
Abstract
Introduction
The revelation in 2000 of a novel ACE homologue, ACE2, with a high catalytic efficiency in the conversion of Ang II to Ang-(1–7), has dramatically altered the landscape in which the function of the RAS system is viewed. Actions of the ACE2–Ang-(1–7) axis are often opposite to the well-documented ACE–Ang II axis (30, 54, 71). Accordingly, several studies suggest that the activation of the ACE2–Ang-(1–7) axis can limit the pathological effects of Ang II in CVD. Further, because chronic treatment with ACE inhibitors or ARBs can increase the plasma Ang-(1–7) levels several fold, at least part of the beneficial effects of these RAS blockers may be due to Ang-(1–7) effects. Thus, the concept that an imbalance in ACE2-Ang-(1–7) and ACE-Ang II axes is critical in the development of CVD has now emerged.
RAS Pathways
The traditional view of the RAS as a linear enzymatic pathway toward the production of a single active end product (Ang II) is incomplete. We now know that the system functions as a hierarchical network of enzymes that produce angiotensin peptide metabolites with potentially opposing functions (Fig. 1). The starting point in the canonical pathway begins with the glycoprotein angiotensinogen produced and secreted into the blood stream by the liver, and also secreted by cells in most organs, including the vasculature, kidney, brain, heart, and lungs. The enzyme renin then plays a pivotal role in the cleavage of angiotensinogen to produce the inactive Ang I peptide, which serves as the rate limiting substrate for Ang II and Ang-(1–7) production. Ang I is cleaved to produce Ang II via ACE and/or chymase. Renin in the blood stream in concert with circulating angiotensinogen and ACE, found in abundance on the surface of endothelial cells, controls the levels of circulating Ang II, which acts as a humoral factor (endocrine effects). However, many tissues have the capacity to produce angiotensinogen, renin (or renin-like enzymes), and ACE to produce Ang II at the local tissue level (autocrine/paracrine effects).

This seemingly straight-forward pathway becomes complicated by the fact that in many tissues, another active angiotensin metabolite, Ang-(1–7), can be generated from Ang II by an enzyme named ACE2 (17, 54). ACE2 was first cloned from 5’ sequencing of a human heart failure ventricle cDNA library in 2000 (17). It shares 42% of the genomic structure of ACE but is not expressed as widely as ACE, localized mainly but not exclusively in the heart, kidney, and testis in humans (17, 30, 71).
The enzymatic control of Ang II and Ang-(1–7) production is further complicated by the fact that ACE2 can also metabolize Ang I into Ang-(1–9). Ang-(1–9) can then be further metabolized by ACE to Ang-(1–7). From this scheme, it is important to note that ACE regulates both the production of Ang II and the degradation of Ang-(1–7); conversely ACE2 regulates the production of Ang-(1–7) and degradation of Ang II. In addition, other endopeptidases in tissues: neprilysin, prolyl endopeptidase, and metallo protease, can cleave Ang I [Ang-(1–10)] to form Ang-(1–7) (8, 22, 61). In the kidney, neprilysin appears to be the major enzyme to process Ang I to Ang-(1–7) (8, 22, 66).
More recently, it has been shown that Ang I, Ang II, and Ang-(1–7) can be formed by an novel angiotensinogen-derived dodecapeptide, proangiotensin-12 (PA12) now called angiotensin-(1–12) [Ang-(1–12)] (69). Ang-(1–12) is an alternative metabolite of angiotensinogen via an renin-independent but otherwise unknown process. Ang-(1–12) can give rise to Ang I via ACE in most tissues or directly converted to Ang II via chymase in the heart (59). Ang-(1–12) also can increase Ang-(1–7) levels, likely due to an indirect effect on enhanced Ang I and Ang II production and ACE2 activity, but a potential direct conversion to Ang-(1–7) by neprilysin or other peptidases has been suggested (69).
The link between angiotensin pathways and NO• production and signaling effects centers upon Ang-(1–7) and activation of its specific receptor Mas, a G-protein coupled receptor encoded by the Mas protooncogene. Mas receptor stimulation activates PI3K-dependent Akt phosphorylation, leading to NO• production by phosphorylation of endothelial NO• synthase (eNOS) (62). Mas activation can also lead to NO• production via neural NO• synthase (nNOS) (77), although the intermediate pathways are not yet defined. Ang-(1–7)/Mas receptor activation can also inhibit mitogen-activated protein kinase (MAPK) phosphorylation, stimulate phospholipase A2 activity and consequently release of arachidonic acid for prostanoid production, and interfere in various ways with Ang II/AT1 receptor mediate signaling pathways (35).
Although, Ang-(1–7) differs from Ang II by only one amino acid, Ang-(1–7)-mediated effects are markedly different from those of Ang II, and it is now widely believed that Ang-(1–7) plays a counter-regulatory role to Ang II (21), mediating a range of effects such as vasodilatation, inhibition of vascular smooth muscle proliferation, and fluid and electrolyte homeostasis. As noted by Ferrario et al. (21), this hierarchical system of proteases and opposing metabolites should provide variable degrees of control over the function of the RAS system within the tissue itself. Thus, local tissue control of expression and activity of ACE and ACE2 and other endopeptidases can determine relative levels of Ang II and Ang-(1–7) production and thus their functional outcomes. This hierarchical interplay also becomes important when interpreting the functional effects of ACE inhibitors and ARBs.
Angiotensin and NO• in the Kidney
Much of what we currently know about the signaling mechanisms of Ang-(1–7) derived from studies on the kidney. The specific binding sites or receptors for Ang-(1–7) were mostly unknown until Santos et al. (64) identified the Ang-(1–7) receptor as the G protein-coupled receptor encoded by the Mas protooncogene. Santos et al. demonstrated that genetic deletion of the Mas receptor abolished Ang-(1–7) binding in the mouse kidney and prevented in vitro and in vivo vascular and renal responses to Ang-(1–7) stimulation. Nevertheless, questions remained regarding the specificity of the Mas receptor for Ang-(1–7). These questions arose from evidence that specific 125I-Ang-(1–7) binding in kidney sections did not differ between wild-type and Mas-knockout mice, and the Ang-(1–7) antagonist A-779 could only displace approximately 40% of labeled Ang-(1–7) binding (58, 64). Despite these reservations, there is good evidence that the Mas receptor mediates most of the known biological and physiological effects of Ang-(1–7) in different tissues (8, 22, 64). In the rat kidney, Mas receptor binding is localized primarily in the inner cortex and the outer stripe of the outer medulla as demonstrated using labeled Ang-(1–7) receptor binding techniques (8, 22, 64). Other studies have reported that Mas receptors are widely expressed in intrarenal afferent arterioles and the tubular epithelium, especially in proximal tubules, where the known biological and physiological effects of Ang-(1–7) have been extensively investigated (8, 22).
Generally, the physiological actions of the renin/ACE/Ang II/AT1 receptor axis are opposed by the actions of the ACE2/Ang-(1–7)/Mas receptor axis. This is largely due to the inhibitory action of MAP kinase- or cyclooxygenase-2-dependent pathways, or stimulation of nitric oxide (NO•)/cGMP-dependent pathway by Ang-(1–7) (36, 63). The activation of the renin/ACE/Ang II/AT1 receptor axis in general terms causes vasoconstriction of renal vasculature, resulting in a decrease in renal blood flow (RBF), a decrease in glomerular filtration rate (GFR) (33), stimulation of proximal tubule sodium reabsorption (85) and a decrease in urine flow (41), with a consequent increase in arterial blood pressure. Alternatively, the actions of the ACE2/Ang-(1–7)/Mas receptor axis vasodilate the intrarenal blood vessels, increase RBF and GFR, inhibit proximal tubular transport of sodium, and induce diuresis (8, 22) to cause a reduction in arterial blood pressure. Given these opposing actions in the kidney, the ACE2/Ang-(1–7)/Mas receptor axis may be a key mechanism that goes awry in disease conditions such as hypertension, and thus may be an effective target of therapeutic intervention. However, evidence in established hypertension in stroke-prone spontaneously hypertensive rat and the monogenetic TGR(mREN2)27 rat models (38) suggest that the ACE2/Ang-(1–7)/Mas receptor axis may not be involved in these hypertensive models. In addition, Ang-(1–7) may be associated with stimulation of growth-stimulatory pathways in human mesangial cells (88) and with increased arterial blood pressure and adverse cardiac remodeling in rats with nephrectomy (70). Ang-(1–7) also has been associated with accelerated strepzotocin-induced diabetic nephropathy (67), and Ang II-induced epithelial-to-mesenchymal transformation (6). Furthermore, similar to activation of the ACE/Ang II/AT1 receptor axis, Ang-(1–7) was reported to stimulate water reabsorption by enhanced water transport in rat inner medullary collecting duct via interactions with vasopressin V2 receptors (50). Thus, the general opposing effects of the two pathways (ACE/Ang II/AT1 receptor axis vs. ACE2/Ang-(1–7)/Mas receptor axis) in relation to similar actions during a given set of conditions indicate a complexity of interaction between the two pathways in health or disease that is difficult to predict. Further insight into this issue may be gained with differentiation of the receptor subtypes and the specific location of these subtypes within the kidney being activated by Ang II or Ang-(1–7).
The renin/ACE/Ang II/AT1 receptor axis is widely implicated in the pathogenesis and progression of acute and chronic kidney diseases, including hypertension and diabetic nephropathy (9, 52). Regardless of the complexities of Ang-(1–7) effects, there is good evidence that impairment of the ACE2/Ang-(1–7)/Mas receptor axis may be associated with maladies caused by the overactivation of the renin/ACE/Ang II/AT1 receptor axis. Indeed, impairment of the ACE2/Ang-(1–7)/Mas receptor axis was associated with increased blood pressure (5) and acute renal ischemic and reperfusion injury (11). In the kidney, ACE2 protein levels are significantly decreased in hypertensive rats, suggesting an antihypertensive regulatory role of ACE2 that is suppressed in the disease state (84). Thus, overexpression of ACE2 to increase Ang-(1–7) production or stimulation of the Mas receptor by Ang-(1–7) in target tissues remains to be considered a therapeutic intervention to counter the pathological role of the renin/ACE/Ang II/AT1 receptor axis in various renal and cardiovascular diseases.
Angiotensin and NO• in the Heart
Activation of RAS with subsequent generation of angiotensin Ang II is an important mediator of myocardial fibrosis and pathological hypertrophy that lead to heart failure. Conversely, there is good evidence that the ACE2/Ang-(1–7)/Mas receptor axis plays an important role in counterbalancing Ang II effects on the heart to maintain normal cardiac function. ACE2 deficiency is known to impair cardiac contractility and upregulate hypoxia-induced genes (28). ACE2 was also found to be upregulated in failing hearts (7), which may suggest a compensatory role of Ang-(1–7) in cardiac failure. Furthermore, while AT1 receptor antagonism attenuated cardiac remodeling and dysfunction in a rat model of myocardial infarction, these changes were associated with a three-fold increase in ACE2 mRNA expression in the left ventricle (37). Consistent with a key role of ACE2 in post-MI remodeling, overexpression of ACE2 similarly ameliorated left ventricular remodeling and dysfunction after myocardial infarction (81). The pathophysiological relevance of suppressed Ang-(1–7) function in the heart is further highlighted by studies demonstrating that chronic infusion of either Ang-(1–7) (29) or its stable non-peptide analog AVE-0991 is cardioprotective in experimental heart failure. Finally, several studies have demonstrated that Ang-(1–7) is protective against cardiac ischemia-induced injury and arrhythmias (69).
The beneficial antiarrhythmic effects of Ang (1 –7) on the failing heart result from the combined effect of the peptide on the sodium pump, hyperpolarization of cardiac cell membranes, and increased conduction velocity (12). The antiarrhythmic effect of low concentrations of Ang-(1–7) is mediated by Mas receptor activation and release of endogenous prostaglandins. However, Ang-(1–7) at supraphysiological concentrations in isolated hearts induced early-after depolarization (12). Conversely, downregulation of ACE2 was associated with pacing-induced atrial fibrillation (48). These studies suggest that optimal tissue concentration of Ang (1–7) must be achieved to permit a protective role of the Ang-(1–7) on cardiac arrhythmias.
The cardioprotective and anti-remodeling effects of Ang-(1–7) are induced by activation of cardiac Mas receptor and are independent of its effects on blood pressure or Ang II receptors in the cardiac tissue (29). Gomes et al. (27) have recently demonstrated that the ability of Ang-(1–7) to reduce or prevent Ang II-induced pathological remodeling is mediated by a NO•/cGMP pathway in cardiomyocytes.
Angiotensin and NO• in the Neural Control of the Circulation
The concept that the RAS functions as a local tissue regulatory system in addition to an endocrine system derived from studies in the brain (10, 86). A number of studies and reviews underscore the important role of central Ang II on autonomic control of cardiovascular function (15, 87). Here we will focus on recent insights into the balance between brain Ang II and Ang-(1–7) in the brain areas that are involved in regulation of cardiovascular function, namely the paraventricular nucleus (PVN), subfornical organ (SFO), and median pre-optic nucleus (MnPO) in the forebrain, and nucleus of the tractus solitarius (NTS), rostral ventro-lateral medulla (RVLM), and caudal ventro-lateral medulla (CVLM) in the brainstem.
All the components of the RAS are expressed in the central nervous system. Angiotensinogen is expressed broadly throughout the central nervous system with high levels in the hypothalamus and brainstem, in areas involved in the regulation of autonomic outflow. Although angiotensinogen was shown to be mainly located in astrocytes, it was also demonstrated in neurons involved in cardiovascular regulation (51). Although renin immunoreactivity is observed in the hypothalamus and medulla, the levels are relatively low. This disparity between the synthesizing enzyme and subsequent product has been an ongoing controversy for the biosynthesis of angiotensin peptides in the central nervous system. Thus, an alternate pathway for the production of angiotensin peptides by non-renin-mediated mechanisms may involve the production of Ang-(1–12) (69), as described above. Local levels of Ang-(1–12) are higher than Ang I and Ang II in brain areas where local RAS exists (53). Although our knowledge of sites or processing pathways for Ang-(1–12) actions in the central nervous system is yet rudimentary, there are supportive results for Ang-(1–12) in regulation of cardiovascular function within the NTS in rats (2). The regulation and processing of endogenous Ang-(1–12) for central cardiovascular regulation is an important and promising area for future investigations.
ACE2 mRNA and protein have been demonstrated in key cardiovascular areas in the brain (18, 32). ACE2 is widespread throughout the mouse brain, present in nuclei involved in the central regulation of cardiovascular function such as the PVN, NTS, and RVLM, as well as in noncardiovascular areas such as the motor cortex and raphe (47). A dense Mas receptor immunoreactive staining was observed in cardiovascular-related areas from the medulla to the forebrain, such as the NTS, RVLM, CVLM, inferior olive, parvocellular and magnocellular PVN, supra-optic nucleus (SON), and lateral pre-optic area, as sites for the action of Ang-(1–7) in the brain (3). Moreover, at the cellular level, Mas receptor was predominantly present in neurons, as evidenced by co-localization of immunostaining for the neuronal marker, NeuN, and the Mas receptor antibody (3).
The physiological activation of the renin/ACE/Ang II/AT1 receptor axis causes desensitization and blunting of the baroreflex, with a concurrent increase in overall sympathetic outflow, both of central (involving action in the CNS) and peripheral origin (acting locally in the periphery), vasopressin release, and stimulation of drinking (blood volume expansion), with a consequence of an increase in arterial blood pressure (57). Conversely, the activation of the ACE2/Ang-(1–7)/Mas receptor axis sensitizes and enhances the baroreflex (34), reduces overall sympathetic drive as measured by reduced plasma levels of norepinephrine (25), and causes a reduction in arterial blood pressure. It has been suggested that the hypotensive effects of Ang-(1–7) in the central nervous system may be mediated by an increase in bradykinin levels (49), potentiating the hypotensive effects of bradykinin (4) and enhancing the release of NO• (26). ACE2 may serve as the principal enzyme for the balance between the renin/ACE/Ang II/AT1 receptor axis and the ACE2/Ang-(1–7)/Mas receptor axis in the brain, since evidence suggests ACE2 acts as a critical enzyme for the function of Ang-(1–7) in the brain (75).
The classical RAS signaling pathway (ACE/Ang II/AT1 receptor axis) in the brain has been well studied. The ACE2/Ang-(1–7)/Mas receptor axis also participates in cellular signaling pathways that regulate broad aspects of brain function, including synaptic plasticity, normal development, and neuronal cell death (60, 73, 75). ACE2 has been suggested to participate in the central regulation of blood pressure and altered sympathetic nerve activity in hypertension. Overexpression of ACE2 in the brain reduces hypertension by improving arterial baroreflex and autonomic function in hypertensive animals (72, 76). Injection of ACE2 inhibitor MLN4760 into the NTS reduces baroreceptor reflex sensitivity for heart rate control in rats (16). Overexpression of ACE2 exerts protective effects in local tissues, including the brain (28). Lentivirus-mediated ACE2 overexpression in the RVLM causes long-term decrease in blood pressure in the spontaneously hypertensive rats (76). Subsequently, Lazartigues et al. also showed ACE2 over expression in the SFO prevented the Ang II-mediated pressor and drinking responses (20). These are the first pieces of evidence for the beneficial/therapeutic effects of ACE2 overexpression in the central nervous system.
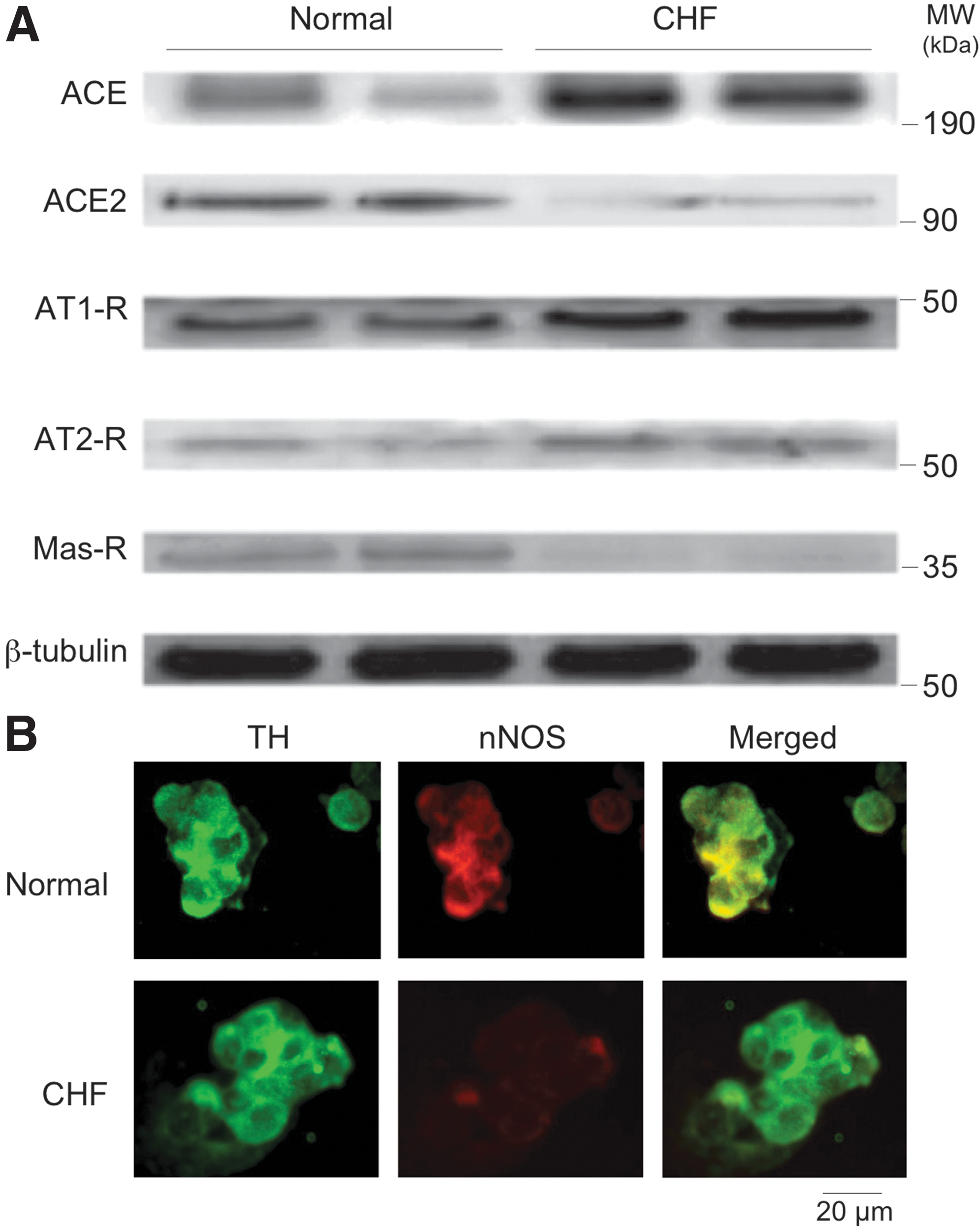
In the central nervous system, it has been shown that NO• reduces, whereas oxidative stress increases, sympathetic activity (24, 79). ACE/Ang II/AT1 receptor axis has been identified with the activation of the reactive oxygen species (ROS) pathway and consequent increase in sympathetic outflow. In the brain, the first evidence of a link between Ang-(1–7) and NO• was from the observation of co-localization of the heptapeptide with NOS in neurons of the SON and PVN (40). Overexpressing ACE2 in the PVN normalized the decreased nNOS protein levels in the PVN in rats with chronic heart failure (CHF) and was accompanied with improved sympathetic nerve activity, suggesting the inhibitory effects of ACE2 on sympatho-excitation involve an NO• mechanism (82). This is corroborated by the studies showing that overexpression of ACE2 in the central nervous system using transgenic mouse model with neuron-targeted overexpression of human (h)ACE2, upregulated expression of NOS immunostaining, and resulted in increased levels of NO• in the cerebrospinal fluid (19). Furthermore, the same study showed that overexpression of ACE2 in the central nervous system abrogated the Ang II-mediated reduction in NOS expression in brain regions known to be involved in regulating arterial blood pressure (19). In addition, ACE2 gene deletion was observed to result in increased levels of superoxide in various brain areas of aged mice (74), suggesting an antioxidant role for ACE2 in the brain. Taken together, these data suggest that the sympatho-inhibitory effects of ACE2 centrally may be linked to switching from the ACE/Ang II/AT1 receptor axis to the ACE2/Ang-(1–7)/Mas receptor axis to increase NO• availability and decrease oxidative stress in the central nervous system.
In a recent study, we showed that there was a concomitant decrease in ACE2 and nNOS within the PVN of rats with CHF (82). These data show that there were decreased levels of ACE2 and Mas receptor in the PVN of rats with CHF, supporting the idea that an imbalance of ACE2 and ACE is involved in the activation of the central renin-angiotensin system during CHF. Adenoviral ACE2 (AdACE2) transfection significantly increased nNOS protein levels in the PVN in CHF rats. AdACE2 transfection also significantly improved the attenuated renal sympathetic nerve activity (RSNA), arterial pressure, and heart rate responses to NOS inhibitor L-NMMA administration within the PVN in the rats with CHF (Fig. 2). These data indicate that ACE2 is involved in NO•-mediated sympatho-inhibition in CHF. How does upregulation of ACE2 within the PVN restore the levels of nNOS in the PVN of rats with CHF? To assess the direct effect of ACE2 on nNOS in an isolated cell culture system, we incubated NG108 cells with AdACE2 and observed a dose dependent increase in expression of nNOS protein (Figs. 3 and 4). Furthermore, co-incubation with A-779 prevented the upregulation of AdACE2-induced nNOS protein expression (82). These data are consistent with the idea that increased ACE2 expression may have caused the increase in nNOS protein expression within the PVN of rats with CHF (82).

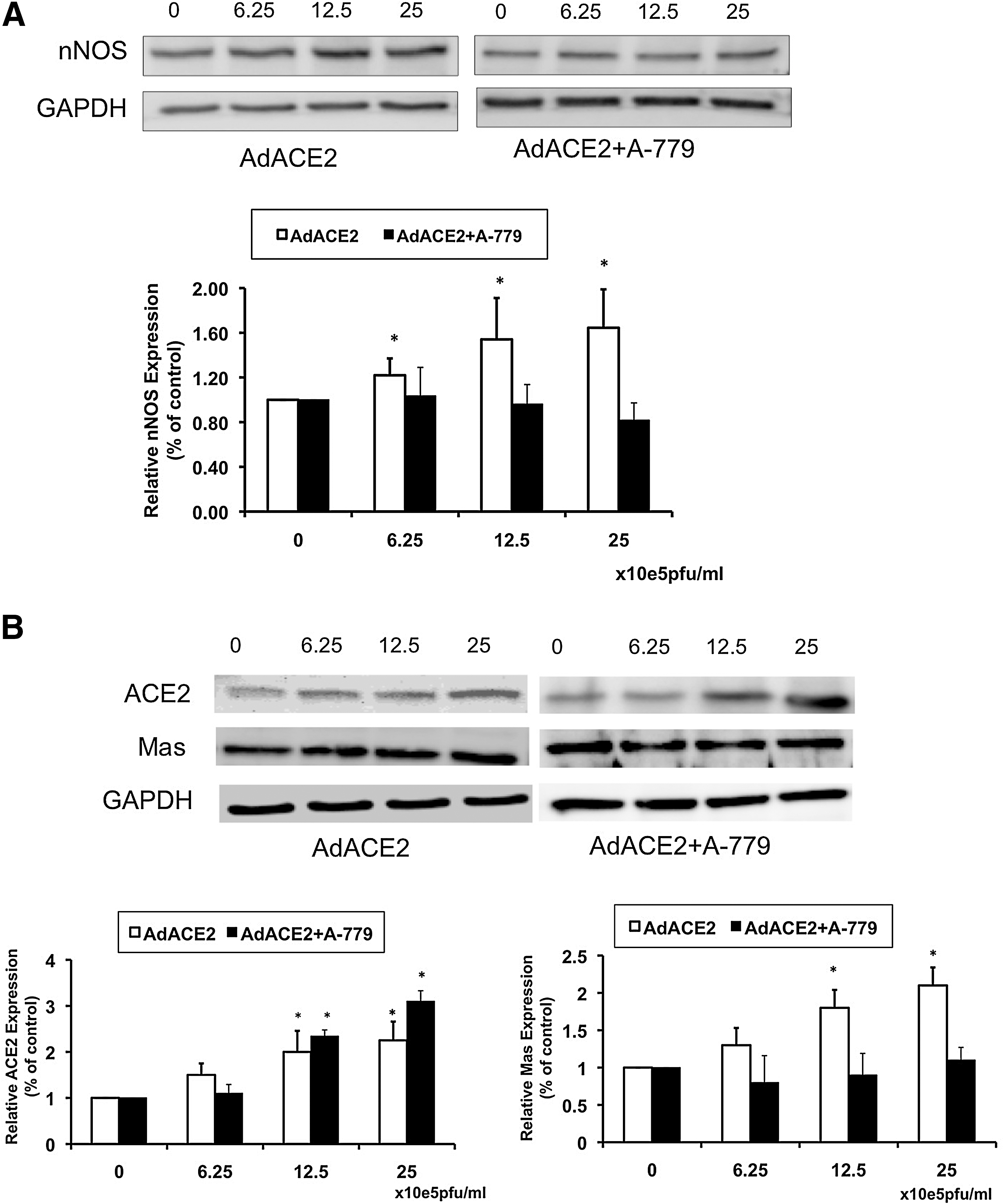

In another study, we addressed the intra-neuronal signaling pathway by which Ang 1–7 may activate neurons (77). For these studies, we used differentiated catecholaminergic (CATH.a) neurons, which express the Ang-(1–7) receptor (Mas-R) and nNOS. Stimulation of CATH.a neurons with Ang-(1–7) increased intracellular NO• levels, as measured by fluorescence confocal microscopy. The stimulatory effect of Ang-(1–7) on NO• production was inhibited by the Mas receptor antagonist (A-779), a nonspecific NOS inhibitor (L-NAME), or a nNOS inhibitor (SMTC), but not by eNOS or iNOS inhibition (L-NIO and 1400W, respectively). We also recorded voltage-gated outward K+ current (I Kv) in the CATH.a neurons. Ang-(1–7) significantly increased I Kv, and this response was inhibited by A-779 or SMTC, but not L-NIO or 1400W. These findings indicate that Ang-(1–7) is capable of increasing nNOS-derived NO• levels, which, in turn, activates hyperpolarizing I Kv in neurons.
The mechanism by which Mas receptor activation by Ang-(1–7) leads to nNOS activation and NO• production is not yet understood. However, Ang-(1–7)/Mas receptor activation has been shown to activate eNOS via a Akt-dependent pathway (62). Ang-(1–7)/Mas receptor activation was shown to phosphorylate Akt via a wortmannin-sensitive process, to differentially phosphorylate/dephosphorylate ser1179 and thr495 sites of eNOS.
Previously, we demonstrated that nNOS gene expression and protein in the PVN are significantly reduced in rats with CHF (55, 80). Stimulation of the PVN with Ang II has been shown to increase RSNA. Ang II also decreases levels of nNOS in neuronal cultures (68). Our data showed that there is concomitant increase in AT1 receptors with a decrease in nNOS within the PVN of rats with CHF. Perhaps Ang II is involved in downregulating levels of nNOS in the PVN in CHF. Ang II induces the expression of the NADPH oxidase subunits p22phox and p47phox, contributing to the enhancement of oxidative stress and reduction of NO• bioavailability (78). Gene transfer of ACE2 strikingly suppressed the expression of NADPH oxidase subunits and increased the NO• bioavailability (83). Recently, Gwathmey et al. showed a marked sensitivity of the intracellular NO• response to Ang-(1–7), which implies that renal nuclei expression of Mas receptor is functionally linked to NO• formation (31). However, a direct effect of ACE2 activation on NOS expression had not been studied until our in vitro data in NG108 cell cultures (82).
The impact of alterations of RAS control of neuronal function in CHF are evident not only in the central nervous system but also in peripheral afferent neural reflex pathways that control cardiorespiratory function. The peripheral chemoreflex plays a contributory role in the activation of the PVN and central activation of sympathetic outflow in CHF (65). An enhancement of the peripheral chemoreflex control of ventilation also is likely to contribute to the genesis of the abnormal breathing pattern, Cheyne-Stokes breathing, and central apneas that occur in CHF.
Similar to that shown in the PVN in CHF, ACE2 and nNOS expression and NO• production are decreased in the carotid body (CB), the sensory organ that mediates the peripheral chemoreflex (Fig. 5) (65). Conversely ACE/Ang II/AT1 receptor signaling are increased in the CB in CHF. Ang II enhances the hypoxic sensitivity of the CB chemoreceptor cells via an interaction with oxygen-sensitive potassium channels to suppress their voltage-gated currents (IKv) in CB glomus cells (45). This effect was shown to be enhanced in CHF to increase the sensitivity and afferent discharge of the CB chemoreceptors in response to hypoxia, which in turn increases central neural activation of sympathetic outflow in CHF.
The intermediate pathways involved in Ang II/AT1 receptor suppression of IKv in CB chemosensory cells are not fully elucidated. The effect is mediated by AT1 receptor activation of NADPH oxidase activation and enhanced production of ROS (43). In addition, a downregulation of antioxidant CuZnSOD and MnSOD function in the CB contributes to enhancing the oxidative stress induced by Ang II (13, 14).
The downregulation of Ang-(1–7)-mediated activation of NO• production in the CB in CHF also contributes to the enhanced CB afferent activity because NO• is inhibitory to CB chemoreceptor activity (44). Because O2 is essential for biosynthesis of NO•, normal basal production of NO• acts as an amplifier of O2 to suppress CB chemoreceptor discharge in normoxic conditions. However, basal NO• production and nNOS expression within the CB (Fig. 5) are depressed in CHF (44). We have found that Ang-(1–7) enhances IKv in CB glomus cells via Mas receptor activation of nNOS and NO• production (Fig. 6), an effect that opposes the inhibitory effect of Ang II on these channels (65).

The opposing effects of Ang II and Ang-(1–7) on CB chemo-sensitivity may provide a mechanism by which CB function can be finely tuned by the balance of factors influencing Ang production and metabolism in the CB. Under normal conditions, Ang-(1–7) may act to maintain NOS expression and NO• restraint of CB chemoreceptor activity. When CB Ang II level is elevated, such as in CHF, its excitatory influence on CB chemoreceptor activity overrides Ang-(1–7) and may even participate in the downregulation of Mas receptor/NO• effects seen in the CHF state. This balance is likely to be controlled by the relative expression of ACE and ACE2 activities in the CB. Consistent with this notion, ACE2 and Mas receptor are suppressed, whereas ACE and the AT1 receptor are upregulated in the CB in CHF.
Thus, a consistent picture is emerging concerning RAS influences on autonomic neurons. Within the central nervous system and peripheral autonomic reflex pathways, these angiotensin pathways result in a balance between oxidative and NO• mechanisms within these regulatory sites that translates into a balance between the sympatho-excitatory and sympatho-inhibitory drive (Fig. 7). An upregulation of Ang II signaling and downregulation of Ang-(1–7) signaling in autonomic neurons leads to enhance sympathetic tone and activation in various disease conditions like hypertension and heart failure condition.

The molecular mechanisms underlying the imbalance of central ACE and ACE2 in CHF are not yet understood. Recent data from a study by Koka et al. (39) have suggested that the p38 MAPK and ERK1/2 signaling pathway is involved in ACE upregulation and ACE2 downregulation. In addition, the pro-oxidant pathways downstream from Ang II–AT1 receptor activation, and nitrosative pathways emanating from Ang-(1–7)–Mas receptor activation also may have controlling and possibly counterbalancing effects on ACE and ACE2 expression. Many transcription factors (1) are redox sensitive, and this may be a transduction mechanism that becomes dysfunctional in CVD to create imbalances in the expression and function of the hierarchical RAS system.
Summary
The accumulative evidence summarized in this review suggests that a balance between the activation of the ACE/Ang II/AT1 receptor axis and the ACE2/Ang-(1–7)/Mas receptor axis plays an important role in the function of the heart, kidney, and autonomic nervous system control of the circulation, and that an imbalance in these opposing pathways toward the ACE/Ang II/AT1 receptor axis leads to CVD. However, a number of major questions remain to be addressed. For example, what are the stimuli in various types of CVD, such as heart failure, hypertension, renal failure, and diabetes, that drive the imbalance of expression of ACE and ACE2 and angiotensin receptors at the level of the heart, kidney, blood vessels, and central nervous system? Furthermore, exercise training has been shown to reverse this imbalance in heart failure (42, 56) and hypertension, for example, but the mechanisms involved in restoring normal RAS function with exercise remain to be elucidated. Elucidating these driving mechanisms in the regulation of the multiple components of the RAS system are key to our ability to develop better therapeutic strategies for CVD.
Footnotes
Acknowledgment
This review was supported by National Institutes of Health Grants P01HL-062222
Author Disclosure Statement
No conflicts of interest, financial or otherwise, are declared by the authors.