
Abstract
Introduction
Innovation
Our study is the first to demonstrate that reactive oxygen species (ROS), which is robustly generated in primary melanoma, has the ability to influence behaviors of tumorassociated macrophages (TAMs). In the melanoma microenvironment, tumor necrosis factor (TNF)-α secretion of TAMs is greatly enhanced by ROS, thereby increasing tumor invasion and even metastasis. Furthermore, this enhancementof TNF-α secretion is proved to be contributed by ROS-increased peroxisome proliferator-activated receptor γ translocation through the MAPK/ERK kinase 1-mediated pathway. These findings accomplish a large step toward defining the role of ROS in the modulation of immune cells in malignant melanoma, and also suggest several approaches for modifying antimelanoma therapies.
The progression and pathogenesis of melanoma strongly depends on the interactions between cancer cells and the various stromal cells in the host (31, 70). Among them, tumor-associated macrophages (TAMs), which are majorly derived from circulating monocytes and are acknowledged as the most abundant leukocytes in melanoma lesions (constituting up to 30% of tumor cells) (24, 56). Although some reports describe their antitumor traits as M1 macrophages (classically activated macrophages) (34), they have been increasingly characterized as M2 macrophages (alternatively activated macrophages) within many kinds of tumors, closely associated with tumorigenesis, tumor development, and tumor metastasis (43, 51). Accumulating evidence has indicated that increased figure of TAMs infiltrating melanoma and high level of macrophage markers expression in melanoma tissue are strongly related with poor prognosis (57, 26). One crucial property of TAMs is their ability to enhance the aggressiveness of cancer cells (41). It has been reported that the secretion of diverse factors such as the epidermal growth factor (19), urokinase plasminogen activator (uPA) (22), and matrix metalloproteinases (MMPs) (33) by TAMs can accelerate tumor cell intravasation and therefore facilitate tumor metastasis. Recently, the tumor necrosis factor (TNF)-α, a wide-known inflammatory cytokine, has been indicated to be responsible for the proinvasiveness characteristic of TAMs. TAM-secreted TNF-α was found to facilitate the invasiveness of MCF-7 and SK-BR-3 breast cancer cells by increasing MMPs in the coculture medium (21). Furthermore, the invasive ability of T47D ductal carcinoma cells was enhanced by TNF-α produced by TAMs (69). In addition, TAM-generated TNF-α can activate the nuclear factor κB (NF-κB)-Snail pathway in different types of tumor cells, promoting their invasiveness (65).
It has been demonstrated that ROS play a substantial role in upregulating inflammatory cytokine secretion of macrophages in several pathological conditions (15, 42). Although the level of oxidative stress has been reported to decrease in TAMs in advanced colon carcinoma and thymoma (13, 14, 67), melanoma TAMs could be one of the potential regulating targets of abundant ROS within the melanoma microenvironment.
Peroxisome proliferator-activated receptor γ (PPARγ) is a multifunction nuclear receptor that has been proved to have anti-inflammation effects in various macrophage models, inhibiting the expression of many proinflammation genes, including the genes of IL-1, IL-6, TNF-α, IL-12 and inducible nitric oxide synthase (iNOS) (2, 3, 45). Until now, several PPARγ regulating processes, which have influence on its activity or expression have been proposed, including ligand-induced activation, proteasomal degradation of activated PPARγ, MAPK-mediated phosphorylation, and newly found subcellular compartmentalization possibly due to MAPK/ERK kinase 1 (MEK-1) binding. Moreover, PPARγ has been also reported to be regulated by oxidative stress (40), providing us another potential target in interrogating the molecular mechanisms in melanoma TAMs.
Considering ROS has been employed as one of the therapeutic targets in increasing number of clinical studies, there is a necessity to thoroughly characterize the role of ROS in the influence of melanoma TAM behaviors. Herein, we are the first to underscore a critical role of oxidative stress in the regulation of TAM-promoted invasion in malignant melanoma. Furthermore, we have also elucidated the relevant molecular mechanisms involving ROS-enhanced TNF-α secretion resulted from PPARγ translocation.
Results
High level of ROS exists in malignant melanoma TAMs
We established subcutaneous melanoma models using B16F1 (a low metastatic variant) and B16F10 (a high metastatic variant) cells in C57BL/6 mice. First, the presence and location of TAMs were examined by H&E staining and immunofluorescence. As shown in Supplementary Figure S1A (Supplementary Data are available online at
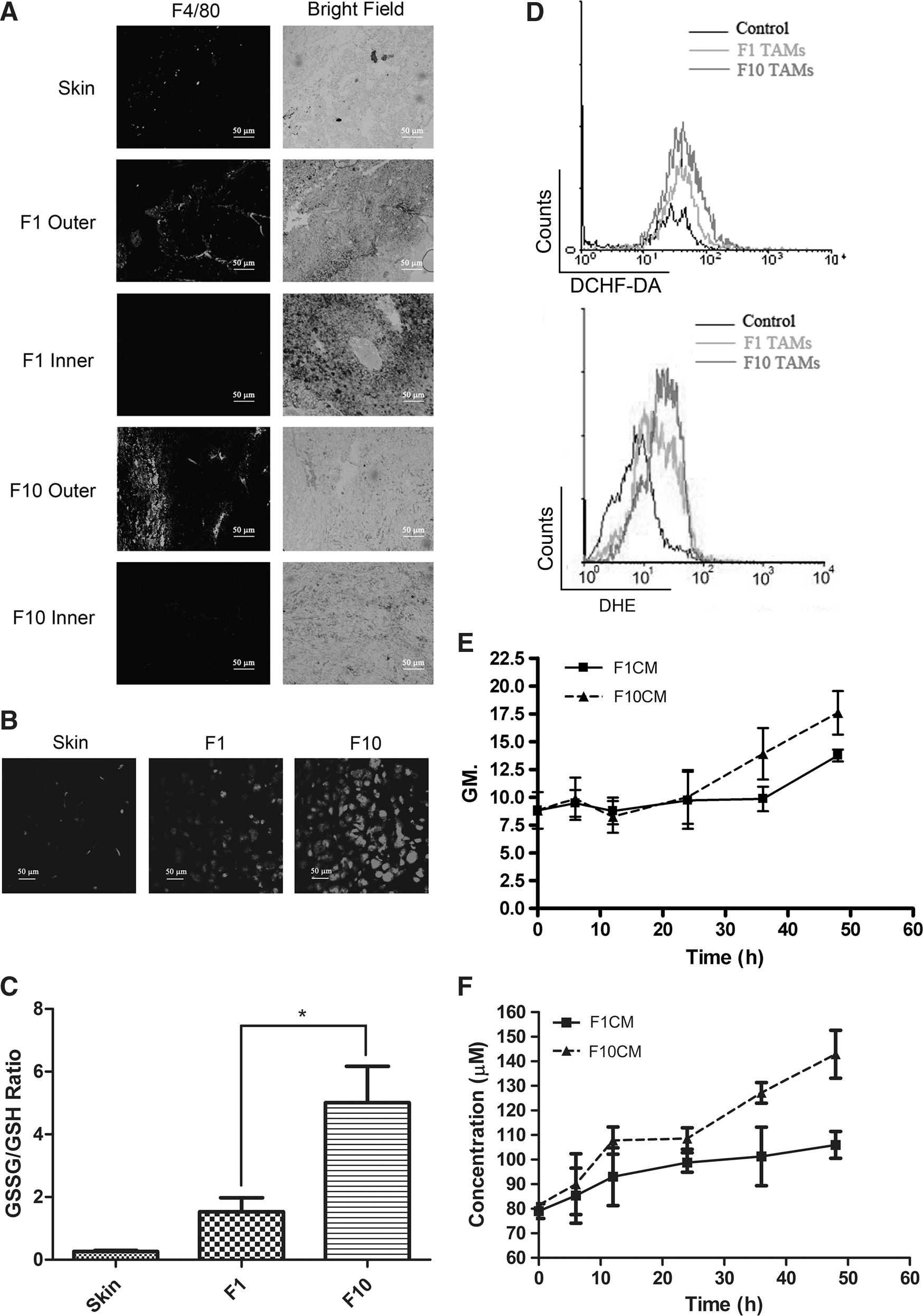
TAMs of malignant melanoma exhibit enhanced antioxidant system, heterogeneous phenotype, and a proaggressiveness signature
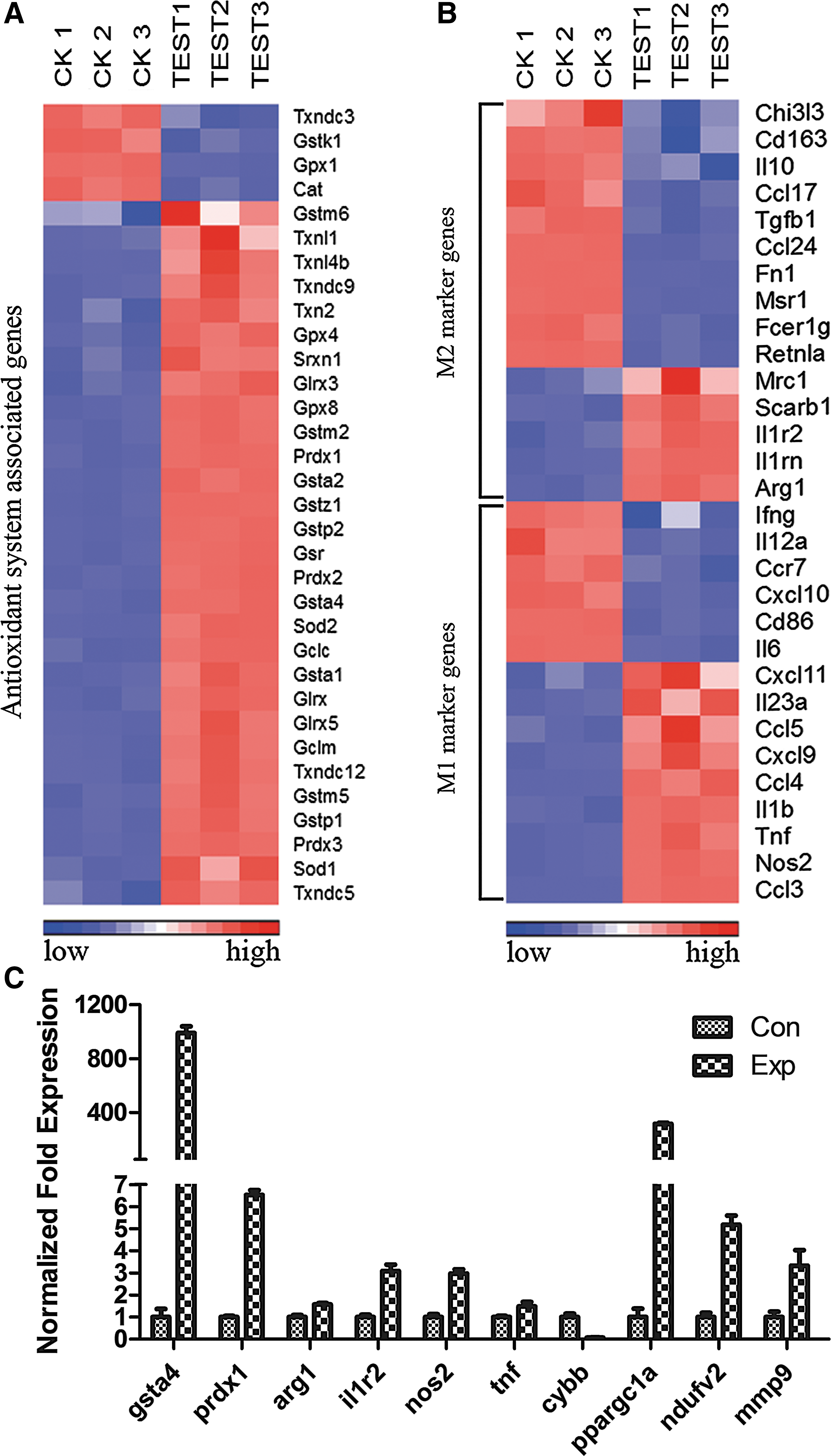
To further explore the redox status and functional phenotype of malignant melanoma TAMs, we performed cDNA microarrays to interrogate the gene expression pattern of B16F10 TAMs by comparing them with normal peritoneal macrophages. A total of 35556 genes were analyzed, among them 621 genes altered by more than fourfold, with 381 upregulated and 240 downregulated (Supplementary Table. S1). To gain deeper insight into the redox condition of melanoma TAMs, we utilized clustering analysis. As seen in Figure 2A, among 33 obviously altered genes (fold-change <−1.5 or > 1.5, p-value <0.05), which are closely related to antioxidant systems, only four of them were downregulated, while others exhibited upregulation, further confirming the results from Figure 1. We continued this study with interrogating the generating mechanism of macrophage intracellular ROS. The NADPH oxidase 2 (NOX2) complex is widely known for massive ROS production during respiratory burst when macrophages confront pathogens (6). Surprisingly, our gene profiling data revealed that all the members in NOX2 underwent apparent downregulation (Table. 1). However, the nuclear cofactor PPARγ coactivator 1-α (PGC1-α) and its downstreaming product transcription factor A, mitochondrial (TFAM), which are inseparately linked to mitochondria biosynthesis (58) were hugely elevated, so as to NADH dehydrogenase (ubiquinone) flavoprotein 2 (NDUFV2) on complex I and the Rieske Fe-S Protein (complex III), which are two main ROS-producing points on the mitochondrial respiratory chain complex (29, 38), indicating mitochondrial ROS generation is one of the main sources of TAMs intracellular oxidative stress. Next, we set out to examine the plasticity of melanoma TAMs. Thirty most renowned and acknowledged M1 and M2 activation markers (15 each, p value < 0.05) (7, 32) were analyzed according to our gene profiling data, clustering figure Figure 2B reveals that 9 m1 genes and 5 m2 genes were increased, while others display diminution of their expressions, suggesting these TAMs had an intermediate phenotype that does not fit the characteristic of either M1 macrophages or M2 macrophages. This conclusion was verified by the measurements of CD206 expression, the secreted TNF-α concentration, the iNOS activity, and the arginase-1 (Arg-1) activity in RAW264.7 TCMEMs (Supplementary Fig. S2). On top of these analyses, pathway analysis was also introduced based on strongly upregulated genes (fold-change >4). Twenty-one pathways were found statistically significant under a p value 0.05 (Supplementary Table. S2). Among them, seven pathways are linked to the metabolism process, showing a much greater energy expenditure of TAMs than peritoneal control. Other signaling pathways such as the toll-like receptor signaling pathway, cell adhesion molecules, focal adhesion, and leukocyte transendothelial migration depict a signature of inflammatory activation and a pro cell migration activity, indicating roles of these cells in tumor invasion and metastasis. Consistent with this finding, several protease genes that contribute tumor invasiveness such as mmp2, mmp9, mmp12, spp1 (osteopontin), ctsl (cathepsin L), and plau (urokinase) were also significantly elevated (Table 1). In general, the data of microarray study indicated that malignant melanoma TAMs, which are under dramatic oxidative stress exhibit a phenotype not fitting the orthodox M1/M2 paradigm and possibly possess a proagressiveness signature.
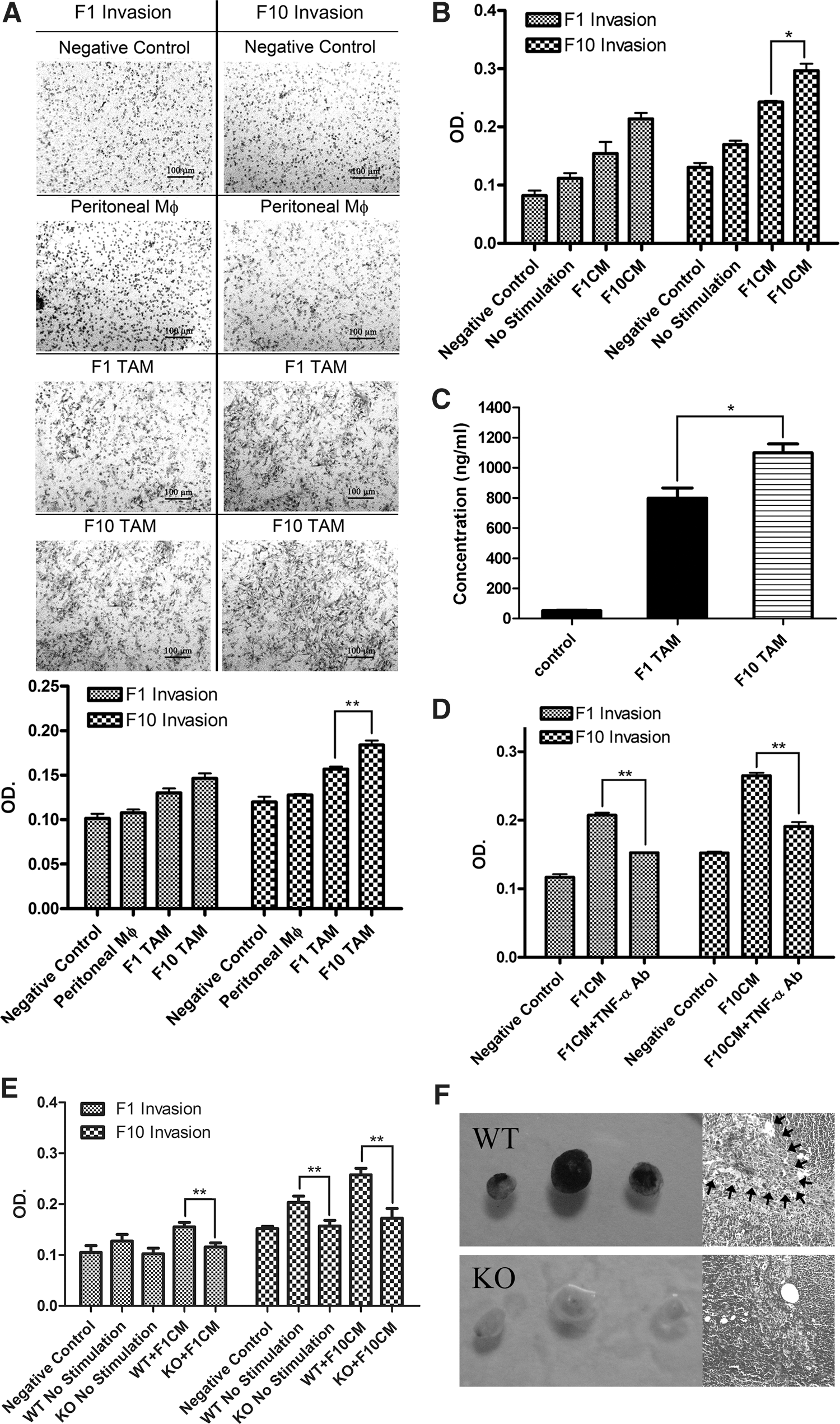
TAMs of malignant melanoma significantly enhance tumor cell invasiveness by TNF-α secretion
Our histology and microarray data have already suggested a vital role of TAMs in promoting tumor cell invasion and metastasis in malignant melanoma. To address this conjecture, we sorted TAMs from both B16F1 and B16F10 subcutaneous melanomas and subjected them to an in vitro assay that was widely utilized previously. As shown in Figure 3A, TAMs from both B16F1 and B16F10 tumors possess more capacity to enhance the tumor cell invasion compared with peritoneal macrophage controls. Interestingly, TAMs isolated from B16F10 tumors gave the greatest rise to tumor cell invasion, no matter cocultured with B16F1 tumor cells or B16F10 cells. Tumor migration ability is also vital in the metastasis process, we measured the promigration capacity of TAMs from two types of melanomas using the wound-healing assay, yielding quite similar enhancing patterns with an invasion test (Supplementary Fig. S3A). We also analyzed the proinvasion capacity of RAW264.7 and ANA-1 TCMEMs with the invasion assay. Likewise, macrophages that were incubated with the B16F10 tumor-conditioned medium (TCM) seemed to have the greatest ability to enhance tumor cell invasion (Fig. 3B and Supplementary Fig. S3B). Next, we set out to elucidate the molecular mechanisms underlying the proinvasion signature of melanoma TAMs. Recently, the inflammatory cytokine TNF-α has been demonstrated to induce tumor invasiveness in several tumor types. Therefore, we hypothesized that it could play the same role in malignant melanoma cells. Our microarray analysis already found more than threefold increase of tnf gene trancription in melanoma TAMs compared to peritoneal controls; this was confirmed with the measurement of TAM-secreted TNF-α by detecting its concentration in the postisolation culture medium with the enzyme-linked immunosorbent assay (ELISA) test. Resembling F1CM and F10CM RAW 264.7 TCMEMs, B16F10 TAMs also produced much more TNF-α than their B16F1 counterpart (Supplementary Fig. S2A, Fig. 3C). To verify whether TNF-α is capable to enhance tumor aggressiveness, we performed an invasion assay with only B16F1 or B16F10 melanoma cells in the presence of exogenous recombinant TNF-α. The results showed that TNF-α can dramatically give rise to their invasiveness in a concentration-dependent manner (Supplementary Fig. S3C). Consistently, the neutralization of TNF-α in a cell invasion assay with an anti-TNF-α antibody significantly decreased TCMEM-promoted tumor invasion (Fig. 3D). To further substantiate the role of TNF-α in the proinvasion trait of melanoma TAMs, we brought in TNF-α knockout mice. Notably, as seen in Figure 3E, the absence of TNF-α nearly completely depleted peritoneal macrophage TCMEM-induced tumor invasiveness compared with wild-type (WT) control. We also observed the development of in vivo metastasis in knock-out (KO) mice. Although there were negligible differences of lung metastasis and inguinal lymphatic gland metastasis on day 21 after foot-pad plantation between KO mice and WT counterpart (Supplementary Fig. S3D, E), much more severe metastasis could be seen in popliteal lymph nodes of TNF-α KO mice (Fig. 3F), indicating a role of TNF-α in promoting lymphatic metastasis of malignant melanoma. Taken together, these data confirm that TNF-α secretion, to a great extent, was responsible for proinvasion and prometastasis signatures of malignant melanoma TAMs.
High level of oxidative stress strengthens the invasion-promoting phenotype of malignant melanoma TAMs
We hypothesized that the large amount of ROS found in malignant melanoma may critically affect the proinvasion signature of TAMs. Therefore, antioxidants N-acetylcysteine (NAC), Vitamin E (Ve), or pro-oxidant H2O2 were introduced in the 48-h education process of TCMEMs to elevate or reduce intracellular oxidative stress. As observed in Figure 4A and Supplementary Figure S4, the intracellular ROS level of TCMEMs was strongly affected by these antioxidants and oxidants. Next, these TCMEMs were subjected to in vitro invasion assay. The result reveals that the proinvasion capacity of TCMEMs whose education process was accompanied with a low ROS level was apparently attenuated. However, the addition of H2O2 in the TCMEM education process could considerably enhance the proinvasion signature of these cells (Fig. 4B). Moreover, the result from the TNF-α ELISA assay seems compatible with the data of invasion assay (Fig. 4C). Likewise, the diminution of intracellular ROS during the education process obviously decreased the promigration capacity of TCMEMs in wound-healing assay (Fig. 4D). Hence, the high level of intracellular ROS in melanoma TAMs is likely to facilitate TAM-enhanced tumor invasiveness by increasing TNF-α secretion.

PPARγ functioned against proinvasiveness signature of malignant melanoma TAMs by downregulating TNF-α production
It has been demonstrated that PPARγ plays a considerable role in regulating the secretion of various inflammatory cytokines in different kinds of macrophages. Hence, we presumed that it also has a similar function in melanoma TAMs. To address this hypothesis, we knocked down PPARγ in RAW264.7 macrophages using small interference RNAs (siRNAs) and performed tumor cell invasion assay in RAW264.7 TCMEMs. Efficient knockdown of the PPARγ protein was shown in Supplementary Figure S5. As we expected, a decrease of PPARγ in TCMEMs led to a significant increase in proinvasion capacity of these cells (Fig. 5A). Consistently, the TNF-α level in the culture medium of PPARγ knockdown TCMEMs was also significantly higher than scrambled control (Fig. 5B). The functions of PPARγ are strongly regulated by its binding ligands. Therefore, we introduced both potent selective agonist and antagonist of PPARγ (GW1929 and GW9662, respectively), in our study to further elucidate the role of this nuclear receptor. As shown in Figure 5C and D, incubation with GW1929 distinctly reduced TNF-α secretion by TCMEMs, resulting in a decreased ability to enhance tumor invasiveness. In contrast, the presence of GW9662 strongly gave rise to the TNF-α production level as well as proinvasion capacity of TCMEMs. To further explore the significance of TNF-α secretion decrease in the downregulation of TAM-enhanced tumor invasiveness by PPARγ, we stimulated TNF-α knockout peritoneal macrophages with melanoma TCMs with the presence of GW9662 or H2O2. Then, we subjected these TCMEMs to invasion assay. Remarkably, the enhancement of TAM-induced tumor invasion by GW9662 or H2O2 was totally abrogated by the loss of TNF-α (Fig. 4E). Hence, these data prove that PPARγ can downregulate proinvasion capacity of TAMs by attenuating their TNF-α secretion.

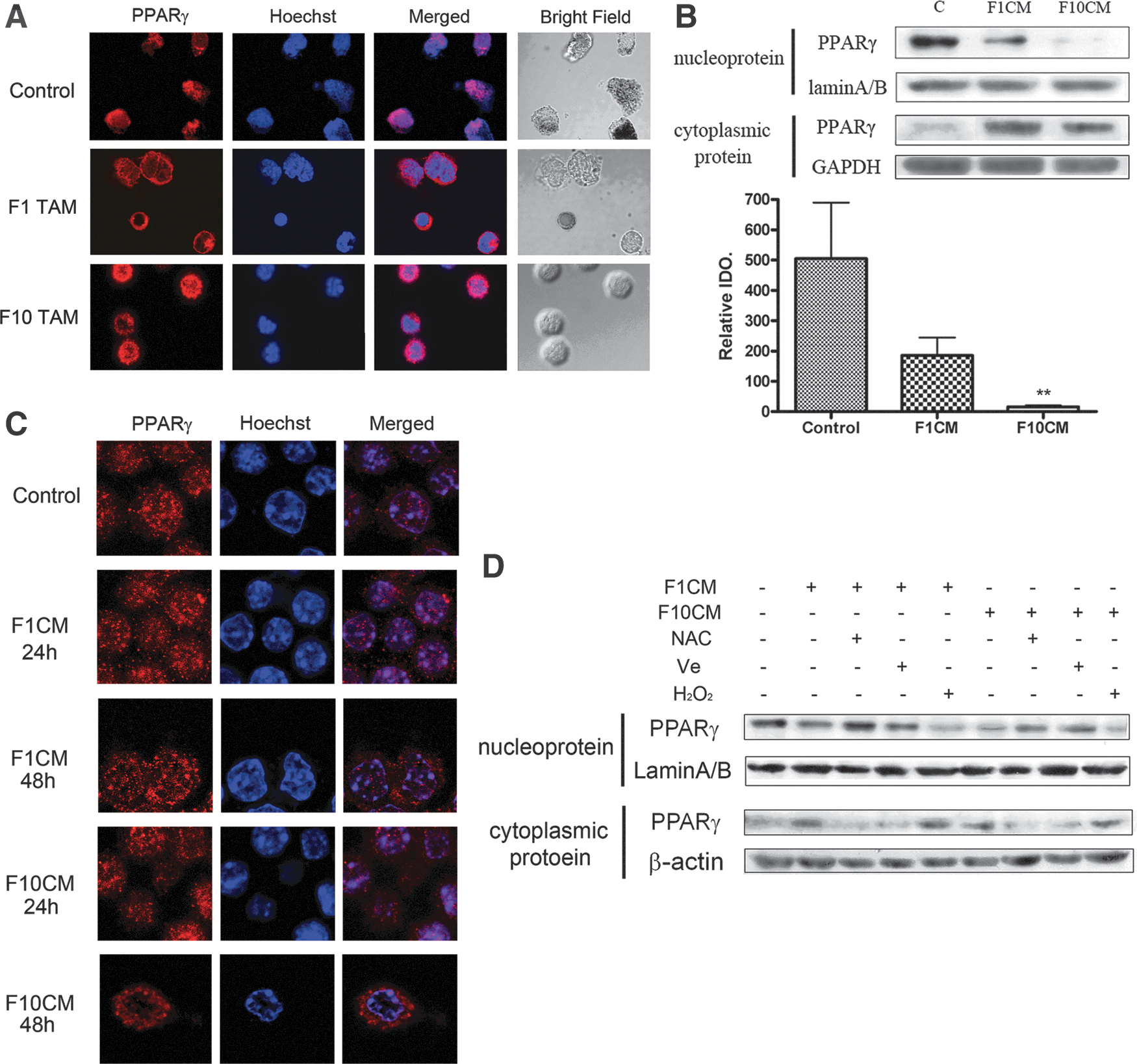
PPARγ translocates from the nucleus to the cytoplasm in TAMs of malignant melanoma
As TNF-α secretion was considerably high in melanoma TAMs, we hypothesized its expression level was downregulated in these cells. However, our data showed that there was no apparent decrease of PPARγ expression in RAW264.7 TCMEMs or even TCMEMs under higher oxidative stress (Supplementary Fig. S6A). Subcellular localization alteration has been implicated to be equally important for the regulation of PPARγ functions. Thus, we turned to examine the subcellular distribution of PPARγ in melanoma TAMs with immunofluorescent staining. Compared with peritoneal macrophages, there was an obvious decrease of PPARγ in the nucleus, while the PPARγ level dramatically rises in the cytoplasm (Fig. 6A). These data indicated a translocation of PPARγ during the activation of macrophages within the tumor microenvironment. We also confirmed this phenomenon in TCMEMs utilizing both Western blot assays and immunofluorescent staining. As seen in Figure 6B and Supplementary Figure S6B, nucleus-to-cytoplasm translocation occurred in both stimulated RAW264.7 and ANA-1 macrophages. Moreover, when examined every 24 h in RAW264.7 cells, a gradual subcellular distribution change of PPARγ was observed (Fig. 6C). To investigate whether the abundant intracellular ROS was linked with PPARγ translocation, we analyzed PPARγ subcellular distribution in TCMEMs under various oxidative states. As shown in Fig. 6D, the presence of antioxidant NAC or Ve could effectively block this translocation. On the contrary, the addition of H2O2 was capable to enhance the nucleus-to-cytoplasm shuttle of PPARγ. Taken together, these data indicate a vital role of intracellular oxidative stress in downregulating PPARγ function by promoting its translocation from the nucleus to the cytoplasm in melanoma TAMs.
Activated MEK-1 is responsible for PPARγ translocation in TAMs of malignant melanoma
To elucidate the mechanism underlying PPARγ translocation in malignant melanoma TAMs, we specifically analyzed MEK-1 that belongs to the MAP kinase family and has been shown to facilitate PPARγ nucleus-to-cytoplasm localization shift after its phosphorylation by directly binding to PPARγ (9). We began with the measurement of MEK-1 subcellular distribution in RAW264.7 TCMEMs. Similar to PPARγ, MEK-1 also showed an apparent translocation from the nucleus to the cytoplasm (Fig. 7A). Next, we examined the localization alteration over time of both PPARγ and MEK-1 to interrogate the linkage between these two proteins. The result displays two very similar changing patterns of the subcellular distribution of these proteins (Fig. S7A). We further examined their distributions in TCMEMs under different oxidative stresses. Again, two identical altering patterns were observed (Fig. 7B), indicating a close correlation between PPARγ and MEK-1 translocations in melanoma TAMs. Then, we continued this study with the MEK-1-specific inhibitor PD98059 to test whether the loss of MEK-1 activation would affect PPARγ translocation. This inhibitor exhibited great effectiveness to suppress MEK-1 phosphorylation without disturbing PPARγ expression compared with the control level (Supplementary Fig. S7B, C). As we expected, the nucleus-to-cytoplasm shifting of PPARγ in RAW264.7 TCMEMs was significantly blocked by the addition of PD98059 during TCM stimulation. Nevertheless, proinvasion capacity of TCMEMs whose 48-h education process was covered with PD98059 was just partially dampened compared with normal TCMEM counterparts (Fig. 7D), suggesting in melanoma TAMs there are other regulating mechanisms that suppress anti-inflammation function of PPARγ besides subcellular translocation. The phosphorylation of PPARγ on several sites like S112 by several MAP kinases (extracellular regulated protein kinases [ERK], p38, c-Jun N-terminal kinases …) can also lead to dysfunction of this nuclear receptor (10). Therefore, we examined the phosphorylation levels of PPARγ (S112) as well as ERK1/2(T202/T204), a downstreaming target of MEK-1, in the RAW264.7 TCMEM nucleus. As shown in Supplementary Figure S7D, the ratio of the phosphorylated protein level to the total protein level significantly increased compared with the control ratio, implicating that the phosphorylation of PPARγ could play a role in inhibiting PPARγ function in melanoma TAMs.

Discussion
Malignant melanoma is more deadly than many other types of cancers due to its high risk for metastasis, which results in a poor survival rate (the 5-year survival rate is under 10%) (5). Moreover, without proper treatment, melanoma cells can spread very rapidly to other organs, resulting in formidable difficulty in melanoma therapies (17, 37). Hence, thoroughly elucidating the mechanisms underlying the complicated metastatic process of malignant melanoma seems quite urgent. In the tumor microenvironment, residing stromal cells bidirectionally interact with tumor cells, generating a permissive/conducive environment that facilitates tumor cells to invade into blood vessels and lymphatic vessels. In this study, we focused on the interplay between TAMs and melanoma cells. Although there have been quite a number of studies depicting various aspects of melanoma TAMs based on plasticity, phenotype, and functions (24), few of them have been aware of the possible role of prevailing oxidative stress in the modulation of these cells, which could also drastically affect the malignancy of melanoma cells. Therefore, our study filled the gap between TAMs and the high level of ROS in the field of melanoma research.
ROS was once recognized as a strong weapon of the immune system to kill tumor cells (23). However, when malignant melanoma cells successfully escape the apoptosis triggered by abundant ROS (30), persistent ROS existence tends to favor melanoma survival, proliferation, and metastasis via activating or enhancing several related pathways (54, 25). We presumed the intracellular oxidative stress of melanoma stromal cells, such as macrophages, could also be affected. Hence, we not only verified the oxidative stress of B16F1 and B16F10 melanoma tissues, but also measured the ROS level of TAMs and TCMEMs, generating positive results (Fig. 1D, E). Consistently, our cDNA microarray analysis also confirmed a great enhancement of genes relevant to the antioxidant system in B16F10 TAMs, which can be explained as the response of macrophage cells intending to quench high oxidative stress (Fig. 2A). Moreover, the result of DHE staining (Fig. 1D) showed a great increase of intracellular superoxide anion in TAMs. Since superoxide anion has a high reactivity and considerably poor membrane permeability (55), this result provides the information that at least part of the intracellular ROS was endogenously generated. This conjecture was further verified by the elevation of intracellular oxidative stress of TCMEMs in the absence of ROS-excreting melanoma cells. The data from microarray and real-time polymerase chain reaction (PCR) verification rule out the possibility of deliberate cellular ROS production by the NOX2 complex or even other NOX enzymes (Table. 1, data not shown). Nevertheless, there is a dramatic increase of the gene level of PGC1-α, which has been reported as a master regulator of inductive mitochondria biogenesis, as well as an important mitochondrial transcriptional factor TFAM, implicating that ROS production from increased number of mitochondria may contribute to intracellular oxidative stress. This assumption is further validated by the gene elevation of NDUFV2 (Fig. 2C), the ROS-generating position of complex one, as well as the considerable increase of metabolic signaling pathways (Supplementary Table. S2). In addition, the mitochondrial ROS generation is also likely to be enhanced by various stresses prevailing in the melanoma microenvironment, such as hypoxia and pro-oxidants. However, this hypothesis still needs our future investigation.
The M1/M2 activation model of macrophages has been widely recognized during the last decade. However, the increasing number of recent studies suggested that this model probably does not entirely describe the complexity and heterogeneity of TAMs. In our study, we found that several genes of both M1 and M2 markers were upregulated in melanoma TAMs. Furthermore, the activities of Arg-1 and iNOS, the expression of CD206 and the secretion of TNF-α were also elevated together in melanoma TCMEMs. These data and the observations of other groups (61, 18a) demonstrated that melanoma TAMs possess a heterogeneous phenotype that does not belong to either M1 or M2 polarized macrophages. Hence, functional studies of TAMs seem to have a larger significance than simply interrogating their plasticity. Our analysis of signaling pathways based on microarray data proposes a proaggressiveness signature of macrophages (Supplementary Table. S2), which is verified by the raise of gene expressions of MMP2, MMP9, MMP12, osteopontin, cathepsin L, and urokinase (Table. 1). This proaggressiveness property was confirmed by both the in vitro invasion test and wound-healing assay.
The experiments conducted by us proposed a vital role of one particular inflammatory cytokine, TNF-α, in TAM-induced tumor invasiveness (Fig. 3). TNF-α was used to be discussed as a potent role tumor killer substantially produced by M1 macrophages to help the immune system inhibit tumor growth and progression (8, 53). Nevertheless, reports in recent years have shown that TNF-α also plays a critical role in tumorigenesis (49), tumor progression (4), and especially tumor invasiveness (66). Existing evidence proposed a relatively upstream position of TNF-α initiating the formation of a permissive/conducive microenvironment for tumor invasion. Kim and his colleagues found TNF-α induce expression of uPA and β-catenin activation in human breast epithelial cells (28). Hagemann et al. proved that coculturing TAMs with tumor cells can enhance the expression of MMPs, especially MMP2 and MMP9, in a TNF-α-dependent manner (21). Wu et al. concluded that TNF-α induces protein stabilization of Snail and b-catenin by inhibiting GSK-3b-mediated phosphorylation through NF-κB and Akt signaling pathways, which contribute to epithelial-mesenchymal transition (EMT) induction and invasion in tumor cells (65). Our experiments underscore the importance of the upstream position of TNF-α by exhibiting nearly complete loss of proinvasion capacity of TCMEMs derived from peritoneal macrophages of TNF-α−/− mice. Moreover, we found the lack of TNF-α can abolish melanoma metastasis to the popliteal lymph node, even though tumor metastasis is a much more complex process involving multiple steps.
The anti-inflammatory effect of the nuclear receptor PPAR-γ has been observed in several human and murine monocyte/macrophage models (45). Our data further corroborated the universality of this effect of PPARγ in TAMs, showing a dramatic increase of TNF-α production after PPARγ silencing or deactivation (Fig. 5B, D). The inflammation-suppressing activity of PPARγ was proved to strongly depend on the regulations of its activation, such as modulation by natural binding ligands. Recently, there has been an increased awareness of the importance of PPARγ modulation by oxidative stress. It has been reviewed that ROS could elicit several cross talks between PPARγ and NRF2, FOXO as well as Wnt/β-catenin signaling pathways, resulting in expression and activation changes of PPARγ itself (40). Our study shed some light on ROS-mediated PPARγ regulation by demonstrating oxidative stress enhanced PPARγ nucleus export in melanoma TAMs. Subcellular compartmentalization of PPARγ has been assumed as a major mechanism modulating protein functions in various kinds of cells. However, the real effects of the alteration of PPARγ subcellular distribution in macrophages still need further investigation. Several distinct intranuclear mechanisms have been reported to contribute to the suppression of inflammatory genes by PPARγ, including transrepressing NF-κB (47), inhibiting the clearance of NCoR corepressor complexes (39) and upregulating IL-1 receptor antagonist (36). Although one study implicated that cytoplasm PPAR-γ may desensitize activated macrophages by attenuating cytosol-to-membrane translocation of protein kinase Cα (PKCα) via direct binding (59), the preponderance of evidence has revealed that nuclear localization is crucial for PPAR-γ interacting with other nuclear factors and transcriptional elements, thus exerting its anti-inflammatory function in macrophages. Hence, we suggest that the nucleus-to-cytoplasm translocation downregulates the inflammatory repression conducted by PPAR-γ in macrophages. This conjecture is strongly supported by our data as howsoever the intracelluar oxidative stress of TCMEMs changed, TNF-α secretion was always negatively correlated with the nucleus expression level of PPAR-γ (Fig. 4A, C; Fig. 7B). One model proposed by Burgermeister et al. to elucidate the mechanisms underlying nucleus export of PPAR-γ focuses on its direct binding with activated MEK-1 (11). Our observations not only corroborated this concept, but also granted the ROS-enhanced MEK-1/PPAR-γ translocation with a pathological significance for the first time.
The almost complete abolishment of PPAR-γ translocation by the MEK-1 inhibitor results in partly repressed proinvasion capacity of TCMEMs, indicating the existence of other molecular events that downregulate the PPAR-γ activity. PPAR-γ phosphorylation has been reported as another process that affects its functions (12). Among various phosphorylation sites, the phosphorylation of S112 (PPARγ2) was widely believed as a potent way to deactivate this nuclear receptor. Our data indicated this S112 phosphorylation may be also included in PPARγ modulation mediated by intracellular ROS in melanoma TAMs. Interestingly, a recent study conducted by Knethen et al. showed that CK-II mediated S16 and S21 phosphorylation of PPARγ could be critical in the shuttling of PPARγ from the nucleus to the cytosol, linking PPARγ phosphorylating status with its subcellular distribution (60). Hence, the phosphorylation of different sites on PPARγ could have various regulating effects. The studies to find and characterize new ROS-regulated phosphorylation sites and the linkage between different regulating mechanisms deserve further attention.
As noted earlier, we demonstrated a remarkable regulation mediated by intracellular ROS in melanoma TAMs. The intracellular oxidative stress promote nucleus-to-cytoplasm translocation of PPAR-γ mediated by MEK-1, resulting in the increase of TNF-α secretion, thus enhance the invasiveness and metastasis of melanoma cells. These molecular mechanisms mediated by ROS are well illustrated in Fig. 8. During the last decade, increasing attention of therapists has been attracted in the development of new therapies against melanoma using ROS as a therapeutic target. Due to the dual role of ROS, both pro-oxidant- and antioxidant-based anticancer approaches have been employed. Although some reports implicated ROS scavenging showed some effects in preventing melanoma growth, much more efforts have been spent on chemotherapy and radiotherapy that elevate ROS generation or inhibit the antioxidant system to directly kill melanoma cells (20, 18). However, nearly all of these studies neglect the critical role of fine-tuned complex interactions between stromal cells and tumor cells. Our study pointed out that inflammatory cytokine TNF-α secreted by TAMs, which is enhanced by ROS can be critical in melanoma lymphatic metastasis (Fig. 3F). Accordingly, how to specifically reduce the oxidative stress in TAMs or how to boost the melanoma ROS level without evoking inflammatory responses in TAMs should be considered in the future modification of melanoma therapies.

Materials and Methods
Reagents and antibodies
RPMI 1640 and the Dulbecco's modified Eagle's medium (DMEM) were purchased from Gibco. All of the chemicals were purchased from Sigma, unless otherwise stated. Anti-mouse PE-CD206 (M1) was purchased from BD Pharmingen. Anti-mouse PE-F4/80 was purchased from eBiosience. Anti-mouse PPARγ (81B8), anti-mouse MEK-1 (30C8), anti-mouse p-MEK-1 (S298), anti-mouse ERK (137F5), anti-mouse p-ERK1/2 (T202/T204), and mentioned loading controls were purchased from Cell Signalling. p-PPARg (S112) was provided by Abcam.
Mice and treatment
Male C57BL/6 mice were purchased from the Model Animal Research Center of the Nanjing University, Nanjing, China and bred in our animal facilities under specific pathogen-free conditions. After a 5-day quarantine, the mice were randomized into groups. Melanoma tumors were induced by subcutaneous injection of 105 B16F1 or B16F10 murine melanoma cells. The control groups were treated with the solvent only.
Cell culture and culture supernatant treatment
B16F1 cells, B16F10 cells (Nanjing Keygen Biotech. Co., Ltd.), RAW264.7 cells (China Center for Type Culture Collection), and ANA-1 cells (Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences), were cultured in the DMEM and RPMI 1640 medium, respectively, supplemented with 10% fetal bovine serum (Gibco), 2 mM
Histology and immunofluorescence assays
Formalin-fixed paraffin-embedded samples were sectioned and stained with H&E for tumor histology analysis. The sections were blocked for 1 h with 3% bovine serum albumin (BSA) and 0.05% Tween 20 (diluted in 50 mM Tris-HCl pH 7.6 containing 150 mM NaCl). The sections were then probed with antibodies against the anti-mouse F4/80 antigen (eBioscience) followed by anti-rat IgG fluorescein isothiocyanate.
The frozen sections of TAMs were stained with DHE purchased from Beyotime. Then, they were observed at the appropriate fluorescence wavelength using a confocal microscope.
The cells were spread on a coverslip treated with polylysine and were fixed with 4% paraformaldehyde for 10 min. The cells were perforated using 0.5% Triton X-100 for 15 min. After being blocked in phosphate-buffered saline (PBS) containing 0.1% Tween-20 with 5% BSA at room temperature for 30 min, the cells were incubated with a primary antibody overnight at 4°C. The cells were then incubated with a secondary antibody for 1 h at room temperature. The cells were then observed at the appropriate fluorescence wavelength using a confocal microscope.
Flow cytometry analysis
The DCFH-DA was purchased from Beyotime (Haimen). The saponin (0.05%) (Sigma-Aldrich) was diluted in PBS and used as a membrane permeator for inner membrane staining. The cells were analyzed on a fluorescence-activated cell sorting (FACS) Calibur cytometer using Cellquest software (Becton Dickinson). The statistics presented are based on 10,000 events gated on the population of interest.
Cytokine assays by specific ELISA
The TNF-α concentration in 150 μl supernatants of 10,000 TAMs or 2 ml supernatants of 1 million TCMEMs was assessed using a standard sandwich ELISA according to the instruction manual. The TNF-α concentrations were expressed in nanograms per milliliter, as calculated from the calibration curves from serial dilutions of murine recombinant standards (eBioscience) in each assay. The sensitivity of the TNF-α assay was 20 pg/ml.
Western blotting
The cell protein was extracted by whole cell lysis or a nuclear protein extract kit purchased from Beytotime, which contained protease and phosphatase inhibitors. The cell debris was removed by centrifugation at 4°C, and the supernatants were collected and stored at −70°C until use. The protein amounts were determined using the bicinchoninic acid protein assay (Pierce), and a dot blot analysis was performed as described previously (11).
Cell invasion assay
The extensively utilized invasion assay in this study is well illustrated in Figure 9 and described in figure legend.
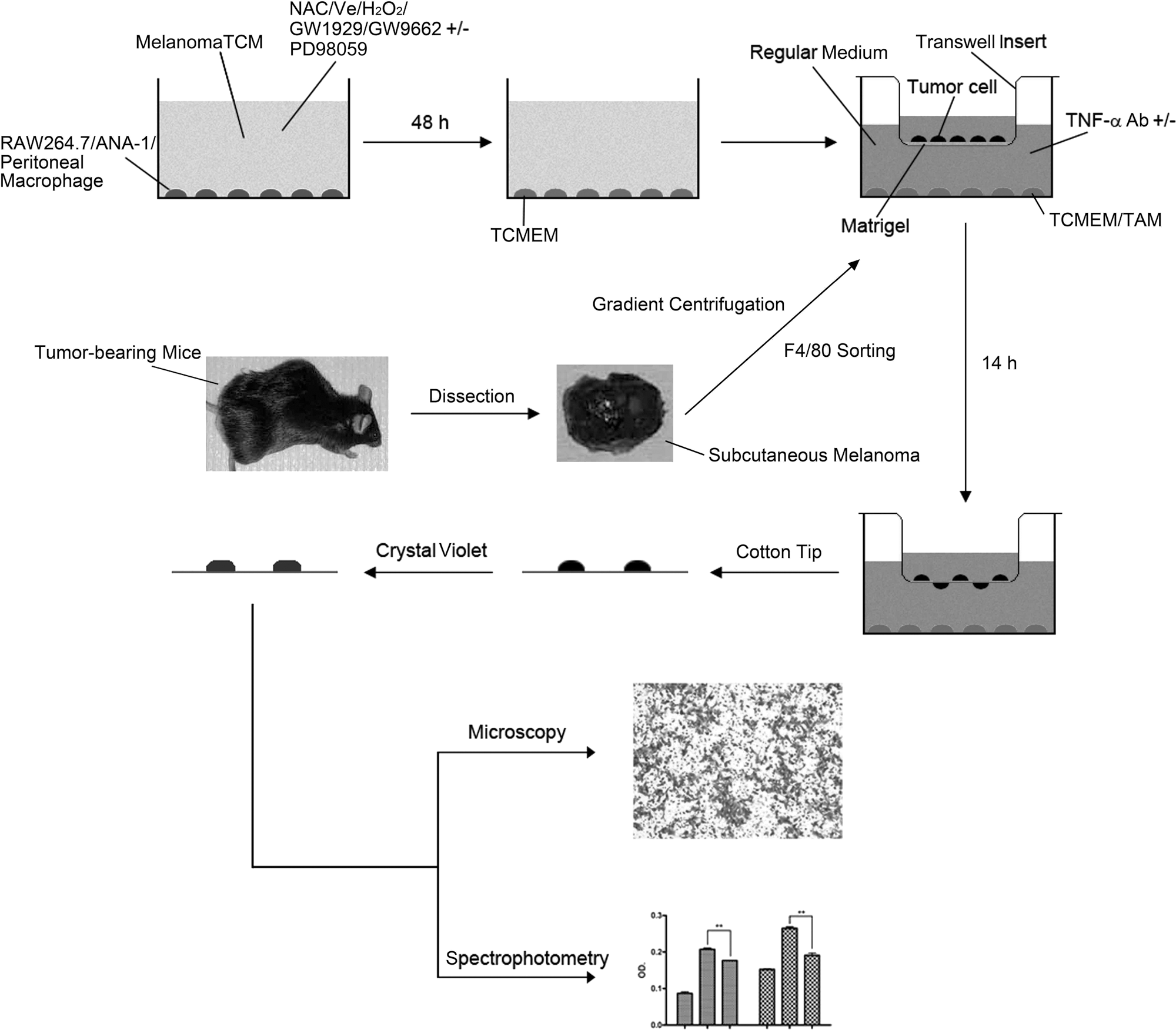
Wound-healing assay
B16F1 or B16F10 cells were cultured in 96-well plates at 40,000/well as confluent monolayers. The monolayers were incubated in the absence of serum for 16 h and wounded in a line across the well with a 10-μl standard pipette tip. The wounded monolayers were then washed twice with serum-free media to remove cell debris and cocultured with 10,000 macrophages in Transwell chambers (0.4-μm pore size) (Costar) in the medium with 10% fetal calf serum (FCS). The microscopic images of the same location of a cell-free wound were recorded at 0 h and 24 h.
Microarray analysis
Total RNA of B16F10 TAMs was prepared from three independent experiments. Microarray experiments were performed by Gene Tech (Shanghai, China) with 20 μg of total RNA, which was labeled with fluorescent nucleotides and hybridized to murine cDNA slides. Hybridized slides were interrogated via an Agilent scanner, data were subjected to background subtraction and normalization, and analyzed with statistical analysis of microarrays software package.
Quantitative real-time PCR verification
The RT reaction was performed on 150 ng of total RNA with avian myeloblastosis virus reverse transcriptase (Promega). Quantitative real-time PCR was performed using QuantiTect SYBR Green I (Qiagen). PCRs were performed in a total volume of 20 μl (1×QuantiTect SYBR Green Master Mix) in the ABI Prism 7000 system (Applied Biosystems). The absolute number of copies of the gene of interest in the experimental cDNA samples was calculated from the linear regression of a standard curve. The expression of the measured genes in each sample was normalized for actin expression.
The details of primers are listed as follows:
arg1:+5′ GAATCTGCATGGGCAACCT 3′ − 5′ CAGGGTCTACGTCTCGCAAG 3′
gsta4:+5′ GATTGCCGTGGCTCCATTTA 3′ − 5′ CAACGAGAAAAGCCTCTCCGT 3′
il1r2:+5′ AAGGATGTGGGTGAAGGGTAAC 3′ − 5′ CTCACAGTGGGATGCGTTTC 3′
nos2:+5′ TGGAGCGAGTTGTGGATTGTC 3′ − 5′ GCCTCTTGTCTTTGACCCAGTAG 3′
mmp9:+5′ CGAAGCGGACATTGTCATCC 3′ − 5′ GTCGTCGAAATGGGCATCTC 3′
ndufv2:+5′ ACAACAGGACCACTATCTCGGTC 3′ − 5′ GGCTCTATGAATGGGCAACAC 3′
cybb:+5′ TGAATGCCAGAGTCGGGATTT 3′ − 5′ CGAGTCACGGCCACATACA 3′
ppargc1a:+5′ CAACAATGAGCCTGCGAACA 3′ − 5′ CATCAAATGAGGGCAATCCG 3′
prdx1:+5′ TGTCCCACGGAGATCATTGC 3′ − 5′ CACAGAAGCGCCAATCACTT 3′
tnf:+5′ CTGTGAAGGGAATGGGTGTTC 3′ − 5′ CAGGTCACTGTCCCAGCATC 3′
siRNA transfection
The RAW264.7 cells (105 cells in 300 μl) were mixed with 100 μl of the electroporation buffer in a cuvette, which contained siRNA oligonucleotides (Stealth siRNA duplex oligoribonucleotides, Invitrogen). The cuvette was immediately transferred to the electroporator and the device was turned on. After 1–2 min, the cuvette was placed on ice, and then cells were plated in six-well plates. The treatments were performed 24 h after the siRNA transfection. The sequences of the PPARγ siRNA1 are 5′-ACAGGCCUCAUGAAGAAUUTT-3′ and 5′-AAUUCUUCAUGAGGCCUGUUG-3′. The sequences of the PPARγ siRNA2 are 5′-UGGAAGACCACUCCCACUCTT-3′ and 5′-GAGUGGGAGUGGUCUUCCATT-3′. For a control, we used a scrambled siRNA, whose sequences are 5′-UUCUCCGAACGUGUCACGUTT-3′ and 5′-ACGUGACACGUUCGGAGAATT-3′.
Primary TAM flow sorting
A mouse subcutaneous melanoma model was established for 3 weeks. The tumors were minced in the DMEM with 0.1% collagenase I (1 h, 37°C), passed through a 19G needle, and filtered using a 40-μm cell strainer (BD Falcon). The cells were centrifuged through density gradient centrifugation and washed with PBS. The cells were then incubated with 10% FCS and an anti-F4/80 antibody for 30 min. The tumor cells were sorted as F4/80−cells. The TAMs were sorted as F4/80+ cells.
Measurement of the intracellular GSSG/GSH
The intracellular GSH content in the melanoma tissue was determined using a GSH and GSSG assay kit purchased from Beyotime (Haimen) according to the manufacturer's instructions.
Statistical analysis
The data are expressed as the mean±standard deviation. The statistical analysis was performed by the Student's t-test when only two value sets were compared. A one-way ANOVA followed by a Dunnett's test were used when the data involved three or more groups. p<0.05, p<0.01, or p<0.001 were considered statistically significant and indicated by *, **, or ***, respectively.
Author Contribution
The project and researchers were funded and supervised by Prof. Pingping Shen. Xuzhu Lin was chiefly responsible for experiment designing and manuscript drafting. Wei Zheng and Jing Lin generated the majority of the data in this manuscript. The rest of the work mentioned in this study was contributed by other authors.
Footnotes
Acknowledgments
We thank Adebusola Alagbala Ajibade (Baylor College of Medicine, U.S.A.) and Jun Cui (the Nanjing University, China) for critical comments and grammatical correction of the manuscript. We also thank Dr. Ning Su (professor, School of Basic Medical Sciences, Southeast University, China) for excellent technical assistance.
This work was supported by the National Natural Science Foundation of China (project No. 91013015, 81273527, 31070764), and the Science Fund for Creative Research Groups (No. 81121062).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.