
Abstract
Introduction
T
In particular, cardiovascular function is largely affected by oxidative stress (51) but also provides multiple sites for redox regulation, including prostanoid synthesis and function (12), vascularization (109), regulation of the activity and function of endothelial nitric oxide synthase (eNOS) (19), as well as the control of the thiol/disulfide state in aging-associated cardiovascular complications (9). In these settings, the transition from redox signaling to oxidative stress is floating and oxidative stress may result from overproduction/impaired detoxification of RONS or impaired/dysregulated redox signaling. The prognostic value of endothelial dysfunction (eNOS dysfunction/uncoupling) in the coronary system was demonstrated by a higher rate of cardiovascular events in individuals with impaired endothelial function (measured by the acetylcholine-dependent vasoconstriction in the coronaries) (111). The relation of oxidative stress to this phenomenon was shown by a higher rate of cardiovascular events in individuals with pronounced improvement of endothelial function by vitamin C infusion (measured by the acetylcholine-dependent increase in blood flow in the forearm), suggesting a higher burden of oxidative stress in their brachial arteries (54). Disappointing for the theory of a “role of oxidative stress in cardiovascular disease,” all large human clinical trials failed to report on beneficial effects of antioxidant therapy [reviewed in Ref. (18)]. Most likely, these negative outcomes are related to the fact that the patients enrolled in these trials already were on antioxidant therapies [e.g., angiotensin converting enzyme (ACE) inhibitors and angiotensin II (AT-II) receptor blockers], cardiovascular complications were already irreversible (e.g., due to remodeling or calcification), systemic antioxidant therapy interferes with protective redox events (e.g., preconditioning) or with important redox signaling pathways (e.g., ROS-mediated cell migration and proliferation), and finally, but most probably, oral antioxidant therapy does not result in local concentrations high enough to affect RONS levels in certain cellular compartments [reviewed in Ref. (18)]. Despite these negative outcomes of the large clinical trials, a large number of trials in small-to-intermediate size cohorts (often using direct infusion of antioxidants resulting in much higher local concentrations than achievable via oral treatment) reported on highly beneficial effects of antioxidants, especially on endothelial function in the periphery [reviewed in Ref. (18)]. Experimental evidence for the impact of RONS in the development of cardiovascular disease originates from studies in genetically altered animals, showing that increased formation of RONS by overexpression of superoxide producing enzymes [e.g., NADPH oxidase isoform 1 (Nox1)] (36) or deletion of important superoxide detoxifying enzymes [e.g., manganese superoxide dismutase (MnSOD, SOD2) and extracellular SOD (ecSOD, SOD3)] (61, 83, 135) aggravates cardiovascular complications. In contrast, deletion of superoxide producing enzymes (e.g., Nox1 or p47phox) (39, 89) or overexpression of superoxide detoxifying enzymes (e.g., MnSOD) (37) improves cardiovascular function. The uncoupling of eNOS is a prominent and well-characterized process in the setting of cardiovascular complications (95) and the depletion of its essential cofactor BH4, among other processes, plays an important role for eNOS uncoupling and the development of endothelial dysfunction (8, 53, 117, 126).
Given the importance of oxidative stress and redox regulation in physiology and pathophysiology, we will highlight the role of mitochondrial ROS (mtROS) formation in these processes and especially focus on the interaction of these mtROS with other sources of oxidative stress, such as uncoupled NOS, Nox isoforms, and xanthine oxidoreductase. After addressing the current state-of-the-art of this “crosstalk” and known mechanisms, we will briefly introduce the different “redox switches” in these enzymatic systems that make them susceptible to redox regulation. Importantly, these “redox switches” are often overactivated in oxidative stress conditions whereas their suppression/downregulation represents an important feedback mechanism of the physiological regulation of redox signaling. Finally we will provide some examples for the involvement of mtROS in cardiac dysfunction as well as inflammatory disease and discuss the new concept of mtROS-driven activation of immune cells leading to migration of these cells to the vascular wall and endothelial dysfunction.
Crosstalk Between mtROS and Other Sources of Oxidative Stress
In two recent review articles, the concept of the crosstalk between mtROS and other sources of oxidative stress was already introduced in detail (25, 34). The interaction of mtROS with the mitochondrial permeability transition pore (mPTP) and activation of mtROS formation by interaction of cytosolic ROS with mitochondrial ATP-sensitive potassium channels (mtKATP) was a major issue in these reviews since these processes represent important pathways for the participation of mitochondria in various physiological [e.g., pre- and postconditioning (33)] and pathophysiological [e.g., initiation and amplification of cellular oxidative stress in various disease states (37, 135)] processes. Opening of the mtKATP channels also represents an important “redox switch” to stimulate mtROS formation (Fig. 1). At the time, when the earlier mentioned review articles by Daiber and Dikalov were prepared, only a small number of publications on the crosstalk between mtROS and other sources of oxidative stress were available. These previous findings are summarized in Figure 1 and will be briefly introduced in the next paragraph, whereas additional and more recent reports will be addressed in the subsequent paragraphs.

Kimura et al. have shown that AT-II-triggered activation of Noxs in the myocardium confers ischemic preconditioning, which was blocked by the Nox inhibitor apocynin (65). In the related editorial, Brandes nicely summarized these findings (17) and provided a scheme depicting the AT-II-dependent activation of the cytoplasmic Nox, followed by cytosolic ROS-triggered opening of mtKATP, loss of mitochondrial membrane potential (mtΨ), opening of the mPTP with subsequent release of mtROS to the cytosol, and activation of kinases [e.g., apoptosis signal-regulating kinase 1 (ASK1) and p38 mitogen-activated protein kinase (MAPK)] leading to preconditioning and cellular protection. These findings probably encouraged Dikalov and coworkers to investigate the role of mtROS in AT-II-induced vascular RONS formation, endothelial dysfunction, and hypertension (42). These authors identified a reinforcing role of mitochondria in the adverse effects of AT-II treatment via initial activation of Nox that triggers mtROS formation (reverse crosstalk), which causes additional Nox activation in a vicious circle. In a subsequent study they provided data that overexpression of the mitochondrial MnSOD or therapy with the mitochondria-targeted antioxidant, mitoTEMPO, completely abolished these adverse effects of AT-II, including the endothelial dysfunction and increase in blood pressure (37). These data were supported by others using rho0 smooth muscle cells [upon chemical depletion of mitochondrial DNA (mtDNA) by ethidium bromide], which lack the increase in Nox activity and Nox1 mRNA induction in response to AT-II treatment, suggesting the need of functional mitochondria for AT-II effects on Nox (136). The feasibility of this reverse crosstalk based on superoxide-driven loss in mtΨ with subsequent stimulation of mtROS formation was demonstrated in silico by a reaction-diffusion model of ROS-induced ROS release in a mitochondrial network and supported by experimental data on superoxide-dependent mitochondrial depolarization obtained in permeabilized cardiomyocytes (141).
Wang and coworkers have identified the molecular mechanisms by which hypoxia-triggered mtROS activates protein kinase Cɛ (PKCɛ) leading to activation of Noxs (104). Pharmacological (apocynin) and genetic (p47phox−/−) inhibition of Nox as well as treatment with the unspecific PKC inhibitor chelerythrine, with a specific PKCɛ translocation peptide inhibitor, or using genetic deletion of PKCɛ (PKCɛ−/− mice) significantly attenuated the hypoxic increase in overall cellular ROS formation. Further support for an essential role of mitochondria-derived ROS in this signaling process came from experiments demonstrating that enhanced mitochondrial and cytosolic H2O2 decomposition by glutathione peroxidase-1 gene overexpression (GPx-1tg) prevented, whereas inhibition of mitochondrial and cytosolic H2O2 breakdown by GPx-1 gene deletion (GPx-1−/−) augmented the hypoxia-induced increase in Nox activity in pulmonary arteries. In a subsequent study they showed that hypoxia-triggered mtROS formation induces accumulation of cytoplasmic calcium (77). Meanwhile, Wang and coworkers have summarized their data on hypoxia-triggered mtROS with downstream Nox activation as well as effects of hypoxia-driven ROS formation on intracellular calcium levels in a comprehensive review (129). Um and coworkers demonstrated that serum withdrawal caused a fast cellular mtROS response followed by a slow Nox1 activation via the phosphoinositol 3 kinase (PI3K) pathway (76). Blockade of the mtROS production increased the viability of cells dramatically, indicating that this early trigger was essential for the adverse effects of serum withdrawal. However, also blockade of the Nox1 pathway conferred protection, indicating that also the late Nox-dependent ROS formation is essential for initiation of cell death.
Our laboratory observed mtROS-triggered activation of Nox and endothelial dysfunction in an animal model of nitrate tolerance (27, 134) and aging (135). Nitroglycerin-induced nitrate tolerance is a suitable model to study vascular oxidative stress and dysfunction since nitroglycerin induces mtROS and peroxynitrite formation as well as endothelial dysfunction and impaired bioactivation of the drug itself (termed nitrate tolerance or tachyphylaxis) (28). In 2005, we observed that 50% deficiency in MnSOD (MnSOD+/− mice) increases nitrate tolerance, a process that is based on impaired bioactivation of nitroglycerin by mitochondrial aldehyde dehydrogenase (ALDH-2), but also endothelial dysfunction, a process clearly related to cytosolic oxidative stress and eNOS dysfunction/uncoupling (27). This suggested an interaction of mtROS (namely, mitochondrial superoxide) with the activity of eNOS localized in the cytosol or plasma membrane. In a subsequent study nitrate tolerance and endothelial dysfunction were blocked by inhibition of nitroglycerin-induced mtROS formation by rotenone, whereas pharmacological (apocynin) or genetic (p47phox−/− or gp91phox−/−) inhibition of the Nox activity only rescued endothelial function, but not nitrate tolerance (134). This indicated that nitroglycerin-induced nitrate tolerance and endothelial dysfunction are spatially distinct processes but a crosstalk between the relevant cellular compartments and the associated RONS formation exists. In a third study, we demonstrated that partial MnSOD deficiency (MnSOD+/− mice) results in increased age-related mtROS formation as well as mtDNA damage associated with significantly impaired endothelial function as compared with MnSOD+/+ littermates (135). These data clearly indicate that mtROS contribute to impairment of cytosolic processes, such as eNOS-derived NO formation, leading to endothelial dysfunction. The details of this crosstalk will be discussed in detail in the subsequent chapters.
In line with AT-II-mediated Nox activation and subsequent stimulation of mtROS formation described previously, Abramov et al. observed a quite similar phenomenon (reverse crosstalk) in astrocytes in response to β-amyloid peptide treatment (3). β-Amyloid peptide challenges caused an increase in cytosolic calcium associated with a depolarization of the mtΨ and both processes were suppressed by antioxidants as well as diphenylene iodonium (DPI) and apocynin, inhibitors of the Nox. A direct crosstalk between mtROS and Nox1 was established by Desouki and colleagues as rho0 (mtDNA-depleted) cells displayed decreased levels of mtROS formation associated with loss of Nox1 expression (32). Vice versa, Nox1 expression increased with antimycin A–stimulated mtROS formation in wild-type cells and it was proposed that this Nox1 induction contributes to tumor cell growth. Coughlan et al. showed that increased glycolysis under hyperglycemic conditions yields high amounts of pyruvate feeding the tricarboxylic acid cycle producing NADH, which in turn stimulates superoxide formation from mitochondrial respiratory complexes leading to increased generation of advanced glycation end products (AGE) and activation of PKC (21). PKC activation will lead to increased Nox activity and incubation of endothelial cells with AGE caused an increase in intracellular H2O2 levels as well as endothelial cell activation envisaged by the expression of tissue factor and vascular cellular adhesion molecule-1 (VCAM-1) on the cell surface, all of which was blocked by DPI. These authors also reported on a reverse crosstalk in the setting of diabetes and hyperglycemia documented by a decrease in mtROS formation by apocynin therapy or deficiency in AGE receptor (RAGE−/−) (21). The increased mtROS generation in the setting of diabetes or hyperglycemia is generally accepted and could provide an important pathway for secondary Nox activation and diabetic oxidative damage (50, 102). Recently, the nitration of GTP and formation of 8-nitro-cGMP by peroxynitrite and/or myeloperoxidase-catalyzed oxidation of nitrite in the presence of H2O2 was proposed as an important electrophilic signaling pathway (6). In a cellular sepsis model, the incubation with lipopolysaccharide (LPS) and cytokines initiated a Nox2-dependent formation of mtROS (reverse crosstalk), which largely contributed to the 8-nitro-cGMP formation. Inhibition of phagocytic Nox activity by deletion of its cytosolic subunit p47phox by small interfering RNA (siRNA) treatment diminished the mtROS and 8-nitro-cGMP formation in response to LPS/cytokines. Likewise, treatment with exogenous H2O2 mimicked the Nox2-dependent ROS formation and induced mtROS production (6). Simultaneous administration of exogenous H2O2 and an NO donor (propylamine NONOate) leads to increased 8-nitro-cGMP levels in a superoxide and mitochondria dependent process since polyethylene glycolated-SOD prevented this formation.
Despite these numerous reports on deleterious effects of mtROS in disease and pathophysiology, mtROS are also involved in redox signaling events, such as the regulation of redox-sensitive transcription factors (e.g., OxyR or Yap1 by H2O2 via disulfide bridge formation) or processes mediated by redox-sensitive protein tyrosine phosphatases/kinases as well as the glutaredoxin and peroxiredoxin systems (106). Another example is the activation of the c-Jun N-terminal kinase (JNK) by mtROS leading to inhibition of the activity of the metabolic enzymes glycogen synthase kinase 3β and glycogen synthase (98). Catalase and N-acetylcysteine inhibited the activation of the JNK pathway whereas antimycin A, a stimulator of complex III–dependent ROS formation, strongly activated the JNK pathway. According to Rigoulet et al., the quality control of mitochondrial biogenesis, the control of preadipocyte proliferation, or regulation of the transcription factor C/EBP homologous protein (CHOP-10) in adipocyte differentiation are other examples of redox signaling pathways controlled by mtROS levels (106). The interplay of mtROS with other sources of oxidative stress and calcium signaling in cardiac dysfunction will be addressed in a separate paragraph below with special emphasis on the clinical importance of these signaling pathways. Also, the role of mtROS in inflammation (mtROS-triggered activation of phagocytic Nox) will be addressed in a separate paragraph below with special emphasis on the clinical importance of these signaling pathways.
Activation of Nox by mtROS
The most important “redox switch” for Nox2 (and Nox1) is probably the previously described RONS (H2O2 or peroxynitrite)–mediated activation of PKC (79) with downstream phosphorylation of cytosolic subunits (e.g., p47phox−/−) and their translocation to the membrane (Figs. 1 and 2) (25, 34). In contrast to p47phox, the Nox1-specific homologue NoxO1 has no regulatory phosphorylation sites and the Nox1-specific activator NoxA1 has several phosphorylation sites that rather contribute to inactivation of the Nox. Moreover, a novel concept consists of the regulation of Nox activity by the redox chaperone protein disulfide isomerase (PDI), the association of which with p47phox leads to the translocation of this subunit with subsequent activation of the Nox complex (74). Recent findings indicate that reduced PDI (with thiols in the sulfhydryl state) associates with p47phox in the cytosol, whereas oxidized PDI (with thiols in the disulfide or sulfenic acid state) causes transfer of this complex to the membrane and Nox-derived superoxide formation (Fig. 2) (31). In addition, numerous reports indicate that the expression of different Nox isoforms is upregulated under oxidative stress conditions [e.g., Nox1 and Nox2 in diabetes (55, 131) as well as Nox1, Nox2, and Nox4 in hypertension (93, 132)], suggesting that their expression is controlled by redox-sensitive transcription factors and/or their mRNA stability is influenced by redox modifications of (de)stabilizing proteins (73, 100). The proof of this concept comes from several reports on an attenuation of Nox-derived ROS production by mitochondria-targeted antioxidants (22, 37). Of special interest may be the Nox4 isoform since it does not share the classical activation pathway with the Nox1 and Nox2 isoforms (translocation of cytosolic subunits to the membrane), is constitutively active, and localizes to the outer mitochondrial membrane (15). Therefore, mitochondrial Nox4-derived ROS may play a decisive role in hypoxia/reoxygenation damage (but also preconditioning) (64), complications during aging (5), myocardial growth and death (88), and in the failing heart (69). Interestingly, potent beneficial properties of Nox4 were recently reported in different cardiovascular disease models (115). The mitochondrial localization of the Nox4 enzyme also facilitates the interaction of ROS coming from the mitochondrial respiratory complexes and this particular Nox.
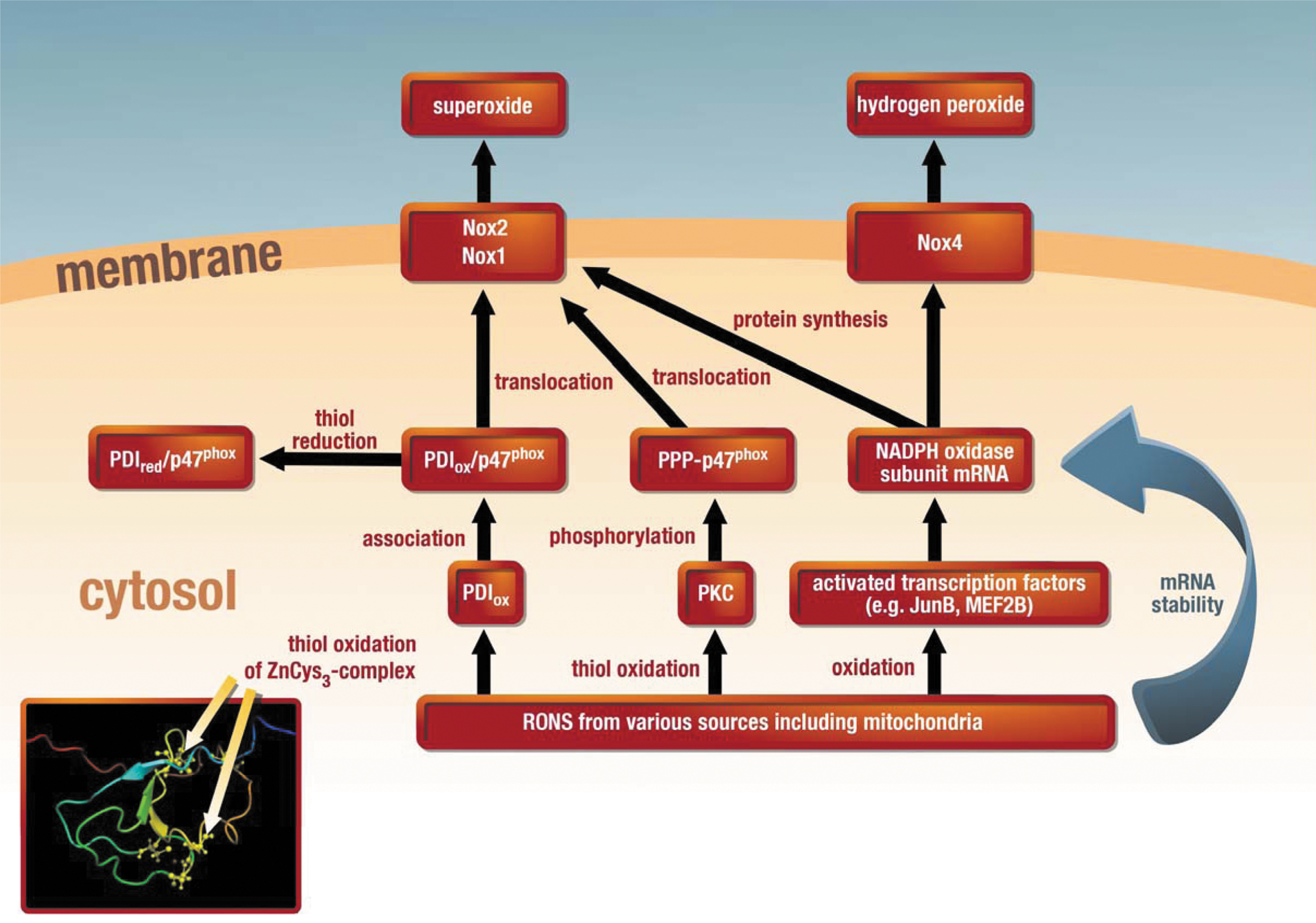
Uncoupling of NOS by mtROS or secondary activated sources of oxidative stress
Besides the classical regulation of enzymatic activity of eNOS (e.g., by calcium/calmodulin, caveolin, heat shock protein 90, palmitoylation, and myristoylation), other regulatory pathways, such as phosphorylation and S-glutathionylation, are directly linked to the formation of redox-active species. These “redox switches” in eNOS confer alterations in enzymatic eNOS activity and may contribute to uncoupling of eNOS as depicted in Figure 3. Uncoupling of eNOS is a process by which electrons leak from the transport in the reductase domain (from NADPH over flavin mononucleotide and flavin adenin dinucleotide) and are transferred to molecular oxygen to yield superoxide instead of NO. This switch is even more detrimental than inhibition of eNOS since uncoupling will switch the enzyme from an NO to a superoxide source, meaning from a protective/beneficial to a harmful/toxic phenotype (45, 95). It should be noted that not only eNOS may be uncoupled and produce superoxide but also neuronal NOS (type 1) (91) and inducible NOS (type 2) (137) are subjected to the uncoupling phenomenon and produce superoxide in the uncoupled state. Among the regulatory pathways (“redox switches”) of eNOS, the concept of the oxidative depletion of BH4 is the most prominent one [reviewed in Refs. (8, 126)]. The first direct evidence of a role of BH4 depletion in eNOS uncoupling and subsequent endothelial dysfunction in vivo was obtained in hypertensive mice in 2003 (70). One year later, the enzymatic source for BH4 synthesis, GTP-cyclohydrolase-1, was identified as an important regulator of eNOS and endothelial function (8), but most interestingly the expression of GTP-cyclohydrolase-1 itself is redox sensitive.

Another direct redox regulatory pathway for eNOS function is the oxidative disruption of the zinc-sulfur complex (ZnCys4) in the binding region of the eNOS dimer resulting in a loss of sodium dodecyl sulfate–resistant eNOS dimers, which has been first described by Zou and coworkers for peroxynitrite-mediated oxidation of eNOS (143). Later it was reported by the same group that hypochlorous acid may also cause disruption of the zinc-sulfur complex in eNOS (138). eNOS dysfunction and altered eNOS dimer stability by hypochlorous acid was also described by Keaney Jr. and coworkers without determination of the zinc content of the enzyme (120). Up to now this concept lacks further in vivo evidence although the concept of peroxynitrite-mediated oxidation of the zinc-sulfur cluster is highly attractive. According to a recent report by Zweier and coworkers, eNOS is adversely regulated and uncoupled (leading to superoxide formation) by S-glutathionylation at one or more cysteine residues of the reductase domain (19). In this initial study, Chen et al. have demonstrated eNOS S-glutathionylation for recombinant purified eNOS by glutathione disulfide (GSSG), redox stress in cultured cells, and finally hypertension in spontaneously hypertensive rats. By site-specific mutagenesis the authors have identified cysteines 689 and 908 in the reductase domain as the targets for this redox modification. Based on our recent observations, eNOS S-glutathionylation is largely increased in nitroglycerin-treated endothelial cells and aortic tissue from nitroglycerin-infused rats (68) as well as in a rat model of streptozotocin-induced diabetes mellitus (116). Therefore, eNOS S-glutathionylation may represent a new “redox master switch” controlling NO versus superoxide production by this enzyme.
Another redox-sensitive regulatory pathway of eNOS represents its phosphorylation. Among all described eNOS phosphorylation sites, the following 3 of them are of particular functional importance: first, the activating phosphorylation at serine1177 mediated by the Akt pathway, which is calcium independent and increases the NO producing activity of eNOS (38); second, the inactivating phosphorylation at tyrosine657 mediated by the protein tyrosine kinase-2 (PYK-2), which inhibits the enzyme without evidence for uncoupling of the enzyme (84); third, the inactivating phosphorylation at threonine495 mediated by PKC, which may contribute to uncoupling and superoxide production by eNOS (44, 80). Since PKC and the protein kinase-2 are redox sensitive (activated by H2O2), these two phosphorylation-based modifications of eNOS may be regarded as redox switches. Another redox regulation of eNOS may be based on asymmetric dimethylarginine (ADMA), which is probably the most potent endogenous inhibitor of eNOS (16) and it is still a matter of debate whether or not ADMA itself may lead to uncoupling of eNOS (123). In particular, the synthesis of ADMA is redox sensitive (127) but also the activity of ADMA hydrolyzing enzymes, such as dimethylarginine dimethylaminohydrolase, is redox regulated. The relationship between ADMA, eNOS activity, and oxidative stress has been reviewed in detail (123).
In summary, all of these mentioned different “redox switches” in eNOS are likely to contribute to uncoupling of this enzyme. However, with regard to their relevance and mechanistic feasibility, it should be considered that some of these “redox switches” induce superoxide formation at the reductase domain level (e.g., S-glutathionylation and Thr495 phosphorylation), which is at variance with the majority of observations on complete inhibition of eNOS-derived superoxide formation by L-NAME, a blocker of its oxygenase but not reductase activity. Therefore, not all pieces in the puzzle of eNOS uncoupling are identified and some mechanistic discrepancies need careful elucidation. Since all of these pathways are based on ROS- and RONS-mediated uncoupling of eNOS, one may assume that mtROS also contribute directly (by interacting with the aforementioned redox switches in eNOS) and indirectly [by activating secondary sources of oxidative stress, such as xanthine oxidase (XO) and Nox] to uncoupling of eNOS. Moreover, numerous examples exist that therapy with mitochondria-targeted antioxidants improves endothelial dysfunction (108).
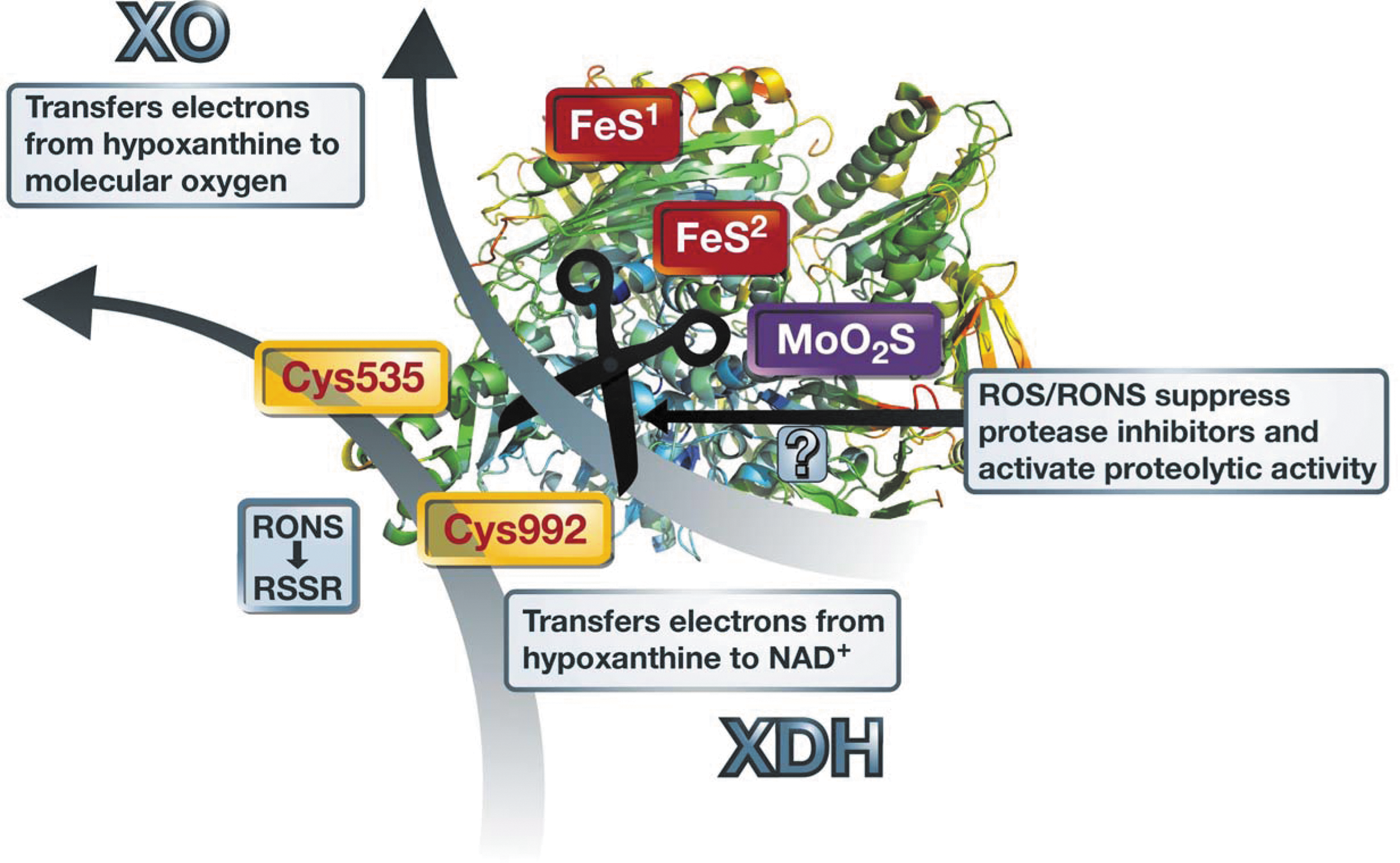
Conversion of xanthine dehydrogenase to the oxidase form by mtROS or secondary activated sources of oxidative stress
The conversion of xanthine dehydrogenase (XDH) to the XO needs oxidation of critical thiol residues (99, 110) as observed in AT-II-induced hypertension (71). The disulfide formation with a subsequent conformational change of the enzyme structure and altered affinity for cofactor (NAD+) as well as molecular oxygen binding is summarized in Figure 4. In addition, protease activity may be increased by oxidative stress due to oxidative suppression of protease inhibitor activity (40, 128) as well as generally increased proteolytic activity under oxidative stress (30). Since AT-II-induced oxidative stress is largely due to activation of mtROS formation [see second paragraph of chapter Crosstalk Between mtROS and Other Sources of Oxidative Stress and Refs. (37, 42, 65)], the conversion of XDH to XO under chronic AT-II treatment or increased levels of this vasoconstrictor (e.g., in diabetes or hypertension) could be mainly driven by mtROS formation. A direct proof for the interaction between mtROS and XO activity was based on the improvement of cardiac complications and XO activation in a model of heart failure by therapy with the mitochondria-targeted antioxidant mitoquinone (48).
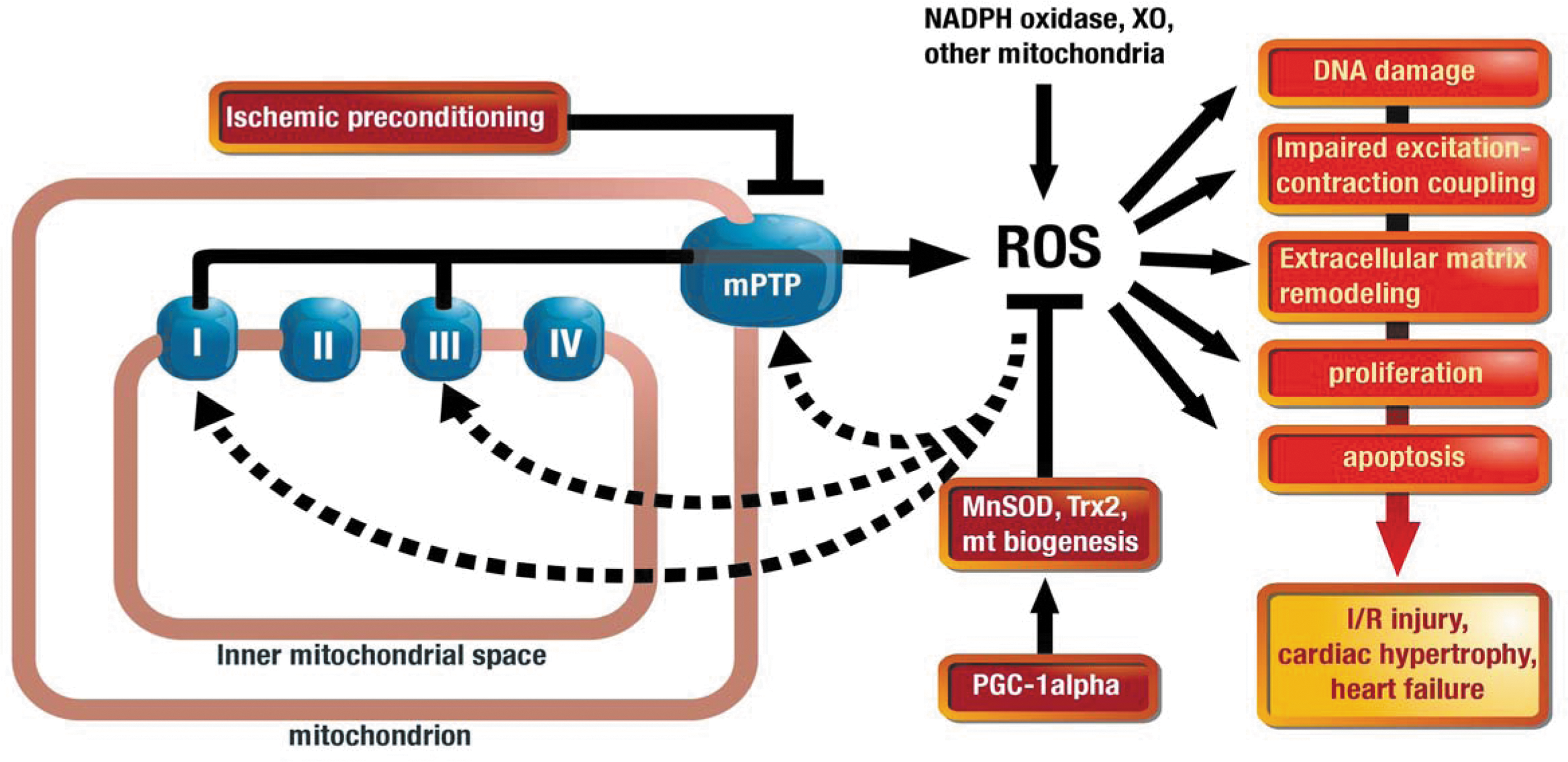
mtROS in Cardiac Disease
The volume density of mitochondria ranges between 22% and 32% in mammalian cardiomyocytes (112). Since the heart mainly uses lipids as fuel, the oxidation of fatty acids in the mitochondria predominates and accounts for ∼90% of total ATP production, resulting in the highest oxygen consumption among all organs [between 7×10−2 ml/(g·min−1) at rest and up to 40×10−2 ml/(g·min−1) during exercise]. As a consequence, cardiac tissue is particularly susceptible to ROS production by mitochondria, which has been implicated in different cardiac disease states, including ischemia/reperfusion (I/R) (75) or cardiac hypertrophy (22).
At the molecular level, mtROS are being produced at complexes I and III of the electron transport chain (ETC) as a consequence of decreased electron flux (e.g., due to altered substrate/cofactor availability or post-translational modifications of the ETC), resulting in electron transfer to molecular oxygen. In addition, ROS can induce the opening of ion channels located at the inner mitochondrial membrane (such as the mPTP, inner membrane anion channel, or mtKATP channels), which results in a depolarization of the mtΨ. To maintain ATP production in this setting, electron flux along the ETC will be increased with the risk of enhanced ROS production, a phenomenon termed “ROS induced ROS release” (142). However, the initial ROS burst that triggers mtROS release may also originate from other ROS sources, such as the Nox (17). In this respect, ROS derived from the Nox isoform Nox4 were shown to stimulate mtROS production, resulting in mitochondrial dysfunction (4). A recent study by Dai and coworkers underlines the importance of mtROS for the development of cardiac hypertrophy. The authors investigated the mitochondrial-targeted antioxidant peptide SS-31 in AT-II-infused mice and found that the cotherapy resulted in a blunted upregulation of Nox4, reduced mitochondrial oxidative damage, and finally prevented cardiac hypertrophy despite the absence of blood-pressure-lowering effects (22), nicely summarized in (87). The causal role of mtROS for the development of cardiac hypertrophy was supported by another study from the same group, in which mitochondria-targeted overexpression of catalase prevented cardiac hypertrophy, fibrosis, and failure (24).
Chronic inhibition of the β-adrenergic receptors has been shown to improve symptoms and outcome in heart failure. In this respect, a recently published study showed that mtROS are necessary for the inotropic response in cardiomyocytes (10). Since catecholaminergic stimulation is mediated by sympathetic neurons and contributes to the progression of heart failure, an interesting study by Yin and coworkers identified mtROS as a key signaling event leading to AT-II-induced neuronal activation (139).
Moreover, mtROS production significantly contributes to I/R injury of myocardial tissue, and the mPTP plays a central role in this process. During ischemia, low ROS levels (e.g., by activation of NAPDH oxidases) may damage the ETC and lead to disturbed electron flux, resulting in an increase in mtROS production. Upon reperfusion, the availability of oxygen will boost mtROS levels, resulting in reperfusion-induced injury (96). Eventually, exacerbated oxidative stress will lead to the opening of the mPTP, disruption of the mtΨ, and cell death (13). These deleterious effects can be further aggravated by pressure overload, which exacerbates oxidative stress and cell death during I/R injury and is mediated by increased opening of the mPTP, resulting in augmented mtROS production (94).
On the other hand, preconditioning describes the phenomenon that transient episodes of ischemia provide protection against subsequent ischemic injury, which has been attributed to the inhibition of mPTP. This process may be initiated by mitochondria (60) or Nox (66) derived ROS, which ultimately results in a decreased susceptibility of mPTP opening and decreased overall oxidative stress (20).
In general, myocardial oxidative stress is determined by the balance between ROS generation and their elimination by antioxidants. Therefore, mitochondrial oxidative stress may also originate from a decreased elimination of ROS in the mitochondrial matrix. Antioxidant enzymes, including MnSOD, peroxiredoxin 3 & 5, thioredoxin, and thioredoxin reductase, constitute the first line of defense against oxidative damage within the mitochondria. A recently published study by Lu et al. identified peroxisome proliferator-activated receptor-gamma coactivator-1α (PGC-1α) as the major regulator of several antioxidant enzyme systems within cardiac mitochondria during chronic pressure overload by transverse aortic constriction in mice (85). Interestingly, deletion of PGC-1α had no effect on myocardial oxidative stress or function under basal conditions, suggesting that this pathway confers cardiac adaptation during pathological stressors such as pressure overload. Initially, PGC-1α was discovered as a major regulator of mitochondrial biogenesis and function. Since cardiomyocytes use predominantly free fatty acids as fuel, the mitochondrial mass is of particular importance to meet the energy demand and to prevent cell death under metabolic stress. In addition, a decreased mitochondrial number may lead to an increased flux of electrons in the remaining mitochondria and subsequent susceptibility to mtROS formation. In accordance with this concept, Dong and coworkers found that high-fat diet–induced cardiac dysfunction was associated with decreased PGC-1α expression and increased oxidative stress, while metallothionein treatment prevented PGC-1α downregulation associated with decreased ROS formation and improved cardiac contractility (41). Taken together, mitochondria constitute the predominant ROS source in cardiomyocytes and can affect their function and structure in many different ways that are summarized in Figure 5.
Activation of Inflammatory Cells and ROS: Role of Nox and Mitochondria
Inflammation has been defined as part of the complex biological response of vascularized tissues to harmful stimuli, such as pathogens, damaged cells, or irritants. Inflammation is a protective attempt by the organism to remove the injurious stimuli and to initiate the healing process. It is a stereotyped and not a targeted response, and therefore it is considered as a mechanism of innate immunity, in contrast to adaptive immunity, which is specific for a certain and discrete pathogen (1). In inflammation, the pathobiology of the (i) vascular wall; (ii) blood cells, like leukocytes and platelets; and (iii) soluble factors, like cytokines and the coagulation cascade, converge in an orchestrated response. In this light, atherosclerosis can be viewed as a typical form of a chronic inflammatory disease, in which this concerted action goes awry and leads to tissue damage, ischemic injury, and death (52, 78).
It is important to note that oxidative stress represents such an “irritant” that leads to an unspecific defense reaction. On the other hand, innate immune cells—especially myelomonocytic cells, like neutrophils and monocytes—are characterized by their ability to produce large amounts of ROS mainly via their phagocyte-type Nox in an attempt to eliminate pathogens or extrinsic structures in an “oxidative burst” (29, 97). A prototypic proinflammatory mediator (121) and an activator of the Nox via PKC-mediated mechanisms (103) is the peptide hormone and effector of blood pressure increase, AT-II.
The AT-II receptor type 1 (AT1R) has been described to be essential for monocyte migration (62) and systemic mobilization in states of inflammation (122). Increased levels of hyperreactive crosslinked AT1R dimers are found on monocytes of hypertensive patients and were described to correlate with an increased AT-II-dependent monocyte activation and adhesiveness (2). AT-II has been described as an essential mediator of atherosclerosis, and it has been shown to directly promote monocyte adhesion to endothelial cells (63) while ROS were demonstrated to play an important role for migration and adhesion (56). AT1R blockade has been shown to decrease Nox activity and progression of atherosclerosis in genetic apolipoprotein E deletion (ApoE−/−) mice (130) and in patients with coronary artery disease (140). The essential role of lysozyme M (LysM)–positive myelomonocytic cells for vascular dysfunction and inflammation has recently been demonstrated in a transgenic mouse model with a cre-inducible diphtheria toxin receptor (iDTR). Toxin-mediated depletion of LysM+ cells prevented AT-II-induced blood pressure increase, vascular dysfunction, and Nox2 expression as well as vascular ROS formation in LysMiDTR mice (132).
Further experimental data suggest that monocyte recruitment in hypertension might be ROS dependent, since not only genetic AT-II receptor type 1 deletion (AT1R−/−) mice, but also SOD overexpressing mice, were partially protected from AT-II-induced monocyte chemoattractant protein-1 (MCP-1)–dependent chemokine receptor (CCR2) on monocytes and from increased vascular monocyte infiltration in response to AT-II (59). The importance of Nox-derived ROS for arterial hypertension has been demonstrated in mice lacking the Nox1 isoform predominantly expressed in vascular smooth muscle cells (89). Vice versa, overexpression of Nox1 exacerbated AT-II-induced hypertension in mice (36).
Additionally, molecular inhibition of the phagocyte-type Nox2 by gp91phox dstat led to an attenuation of AT-II-induced blood pressure increase in mice (72), and Nox2 is not only important for AT-II-induced neutrophil activation (43), but also for activation of monocytes/macrophages in several disease models (101). Indeed, the monocyte progeny of myeloid cells from patients with X-linked chronic granulomatous disease regained their functionality after retroviral gene transfer of gp91phox in the myeloid cells, indicating the central role of Nox2 not only for the respiratory burst, but also for lineage commitment and viability of monocytes (86).
To test the impact of a functional Nox and intact AT-II signaling in monocytes, we reconstituted the depleted LysMiDTR mice with monocytes from the circulation of AT1R−/− and gp91phox-/y mice. The defective monocytes failed to reestablish vascular dysfunction and oxidative stress in response to AT-II. Most compelling, reconstitution of depleted mice with AT1R−/− and gp91phox-/y monocytic cells did not restore AT-II-mediated systolic blood pressure increase (132). These findings clearly indicate that in our study circulating monocytic cells that harbor a Nox2-containing Nox and contain an AT1R are a prerequisite for blood pressure increase in response to AT-II.
A novel concept was introduced to the field, when Doughan and coworkers demonstrated, that AT-II was also a promoter of ROS formation from the mitochondrial respiratory chain (42). They suggested a feed-forward mechanism in AT-II-induced vascular oxidative stress, when they demonstrated in endothelial cells that mtROS formation in response to AT-II could be blocked by the NAPDH-oxidase inhibitor apocynin or by knockdown of the Nox subunit p22phox, and that Nox activation by mtROS could be blocked by the mtKATP channel blocker 5-hydroxydecanoate. In a second approach from the same group, Dikalova et al. were able to prevent AT-II-induced hypertension and vascular dysfunction by in vivo treatment with mitochondrial targeted antioxidant, mitoTEMPO, and even therapeutically decreased high blood pressure in mice with established hypertension (37). Likewise, MnSOD overexpressing mice were partially protected from AT-II-induced vascular dysfunction, suggesting that mtROS might trigger Nox-derived ROS in a SOD sensitive manner. In a similar way, our laboratory demonstrated that this crosstalk is also involved in the pathogenesis of nitrate tolerance and nitroglycerin-induced vascular dysfunction (134). Recent data by Dikalov and coworkers supported the mtROS-mediated activation of phagocytic Nox (Nox2) in human lymphoblasts and whole blood (35).
Taken together, experimental and clinical data suggest that (i) innate immune cells like inflammatory monocytes are mechanistically involved in vascular dysfunction and oxidative stress and that (ii) mitochondrial and Nox derived ROS “crosstalk” with each other in vascular disease (Fig. 6). It remains a yet unsolved question, whether activation of Nox-derived ROS by mtROS within leukocytes contributes to vascular dysfunction in vivo. Myelomonocytic cells contain mitochondria (7), and mitochondrial apoptotic pathways induced by reactive intermediates like AGE in diabetes have been shown to promote platelet–leukocyte interaction and vascular inflammation (47). Deficiency of uncoupling protein 2 (UCP2) leads to increased mtROS formation especially in macrophages (11), and chimeric mice with genetic uncoupling protein 2 deletion (UCP2−/−) bone marrow transplanted into low-density lipoprotein receptor knockout (LDLR−/−) mice that are fed a high-fat diet show accelerated atherosclerotic lesion formation, suggesting a proinflammatory role of mtROS from leukocytes in vascular disease (14). These data suggest a hierarchical nature of ROS formation in leukocytes in atherosclerosis with mtROS preceding ROS formation from Noxs, since atherosclerotic ApoE−/−gp91phox −/− mice show less superoxide formation, but no reduced lesion size (67). Preliminary data from our laboratory show that mtROS derived from the respiratory chain indeed lead to an activation of Noxs by promoting multimerization of the enzyme in isolated peripheral blood mononuclear cells. Current studies focus on therapeutic options to block mtROS formation in leukocytes in vivo with the aim to attenuate AT-II-induced vascular dysfunction.

Conclusions and Outlook
The interconnection between different sources of oxidative stress may explain the observation made in several studies that inhibition of only one source of RONS is able to completely normalize a cardiovascular disease state (25, 34). This was shown for the inhibition of XO-derived ROS by allopurinol in experimental diabetes, hypertension, and pulmonary arterial hypertension; for the prevention of mtROS formation by mitochondria-targeted antioxidants in hypertension and I/R; or for the inhibition of Nox by apocynin in diabetes, hypertension, and I/R.
Especially the activation, adhesion, and infiltration of white blood cells obviously plays an essential role for the development and progression of cardiovascular disease (Fig. 7) and recent data clearly show that this process is triggered by ROS in general and mtROS in particular. Also in sepsis, the inhibition of mtROS formation by mitochondria-targeted antioxidants represents a novel strategy to overcome septic complications (46). Since patients with chronic heart failure displayed elevated numbers of RONS-positive white blood cells and platelets (measured by cytosolic and mitochondrial RONS-specific probes) in their circulation (57), mitochondria-targeted antioxidants may also represent an attractive new strategy in this cardiovascular complication.

Footnotes
Acknowledgments
The authors thank Thilo Weckmüller for graphical assistance. The present work was supported by a grant from the Federal Ministry of Education and Research (BMBF 01EO1003) [to P.W., T.M., and A.D. within the Center for Thrombosis and Hemostasis (CTH) Mainz], from the Stiftung Rheinland-Pfalz (to E.S.), from the German Research Foundation (DFG WE 4361/3-1 to P.W.), and the Stiftung Mainzer Herz to all authors.