
Abstract
Introduction
Innovation
Bodies of evidence are now cumulating that cell fate depends on its redox status. Here, we show that oxidative and mild metabolic stresses are able to activate the same nitric oxide-dependent redox signaling pathway. Our study pinpoints a crucial intermediate in the process of redox control, that is, the p53-dictated PGC-1α antioxidant response. We show that the transcriptional complex in charge of antioxidants expression, the nuclear factor (erythroid-derived 2)-like2, is a key component of this system.
The tumour suppressor p53 was firstly recognized as having a key role in the control of cell cycle and DNA repair pathways, thus maintaining genomic stability (9, 31). More recent discoveries reveal new and opposite functions for p53 in the regulation of various aspects of cellular metabolism, senescence, and apoptosis (7, 13, 36, 46). Among the genes that are directly modulated by p53, antioxidants, mitochondrial, and glycolytic genes are included (16, 17, 36, 42). Another important key modulator of cellular metabolism is nuclear factor (erythroid-derived 2)-like2 (NFE2L2 or NRF2), a bZIP transcription factor controlling the expression of genes coding for detoxifying and antioxidant enzymes, thus maintaining ROS homeostasis (10). For these reasons, p53, NFE2L2, and PGC-1α may have overlapping functions as regulators of the antioxidant defense system. Intriguingly, NFE2L2, PGC-1α, and p53 can be activated by oxidative stress through the same redox-dependent pathways, including those managed by the nitric oxide (NO)/cGMP (1, 8, 23). Then, it could be hypothesized that a cross-talk exists between them upon redox imbalance.
We recently demonstrated that the disruption of the cellular redox buffer controlled by glutathione (GSH) increases endogenous physiological flux of NO in neuronal cells, leading to an upregulation of p53 by the NO/cGMP signaling pathway (1). GSH is the major cellular nonenzymatic antioxidant (21, 48) playing important roles in nutrient metabolism, and regulation of many cellular events (48). Decrement of GSH levels contributes to oxidative stress associated with ageing and many pathological states, including neurodegeneration, inflammation, and infections (6, 22, 25). PGC-1α expression is enhanced by energetic stress (i.e., fasting/calorie restriction and physical exercise), a condition that may also affect GSH content (30, 37, 45). On the basis of this evidence, in the present report, we investigated whether redox-mediated p53 activation could intersect with PGC-1α-regulated pathways and modulate cellular adaptive response to redox and metabolic imbalance.
Results
NO increase induced by GSH deficiency is associated with p53 and PGC-1α upregulation in SH-SY5Y cells
We have previously showed that in SH-SY5Y neuroblastoma cells GSH depletion obtained by
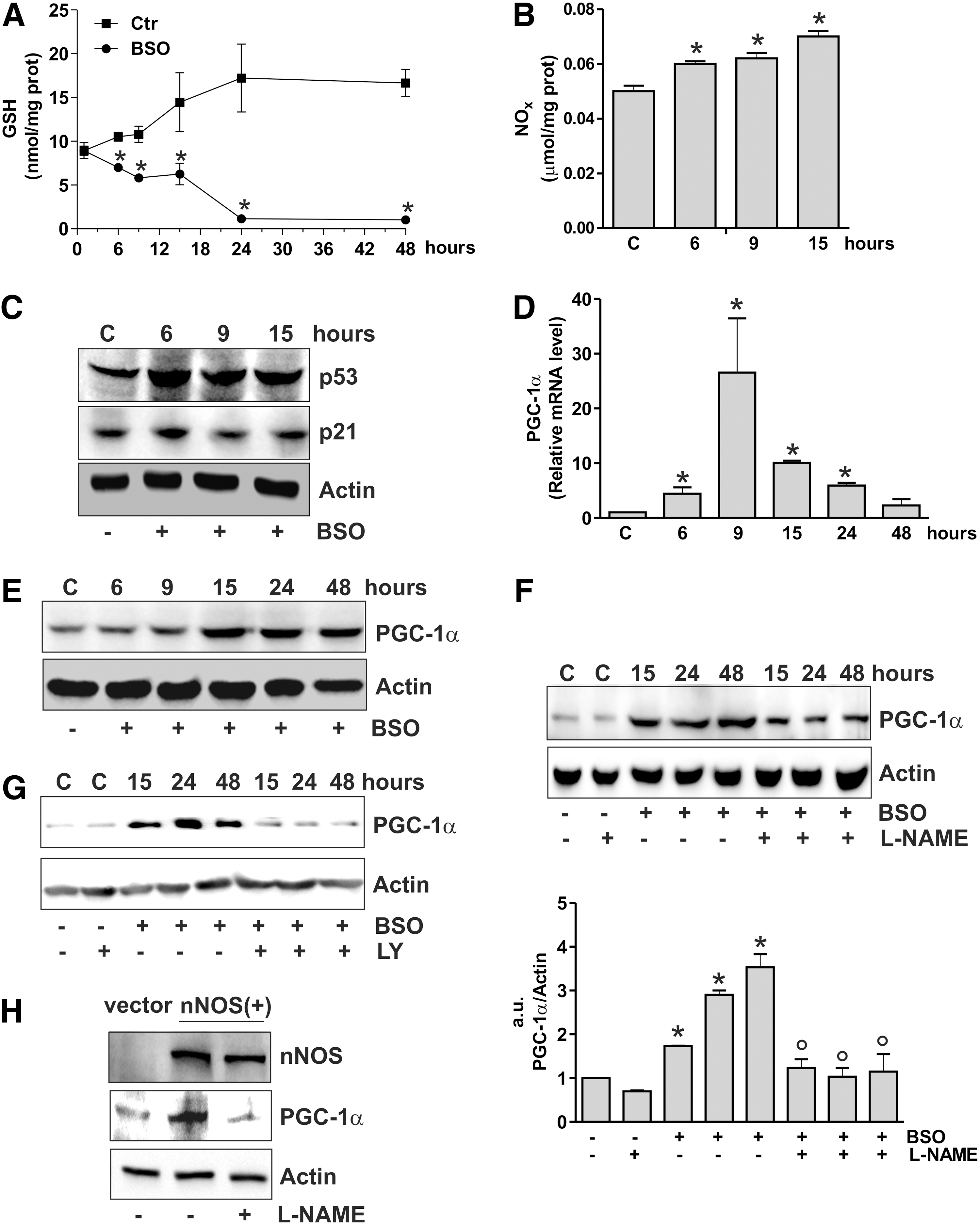
PGC-1α expression is associated with increased binding of p53 to human PPARGC1A promoter
The data obtained show that the increase of p53 precedes PGC-1α induction. Thus, we further investigated the role of p53 as upstream modulator of PGC-1α expression by downregulating p53 through RNA interference (RNAi). p53 downregulation was efficiently achieved in SH-SY5Y cells and proved to be successful in inhibiting the BSO-mediated increase of PGC-1α mRNA and protein levels (Fig. 2A). The same results were obtained by treating cells with pifithrin α, a chemical compound having the ability to suppress p53-mediated transactivation (20) (Fig. 2B). We then used human lung cancer NCI-H1299 cells that are null for p53 and are able to produce NO via endothelial NOS (29). GSH depletion in these cells failed to induce PGC-1α unless wild-type p53 was transfected (Fig. 2C and Supplementary Fig. S1A; Supplementary Data are available online at
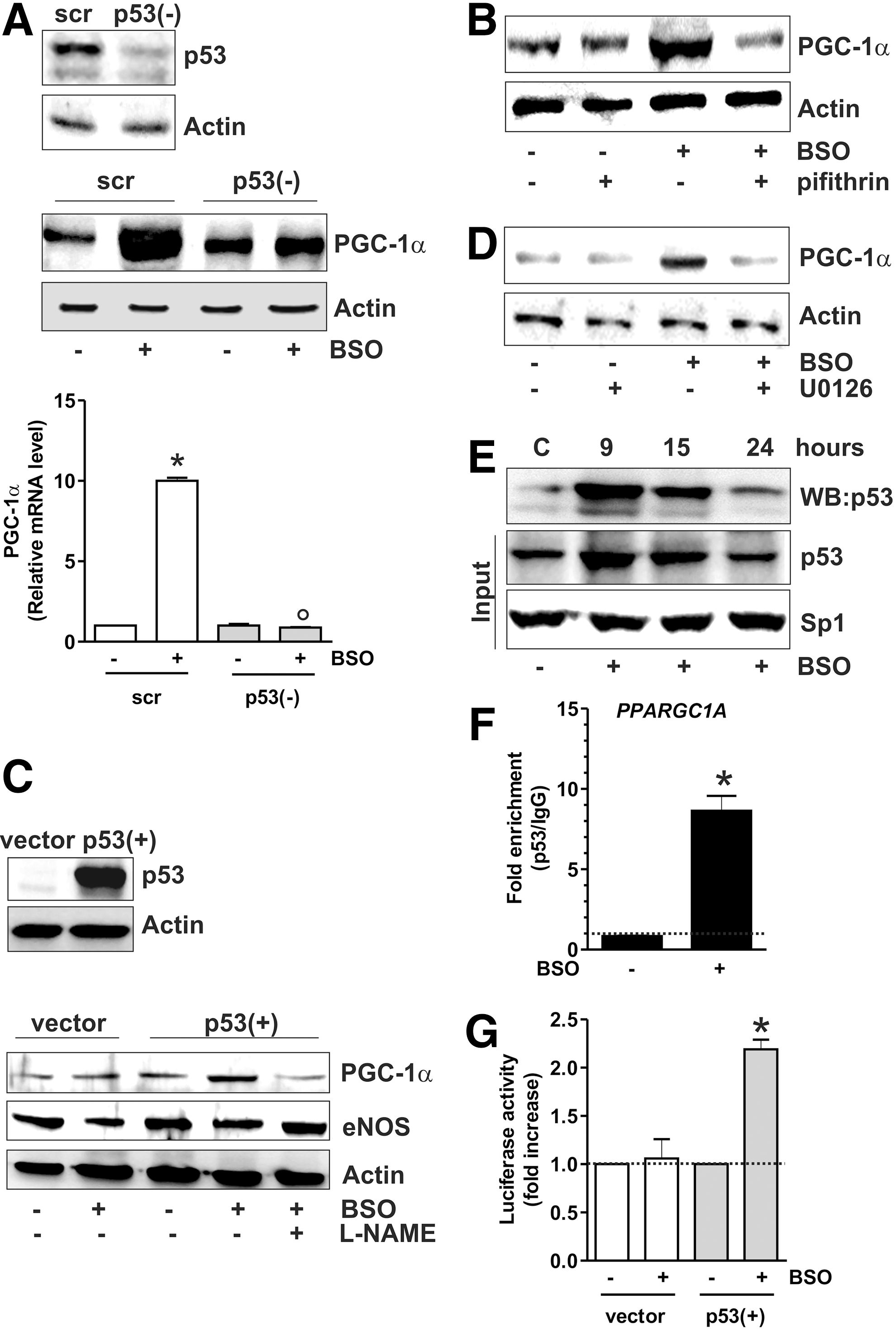
By analyzing the human PPARGC1A promoter, Irrcher et al. identified a unique putative p53-response element (p53RE) at −1237 (19). Our effort was therefore directed to study whether p53 could bind this promoter region. SH-SY5Y nuclear extracts were incubated with a biotinylated oligonucleotide representing p53RE, and Western blot analysis of p53 was carried out after the oligo-pulldown. As reported in Figure 2E, p53 protein significantly accumulated within the nucleus, and this event was associated with its increased DNA-binding activity, especially after 9 h from BSO addition. We performed a chromatin immunoprecipitation assay (ChIP) assay to confirm the increased binding of p53 to such p53RE. qPCR analysis revealed that the PPARGC1A promoter is not bound by p53 in control cells. However, a 10-fold increase in the occupancy of p53RE in BSO-treated cells was observed (Fig. 2F). The capacity of p53 to bind the PPARGC1A promoter was also investigated by performing a gene reporter assay. In particular, the PPARGC1A promoter region spanning from +1 to −1600 bp, and comprising the p53RE located at −1237, was cloned into a pGL3 vector upstream the luciferase gene and transfected either in NCI-H1299 p53-null or NCI-H1299-p53(+) cells. Figure 2G shows that BSO is not able to increase luciferase activity in NCI-H1299 cells that are null for p53. Conversely, in NCI-H1299 p53(+) cells, about a twofold increase in luciferase activity was observed after BSO administration, confirming that PPARGC1A promoter expression is under the control of p53.
p53-mediated PGC-1α expression results in NFE2L2-mediated antioxidant response
Our previous data indicated that depletion of GSH was not associated with increased mitochondrial biogenesis; indeed, the content of mitochondrial proteins such as Hsp60, cytochrome c oxidase, and cytochrome c remained unchanged after BSO treatment (1). To deeply investigate on this matter, we looked at canonical markers of PGC-1α-induced mitochondrial biogenesis, that is, nuclear respiratory factor-1 (NRF-1) and mitochondrial transcription factor A (TFAM) (38). Figure 3A and B show that NRF-1 and TFAM levels were not altered both in terms of protein and mRNA, supporting the evidence that mitochondrial biogenesis is not operative upon GSH depletion. PGC-1α has also a pivotal role in coordinating a large part of the antioxidant defense system and has complementary and often-overlapping functions with NFE2L2 (10). The analysis of the mRNA and protein levels of NFE2L2 (Fig. 3C) demonstrated that it was significantly induced by BSO treatment starting at 15 h. We then evaluated whether the PGC-1α- and/or NFE2L2-governed antioxidant responses were enhanced. Figure 3 shows that the protein (Fig. 3D, E) and mRNA (Fig. 3F, G) levels of manganese superoxide dismutase (SOD2) and γ-glutamylcysteine ligase (γ-GCL) were significantly increased starting from 15 up to 48 h after BSO administration. Conversely, the level of Trx1, catalase, and SOD1 was not affected (Fig. 3D). To verify whether the binding activity of NFE2L2 was modulated by BSO, NFE2L2 association with the promoter of the human GCLC gene, coding for the catalytic subunit of γ-GCL, was assayed. Both oligo-pulldown (Supplementary Fig. S2A) and ChIP analyses (Fig. 4A) indicate that NFE2L2 binding activity was enhanced, with an eightfold increase in the occupancy of NFE2L2RE in the GCLC promoter at 15 h.

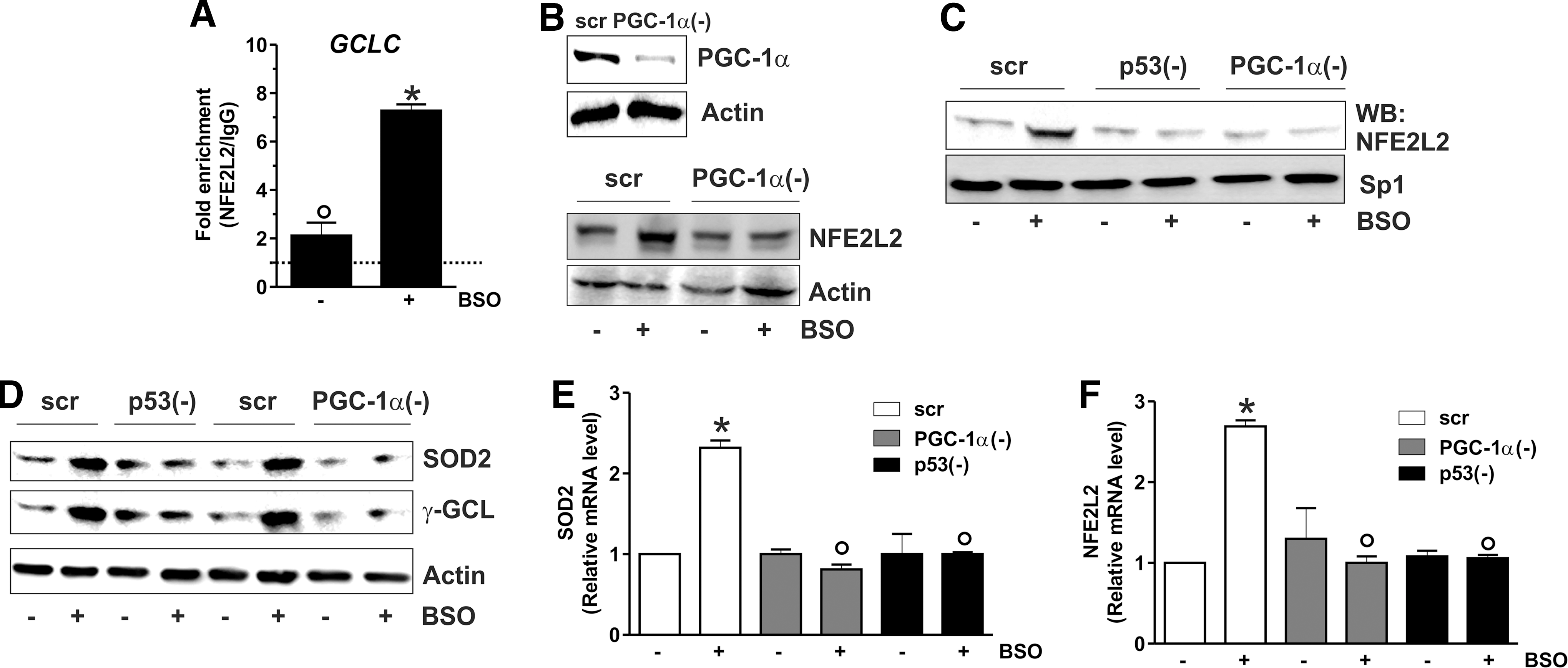
To characterize the sequence of the signaling events leading to the antioxidant response, we monitored the level of NFE2L2 after downregulation of PGC-1α through RNAi. Figure 4B shows that PGC-1α is efficiently downregulated (upper panel), and this results in the abrogation of BSO-induced NFE2L2 expression (bottom panel). Moreover, the silencing of either p53 or PGC-1α led to a significant abrogation of NFE2L2 binding to the GCLC promoter upon GSH depletion as demonstrated by the oligo-pulldown assay reported in Figure 4C. A parallel decrease in the SOD2 and γ-GCL protein content was also observed upon downregulation of p53 or PGC-1α by RNAi (Fig. 4D). Figure 4E and F show that either p53 or PGC-1α downregulation was effective in inhibiting NFE2L2 as well as SOD2 mRNA expression, demonstrating that they are upstream modulators of the NFE2L2-mediated antioxidant response.
We previously reported that BSO was able to increase the level of ROS/RNS in SH-SY5Y cells (3), and prolonged treatment with BSO (48 h) resulted in the induction of a caspase-independent apoptotic program (1); therefore, we wonder whether the antioxidant response downstream of the p53/PGC-1α pathway could modulate cell fate. To this end, we downregulated p53, PGC-1α, or NFE2L2 protein by RNAi (Fig. 5A), and we monitored the production of ROS/ONOO˙− and the rate of dead cells after BSO treatment. Figure 5B shows an augmented production of ROS/ONOO˙− in BSO-treated cells, which is further enhanced after downregulation of p53, PGC-1α, or NFE2L2. As expected, p53, PGC-1α, or NFE2L2 downregulation strongly enhanced cell death induced by BSO at 48 h (Fig. 5C). The role of NFE2L2 in promoting the antioxidant response is evident in NFE2L2-downregulating cells. In particular, upon NFE2L2 downregulation through RNAi, BSO treatment was not more able to elicit SOD2 upregulation (Fig. 5D). In line with the protective role of PGC-1α against oxidative stress, overexpression of PGC-1α completely abrogated the increase of ROS/ONOO˙− levels after BSO treatment (Fig. 5E), resulting in a significant reduction of cell death commitment (Fig. 5F).

p53 binding to the mouse ppargc1a promoter is positively related to PGC-1α increase in GSH-depleted murine models
Recently, three p53 putative binding sites on the mouse ppargc1a promoter were identified. The sites located at −954 and −564 were demonstrated to function as p53 repressive regions that inhibit PGC-1α expression (36). Therefore, we investigated whether p53 could modulate PGC-1α expression also in murine cells. To this end, C2C12 myoblasts were treated with BSO that was efficient in decreasing GSH content (Fig. 6A). Also, in this case, GSH decrement results in the increase of PGC-1α and SOD2 content both in terms of protein (Fig. 6B) and mRNA (Fig. 6C, D). L-NAME completely abolished such event (Fig. 6B), confirming that NO increase was the triggering factor. Moreover, p53 inhibition by pifithrin-α was able to block PGC-1α and SOD2 upregulation (Fig. 6E and Supplementary Fig. S1B).
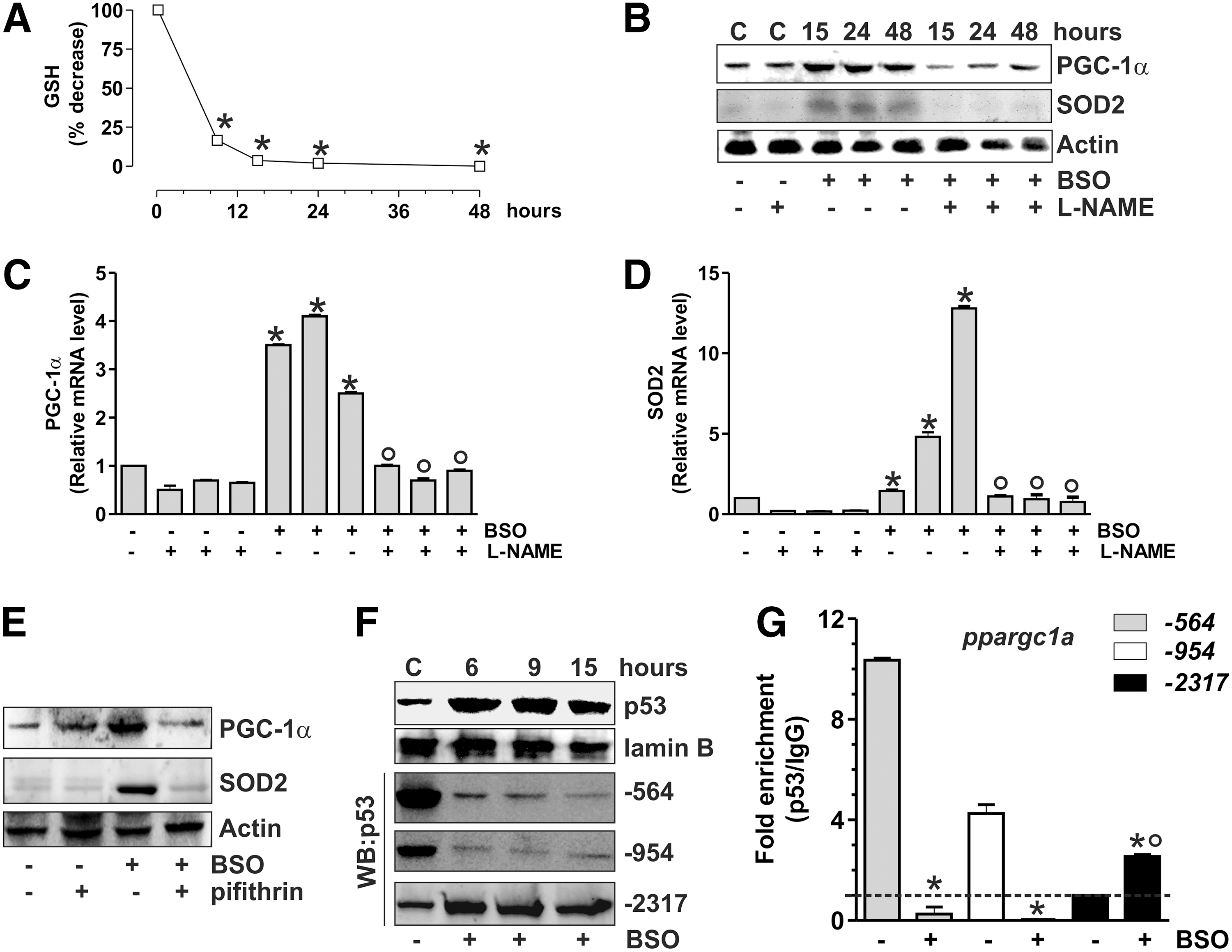
We performed oligo-pulldown assays by using the two identified p53 repressive regions (−954 and −564) and a third region located at −2317, which was hypothesized to be an additional p53-binding site (36). Consistent with the PGC-1α upregulation that we observed after BSO treatment, the two repressive regions were rapidly released by p53 (Fig. 6F). The oligo-pulldown assay reported in Figure 6F reveals that the p53 binding to the −2317 region was concomitantly enhanced. ChIP analyses of all the three p53-binding sites were carried out to confirm the regulatory role of p53 on the murine ppargc1a promoter. The qPCR analysis shows that the −2317 region is not bound by p53 at the basal level, in line with the results reported by Sahin et al. (36). However, a significant increase (threefold) in p53 occupancy of the −2317 region was observed after BSO treatment, while the −564 and −954 regions show a complete loss of p53 binding (Fig. 6G).
The proof that the signaling pathway discovered in human cells is also operative in murine cells prompted us to move to study the in vivo system. In particular, we induced GSH deficiency in mice by adding BSO in drinking water. Mice were sacrificed after 5 weeks of treatment, and GSH content was analyzed by HPLC in the brain and skeletal muscle. Among the metabolically active organs, the brain was selected on the basis of its reported resistance to GSH depletion by BSO (12) and protection from the energetic imbalance caused by fasting (14). Skeletal muscle was chosen because of the well-known dependence of its metabolism on PGC-1α activity (33). As shown in Figure 7A, a significant GSH depletion in tissues from BSO-treated mice was obtained that was more marked in skeletal muscle with respect to the brain. Densitometric analyses of p53, PGC-1α, and SOD2 levels (Fig. 7B) detected by Western blot (Supplementary Fig. S3) demonstrated a significant increase of these proteins after BSO treatment. Concomitant administration of L-NAME completely abolished the response to GSH deficiency (Fig. 7B and Supplementary Fig. S3). In line with these results, an increase of PGC-1α and SOD2 mRNA was also observed upon BSO supplementation, and L-NAME was able to abolish such increase (Fig. 7C).
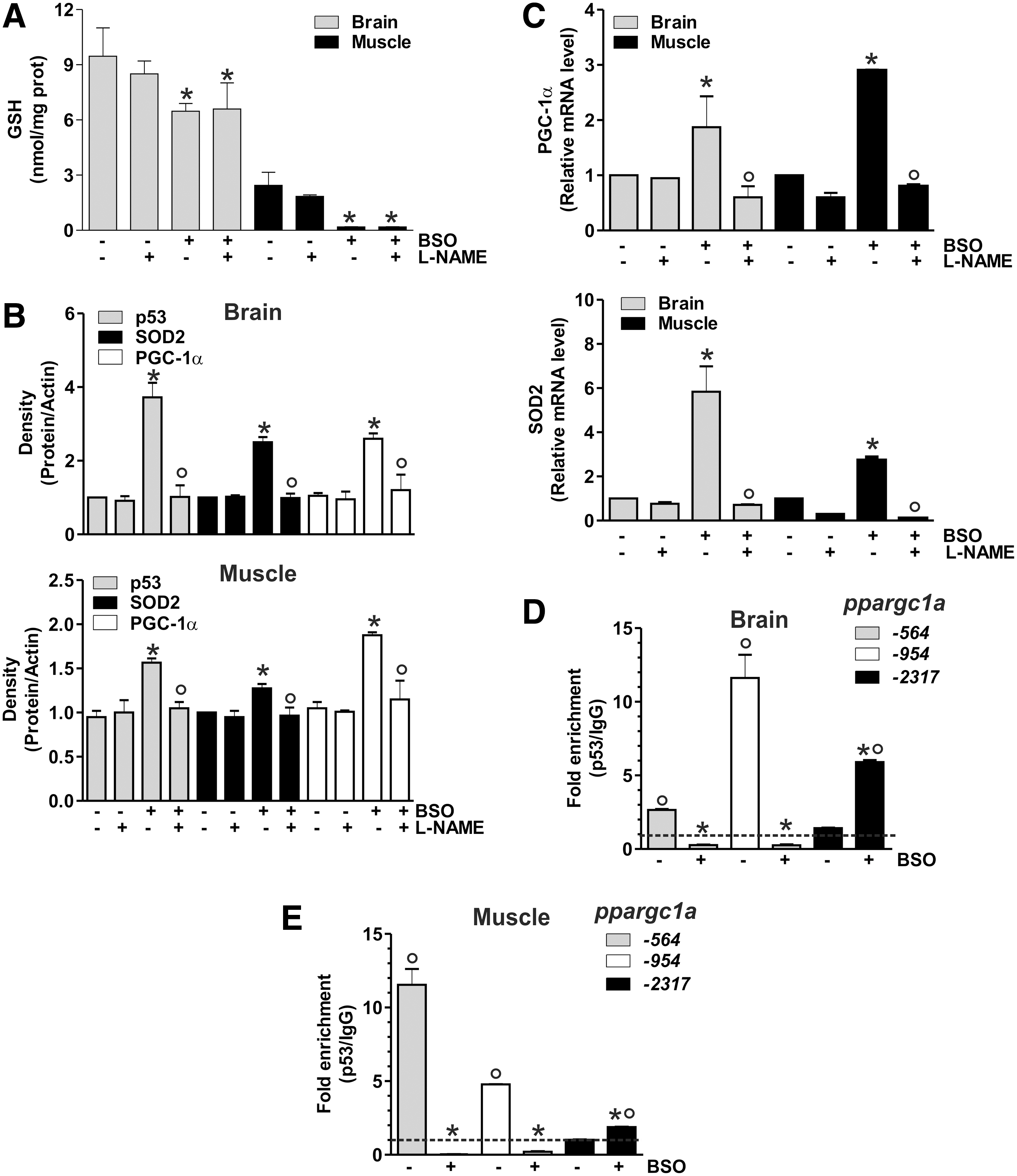
ChIP assay was carried out to analyze the binding of p53 to the ppargc1a promoter. As reported in Figure 7D and E, BSO induced a release of p53 from −564 and −954 regions, and a concomitant increase of p53 binding to −2317 region both in brain and skeletal muscle, respectively. Different degrees of p53 binding to these promoter regions are operative under basal conditions, with the binding of p53 more efficient at −954 in the brain, and −564 in skeletal muscle and C2C12 cells.
GSH decrease promoted by fasting is accompanied by the upregulation of the p53/PGC-1α pathway
PGC-1α is regulated transcriptionally by different pathways upon starvation (11, 24). Therefore, we asked whether the binding of p53 to the ppargc1a promoter could be influenced upon fasting. We first investigated whether fasting was able to affect GSH content in the mouse brain and skeletal muscle. Mice were starved for 24 h, and GSH level was assayed in tissue homogenates. Figure 8A shows that GSH was significantly decreased in the brain and skeletal muscle, and this phenomenon was associated with an increase of p53 protein (Fig. 8B, C). Fasted mice also displayed augmented protein (Fig. 8D, E) and mRNA (Fig. 8F) levels of PGC-1α and SOD2 in both the brain and skeletal muscle. Nuclear extracts from the brain and skeletal muscle of ad libitum fed or fasted mice were subjected to analysis of p53 binding to the ppargc1a promoter. Oligo-pulldown (Supplementary Fig. S2B, C) and ChIP analyses (Fig. 8G) demonstrated that fasting was associated with the release of p53 from the repressive regions (Supplementary Fig. S2B, C) and the increase of p53 binding activity on the −2317 site. ChIP analysis also confirmed that the −2317 region is not bound by p53 under resting condition (as the same value was observed in sample immunoprecipitated with anti-p53 with respect to IgG isotype control). On the contrary, the p53 binding to such region is significantly enhanced during fasting (2-fold and 1.3-fold with respect to IgG isotype control in the brain and muscle, respectively) (Fig. 8G).

Discussion
Here we show that p53, PGC-1α, and NFE2L2 are part of the same redox signaling pathway that positively coordinates the expression of SOD2 and γ-GCL in response to GSH decrement. GSH depletion—obtained by BSO-mediated inhibition of the rate-limiting enzyme of its synthesis (γ-GCL)—represents the primum movens of such a cellular response. The most important finding is that p53 is able to bind the −1237 p53RE on the human PPARGC1A promoter, and is fundamental to assure the expression of PGC-1α. Indeed, the downregulation of p53 was associated with the failure of PGC-1α mRNA increase. We also show for the first time that NFE2L2 expression is controlled by the p53/PGC-1α axis, thus giving a mechanistic effort to their overlapping functions and/or synergistic actions in regulating the antioxidant system (10, 47). During the progressive decline of GSH, the activated PGC-1α/NFE2L2 pathway stimulates a sort of adaptive response that buffers the potential harmful effect of ROS/RNS, resulting in mild oxidative stress and protection against cell death. The statement that PGC-1α plays a central role in modulating the NFE2L2-mediated antioxidant response comes from the data demonstrating an enhanced ROS/RNS concentration and cell death and a failure in the upregulation of SOD2 upon p53, PGC-1α, or NFE2L2 downregulation. In this context, we have strong evidence that PGC-1α is in some way directly involved in the regulation of the NFE2L2 expression/activity. Even though a molecular interaction between NFE2L2 and PGC-1α has not been investigated in the present work, it could be interesting to establish whether besides modulating transcription factors such as NRF-1, -2 and CREB, PGC-1α could also coactivate NFE2L2, thus assisting the induction of antioxidant genes. This matter is currently under investigation in our laboratory. The NO-mediated p53 upregulation culminates in the expression of PGC-1α and SOD2 also in murine cells and mice, revealing that the transduction of the redox signaling triggered by GSH deficiency is of general application. Even if PGC-1α is significantly upregulated, mitochondrial biogenesis is not induced, according to a recent report showing that GSH is a pivotal factor in the activation of mitochondrial biogenesis. In particular, GSH positively regulates the activity of the most important modulator of mitochondrial biogenesis in yeast, that is, the transcriptional coactivator Hap4p (49). It is likely that a reducing environment is fundamental for the function of PGC-1α as a coactivator of mitochondrial genes. Conversely, it is possible that a decreased redox state triggered by GSH deficiency may switch PGC-1α activity toward the antioxidant response.
We found that, in GSH-deficient murine cells and mice, PGC-1α and SOD2 upregulation is associated with p53 release from the two repressive regions in line with the suggested negative role exerted by p53 on ppargc1a transcription (36). Interestingly, we have also demonstrated that a third p53RE on the mouse ppargc1a promoter, which is not bound by p53 under resting conditions, is engaged by p53 upon GSH deficiency. On the basis of all these observations, we can suggest that in mice, PGC-1α transcription is controlled by p53 at two different levels. The first level consists in the negative regulation by p53 through its binding to the two repressive regions as previously suggested (36); the second level is represented by the binding of p53 at a unique activating region. In our mouse experimental model, the enhanced expression of PGC-1α is likely the result of the release of p53 from the repressive regions and the parallel increased binding of p53 at the activating site. The diverse behavior of p53 in recognizing the p53REs on the mouse ppargc1a promoter may be dependent on redox modification (e.g., S-nitrosylation) of critical p53 amino acids that differently affect (enhance/inhibit) its DNA-binding activity to specific consensus sequences. The regulation of PGC-1α expression appears to be more intricate in mouse than in human, wherein the two p53 repressive regions have not been conserved during evolution. Indeed, PPARGC1A is one of the 49 identified human accelerated regions, which means that such gene is conserved throughout vertebrate evolution, but is strikingly different in humans (28). Indeed, the pairwise alignment of the PPARGC1A promoter region with that from mouse results in a poor identity (49%). Even if no apparent structural similarities can be observed between the mouse and the human p53RE, our evidence indicates that the −2317 region on the mouse ppargc1a promoter is the functional homolog of the −1237 on the human PPARGC1A promoter, and its binding by p53 positively correlates with enhanced PGC-1α transcription.
We show that 1-day fasting in mice is able to reduce GSH levels in the brain and skeletal muscle, and this event is associated with the detachment of p53 from the repressive regions and the increased binding activity of p53 to the activating region on the ppargc1a promoter. We also documented the effective activation of the antioxidant response via the upregulation of SOD2 during fasting. It was proposed that the increase of NO levels was essential for many beneficial effects of dietary limitation (26, 27), and the decrease of GSH content could further enhance NO imbalance. Although the effective increase of NO concentration and the strict connection between GSH decrement, oxidative stress, and the activation of p53/PGC-1α remain to be investigated in our experimental model of fasting, the observation that the ppargc1a promoter undergoes the same regulatory mechanisms with respect to chemical GSH depletion strongly indicates the intervention of NO/cGMP signaling.
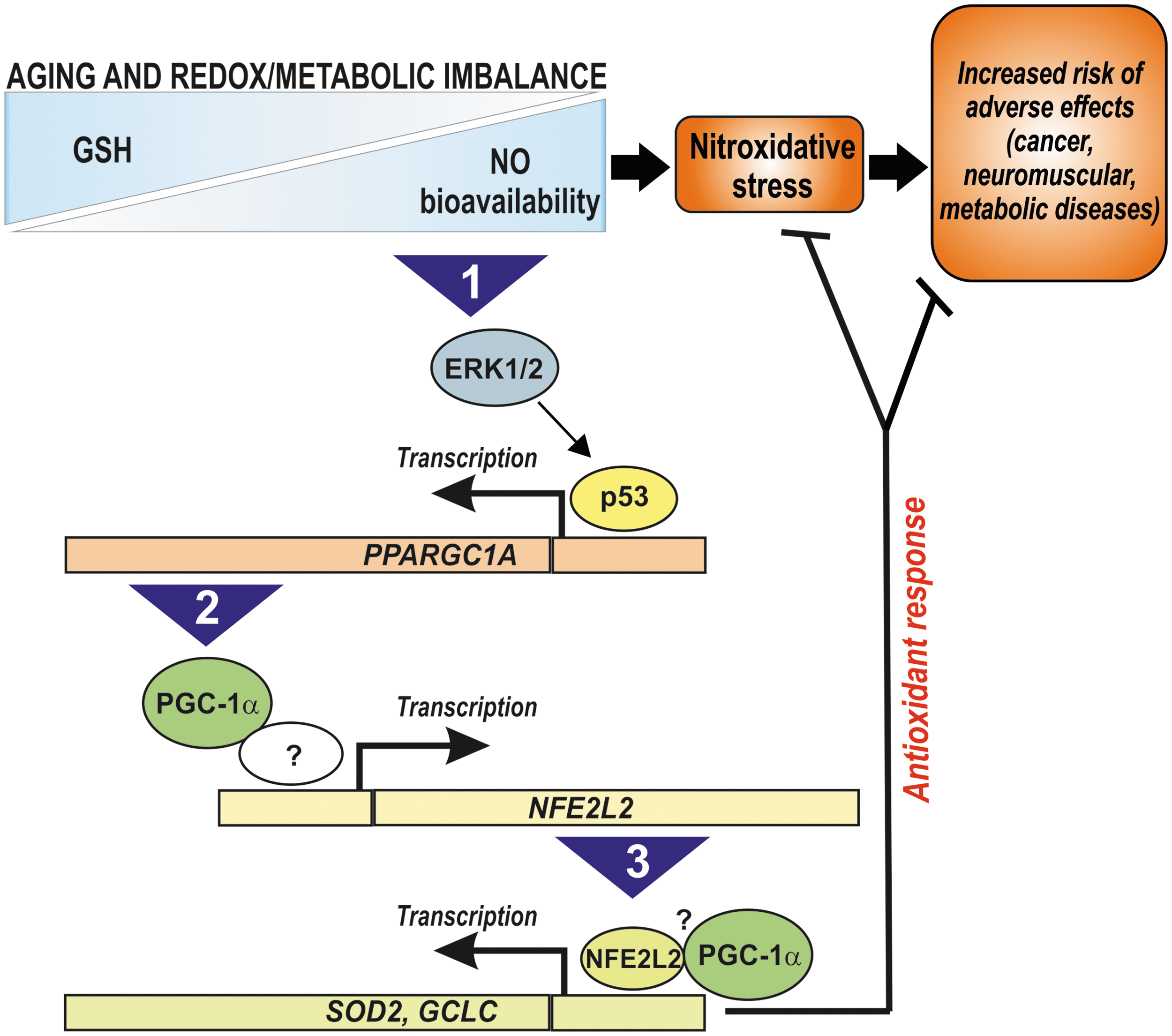
We previously showed that increased NO bioavailability is the primary mediator of neurotoxicity upon GSH depletion in vitro, resulting in a substantial nitration and S-nitrosylation of cellular proteins (1). However, the results obtained in the present work show that a parallel and compensative NO-dependent induction of antioxidants via a p53/PGC-1α/NFE2L2 pathway is induced, which is directed to dampen the detrimental effect of GSH decrement. Then, it can be hypothesized that the decline of GSH level occurring with normal ageing in many organisms (32) is a beneficial rather than detrimental event and may serve as a molecular signal inducing an endogenous antioxidant adaptive response that culminates in increased resistance to environmental/pathological stress and possibly in the extension of lifespan. Dramatic pathological variations of GSH content and/or the inability to activate such stress response could thus increase the risk of adverse effects leading to age-related pathologies (e.g., cancer, neuromuscular, and metabolic diseases). Therefore, the pharmacological targeting (e.g., through BSO treatment) or metabolic modulation (e.g., through fasting or exercise) of the NO/p53/PGC-1α/NFE2L2 pathway promises to be an effective therapeutic approach that can safeguard health, therefore favoring longevity (Fig. 9).
Materials and Methods
Cell cultures, transfections, and ROS/RNS assay
Human SH-SY5Y neuroblastoma cells and murine C2C12 myoblasts were purchased from the European Collection of Cell Cultures (Salisbury, United Kingdom) and cultured as suggested by the supplier. NCI-H1299 cells lacking of p53 were a kind gift of Prof. Cesareni (Department Biology, University of Rome Tor Vergata).
BSO, a highly selective and potent inhibitor of the enzyme γ-GCL, was added in the culture medium at a concentration of 1 mM. The NOS inhibitor L-NAME and the guanylate cyclase inhibitor LY-83583 were used at a concentration of 0.1 mM and 0.002 mM, respectively. Treatments with cell-permeable MEK1/2 inhibitor U0126 and p53 inhibitor pifithrin α were done at a concentration of 260 nM and 0.02 mM, respectively.
Plasmids were transfected by electroporation as previously described (2) using pSV-PGC-1α (Addgene, Cambridge, MA), pcDNA3.1-nNOS, or pcDNA3.1-p53 (kindly donated by Dr. Yvonne Sun, The Cancer Institute of New Jersey, NJ). PGC-1α and p53 siRNA oligonucleotides are reported in Table 1. ROS/RNS levels were assayed by means of DCF-DA staining as previously reported (44).
PGC-1α, peroxisome proliferator-activated receptor-γ coactivator-1α.
Animals
C57/BL/6J and CD1 (5 weeks of age) were obtained from Harlan Laboratories S.r.l. (Urbino, Italy). Twenty C57/BL/6J mice were randomly divided into four experimental groups: mice treated with BSO (20 mM); mice treated with L-NAME (4 mM), mice treated with BSO together with L-NAME, or with unsupplemented water. BSO and L-NAME were administered for 5 weeks in drinking water. Mice had free access to food and water.
For fasting experiments, eight mice were divided randomly into two groups: ad libitum fed and fasting (24 h without food). We conducted all mouse experiments in accordance with the accepted standard of humane animal care and with the approval by relevant national (Ministry of Welfare) and local (Institutional Animal Care and Use Committee, Tor Vergata University) committees.
Determination of GSH
Intracellular GSH level was measured by HPLC as previously described (4).
Preparation of cell lysates and Western blot analysis
Cell lysates were obtained and Western blot analyses were performed as previously described (1).
RT-qPCR analysis
Total RNA was extracted using TRI reagent (Sigma-Aldrich, St. Louis, MO). Three micrograms of RNA was used for retrotranscription with M-MLV (Promega, Madison, WI). qPCR was performed in triplicates by using validated qPCR primers (BLAST), Ex TAq qPCR Premix (Lonza Sales, Basel, Switzerland), and the Real-Time PCR LightCycler II (Roche Diagnostics, Indianapolis, IN). mRNA levels were normalized to actin mRNA, and the relative mRNA levels were determined by using the 2−ΔΔCt method. The primer sequences are listed in Table 2.
NRF-1, nuclear respiratory factor-1; SOD2, manganese superoxide dismutase; NFE2L2, nuclear factor (erythroid-derived 2)-like2; TFAM, mitochondrial transcription factor A.
Oligo-pulldown assay
Oligo-pulldown assay was carried out as previously described (5) by using the p53RE at −1237 on the human PPARGC1A, or at −2317, −954, or −564 on mouse ppargc1a promoter, and ARE sequence on the human GCLC promoter (Table 3). Oligo-pulldown specificity was demonstrated with mutant oligonucleotides used as negative controls (data not shown).
Chromatin immunoprecipitation assay
ChIP was carried out according to the protocol of Im et al. (18) with some modifications. Briefly, after cross-linking of nuclei extracted from SH-SY5Y cells were fragmented by ultrasonication using 4×15 pulse (output 10%, duty 30%). Samples were precleared with preadsorbed salmon sperm Protein G agarose beads (1 h, 4°C), and overnight immunoprecipitation using anti-p53 or control IgG antibody was carried out. After de-cross-linking (1% sodium dodecyl sulfate at 65°C for 3 h), qPCR was used to quantify the promoter binding with 30 cycles total (95°C, 1 s; 60°C, 30 s; 72°C, 60 s). Results are expressed as fold enrichment with respect to IgG control. The primers used are reported in Table 1.
Luciferase-based transcription assay
The PPARGC1A promoter region spanning from +1 to −1600 bp was cloned into a pGL3 Basic Vector (Promega). Reporter gene construct was transfected in NCI-H1299 cells together with pcDNA3.1-p53 [p53(+)] or pcDNA3.1 empty vector (vector) as above reported. Luciferase reporter gene activities were determined by using Luciferase Assay System (Promega). Light intensity was measured using a microplate luminometer (Perkin Elmer, Milan, Italy). Light intensity values from cell cultures transfected with pGL3 Basic Vector were used to correct for background. Normalized luciferase activities were expressed as fold increase with respect to the values from control (BSO-untreated), which were set arbitrarily at 1.
Statistical analysis
The results are presented as means±SD. Statistical evaluation was conducted by ANOVA, followed by the post-Student-Newman-Keuls. Differences were considered to be significant at p<0.05.
Footnotes
Acknowledgments
We thank Dr. Marcello Giorgi (Dept. of Biology, University of Rome Tor Vergata) for his technical assistance in performing gene reporter assay experiments. We are also grateful to Prof. Fabrizio Loreni (Dept. of Biology, University of Rome Tor Vergata) for providing intellectual support. This work was partially funded by grants from MIUR and Fondazione Roma (Research Grant 2008).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.