts (104). In spontaneously hypertensive stroke-prone rats, expression and activity of MnSOD were unchanged, but those of copper–zinc SOD (Cu/ZnSOD) were decreased, an effect that was attenuated by treatment with an ACE inhibitor and abolished by ARB treatment (125), suggesting that AngII inhibits Cu/ZnSOD, but not MnSOD. However, in AngII-treated rat cardiac fibroblasts, the activity of both MnSOD and Cu/ZnSOD was inhibited, and expression of MnSOD, but not Cu/ZnSOD, was decreased (59). Similarly, in rats subjected to MI, MnSOD expression in the infarct area and activity of both MnSOD and Cu/ZnSOD were reduced, and these decreases were suppressed by treatment with RAS inhibitors (127). In addition, nuclear SIRT1, which is increased in cardiomyocytes in several models of HF, upregulates MnSOD expression, leading to a reduction in ROS levels and apoptosis in AngII-treated C2C12 cells, as well as improved cardiac function and survival in hamsters with chronic HF (126). In rats with MI-induced congestive HF, the activities of SOD, catalase, and GPx were all reduced, but only the GPx activity was improved by losartan treatment (49), suggesting that AngII is involved in downregulating GPx, but not the other antioxidants, in this model. Likewise, treatment with an ACE inhibitor increased GPx activity in rats with chronic MI (127). On the other hand, GPx expression and activity have been shown to be either unchanged or increased in AngII-stimulated rat cardiac fibroblasts and HF patients (6, 11, 20, 59, 104), and GPx mRNA expression is increased rather than decreased during the transition to congestive HF in hypertensive Dahl salt-sensitive rats, an effect that is reversed by treatment with an ARB (122). The data on catalase are even more difficult to interpret, with different studies indicating that catalase expression and activity are alternately upregulated, downregulated, or unchanged in the hearts of patients with end-stage HF (6, 11, 20, 104). Some of this discrepancy may be due to differences in the patient populations, including ischemic versus dilated cardiomyopathy, but further investigation is necessary to clarify this issue. In support of a role for catalase downregulation in mediating the effects of AngII, however, is the fact that catalase expression was shown to be downregulated in the hearts of AGT transgenic mice and in cardiomyocytes treated with AngII (85). Interestingly, although thioredoxin (Trx) can be oxidized (and thereby inhibited) by ROS, it is actually upregulated by AngII and in patients with chronic HF (21, 37, 144). Trx is protective against apoptosis by promoting ubiquitination and degradation of apoptosis signal–regulating kinase 1 (61, 148), prevents oxidation-dependent nuclear export of HDAC4 (3), and inhibits AngII-induced cardiac hypertrophy through upregulation of miR-98/let-7 microRNAs and downregulation of cyclin D2 (140, 144), suggesting that it acts as a negative feedback regulator of AngII (Fig. 5). Thus, while some studies appear to support a role for AngII in the regulation of antioxidants, others seem to negate the existence of such a mechanism or suggest that antioxidant activity may actually be upregulated in order to compensate for increased AngII-induced ROS generation. The solution to this riddle is probably context-dependent, and further investigation is required to fully establish the nature of the relationship between AngII signaling and antioxidant expression and activity.
Mitochondria
Mitochondrial leakage is considered the major source of ROS under normal physiological conditions, but the amount of ROS produced in this way is generally low and is kept in check by antioxidants such as mitochondrial MnSOD and catalase. However, under pathological conditions, mitochondrial damage and dysfunction can lead to blockage of the electron transport chain and increased ROS production, while downregulation of antioxidants may result in decreased scavenging capacity, causing an overall increase in mitochondrial ROS levels. Because of its high energy demand, the heart has a higher mitochondrial volume than many other tissues, and its proper function is especially sensitive to changes in mitochondrial function and maintenance of the energetic balance. Whether or not mitochondrial sources contribute significantly to AngII-induced increases in ROS in the heart has recently come under investigation. Increased ROS generation in cardiomyocytes as a result of AngII treatment was shown to occur primarily in mitochondria, resulting in a decrease in mitochondrial membrane potential (17). Although AngII stimulation of cultured neonatal rat cardiomyocytes did not affect the mitochondrial DNA (mtDNA) copy number (118), AngII-treated mice did exhibit a significantly decreased mtDNA copy number and increased mtDNA deletions, as well as decreased mitochondrial respiratory capacity and increased mitochondrial damage (17). These effects were accompanied by hypertrophy, fibrosis, and diastolic dysfunction, and were inhibited by mitochondrial catalase, suggesting that mitochondrial ROS play a major role in AngII-induced hypertrophy and dysfunction. Studies using metabolomic profiling and electron microscopy have demonstrated that cardiac substrate use and mitochondrial biogenesis and morphology are altered in the presence of increased RAS component expression (17, 76, 138), and mitochondrial autophagy is increased by AngII-induced ROS (17, 103). In addition, AngII can increase the expression of uncoupling proteins and decrease the expression of mitochondrial respiratory chain proteins under certain circumstances (27, 103). How AngII induces mitochondrial ROS generation is not yet known, however. ROS from other sources may cause ROS-induced ROS release (154), through damage to mitochondrial components and oxidation of the membrane permeability transition pore. Indeed, our laboratory has recently shown that overexpression of Nox4 leads to an increase in O2 •− production that can be partially inhibited by rotenone as well as DPI, and is accompanied by increased oxidation of mitochondrial proteins, mitochondrial dysfunction, and decreased mitochondrial biogenesis (2). Conversely, cardiac-specific deletion of Nox4 was protective against pressure overload-induced mitochondrial damage (54). Since Nox4 is upregulated by AngII and is expressed in mitochondria (2), it is possible that AngII stimulates mitochondrially localized ROS generation by Nox4, which in turn causes further ROS generation by the mitochondria themselves. Alternatively, mitochondrial ROS generation could be triggered by elevated ROS levels caused by an AngII-induced decrease in mitochondrial antioxidant expression, as suggested by the fact that AngII signaling resulted in decreased MnSOD expression, accompanied by a significant increase in mitochondrial O2 •− levels (59). On the other hand, eNOS uncoupling, XO, and p47phox expression and translocation can all be activated by ROS, and AngII-induced Nox2 upregulation is inhibited by catalase (17), suggesting that it, too, is ROS dependent. Thus, it is also possible that ROS originating from the mitochondria could trigger additional ROS generation from these other sources, resulting in the creation of a positive feedback loop. Whether a direct mechanism exists by which AngII signaling leads to mitochondrial ROS generation has yet to be determined.
Conclusions
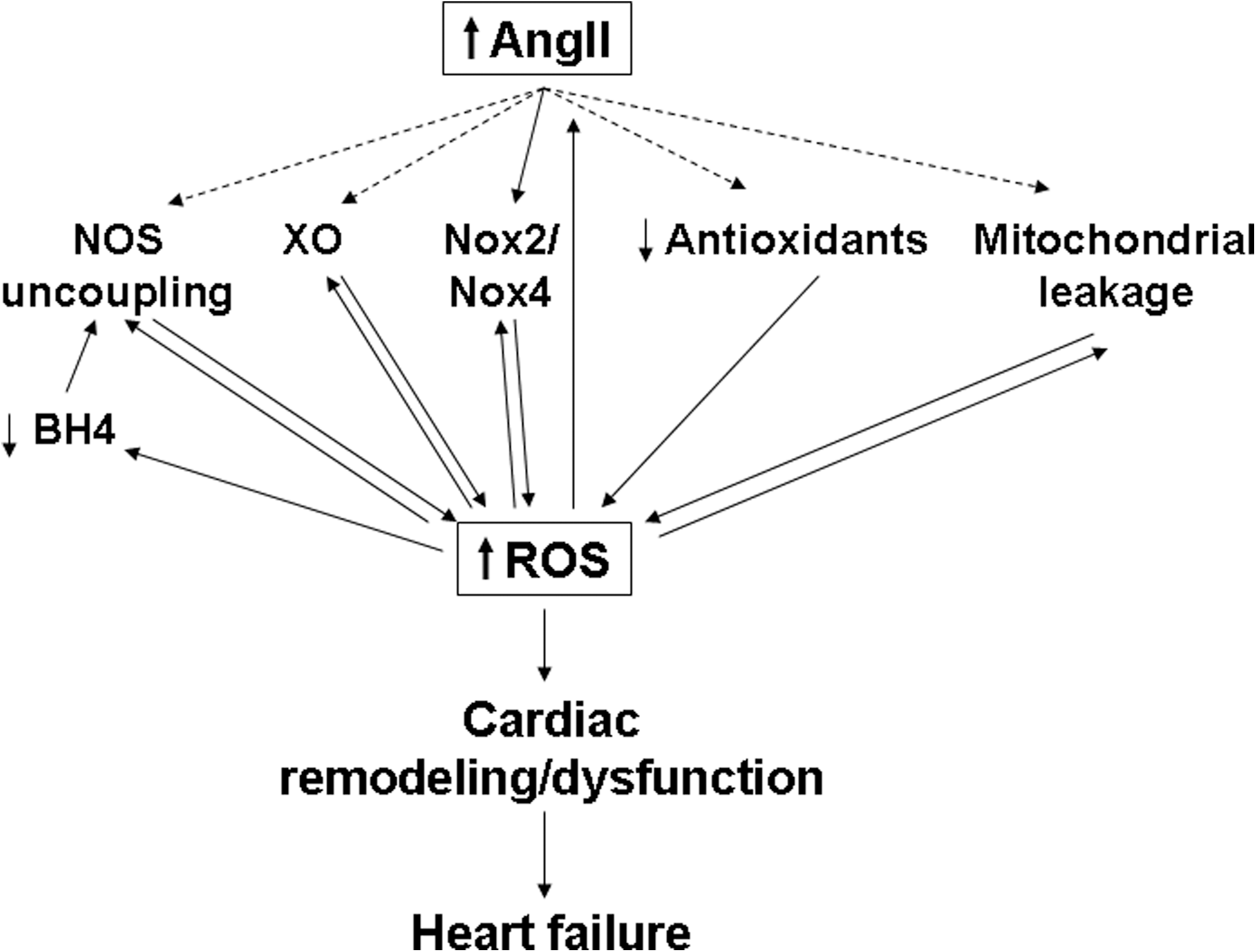
Despite recent medical advances, morbidity and mortality due to CVD and HF remain high. Inhibition of the RAS and AngII signaling has been shown to increase longevity in experimental animal models (5, 9), and RAS inhibitors have been clinically shown to be therapeutically beneficial for patients at risk for and suffering from CVD and HF. Furthermore, ROS generation and oxidative stress are now known to play a critical role in mediating the effects of AngII signaling and in the pathology of the failing heart. However, our current understanding of the signaling pathways and mechanisms connecting AngII signaling, ROS generation, and HF remains incomplete. Several different potential sources of ROS may contribute to the downstream effects of AngII in the heart, and the relative importance of these various sources may vary depending on the cell type and context. In addition, the different ROS pathways may be interdependent, with ROS generated from one source triggering further ROS generation from another source. Such stepwise generation of ROS could theoretically lead to a vicious cycle in which ROS production becomes self-amplifying, and this might contribute to the changes observed in the heart during decompensation and the transition to HF (Fig. 6). Further investigation and a better understanding of how AngII induces ROS generation and how AngII-induced ROS affect cardiac remodeling and function should lead to the development of improved therapies and better outcomes for those suffering from CVD and HF.
Footnotes
Acknowledgments
We thank Dr. Sebastiano Sciarretta for critical reading of the article. This work was supported in part by U.S. Public Health Service Grants HL59139, HL67724, HL69020, HL91469, HL102738, and AG27211 and the Foundation of Leducq Transatlantic Network of Excellence.