
Abstract
Introduction
Life Cycle of T. cruzi and Progression of CD
The life cycle of T. cruzi involves the invertebrate host (triatomid hematophage arthropod), where the noninfective epimastigotes replicate and differentiate to the nonreplicative infective stage (metacyclcic trypomastigotes) at the rectum of the insect vector. During the differentiation process from noninfective epimastigotes to infective metacyclic trypomastigotes (a process called metacylocgenesis), the parasite undergoes complex morphological and biochemical changes in order to effectively infect and survive in the hostile environment of the vertebrate host. Metacyclic trypomastigotes should gain access to the internal medium of the vertebrate host via the mucosal and/or skin wounds invading different cell types, including macrophages, cardiomyocytes, fibroblasts, and smooth and striated muscle cells (4). The acute phase of the disease lasts between 4–8 weeks, generally with mild flu-like self-limiting symptoms, although 10% of symptomatic cases develop severe myocarditis and/or meningoencephalitis. Organ and tissue damage during acute T. cruzi infection is caused by the parasite itself and by the host's acute inflammatory response, elicited by the presence of the pathogen. Between 30% and 40% of patients will develop chronic CD either in its cardiac, digestive, or cardio-digestive manifestation between 10 and 30 years after the initial infection. The pathological findings observed in chronic chagasic cardiomyopathy include widespread destruction and dysfunction of mitochondrial myocardial cells (68, 69), myocardium inflammation, fibrosis, and hypertrophy along with scarce parasite numbers. Exacerbation of the chronic stage with increased blood parasitemia and intracellular parasite replication is found in individuals with immunosuppressive treatment, indicating the pivotal role of the host-immune mediators in the control of parasite proliferation and persistence (57). The pathogenesis of chronic CD myocarditis is complex and involves parasite immune evasion strategies, genetically determined host-immune homeostasis defects, and autoreactive phenomena in the presence of autoantibodies. Damaged tissue in the chronic phase is also accompanied by genetic material from T. cruzi, indicating an active role of parasite persistence in pathology (7, 57). Indeed, in spite of a robust immune response, the host fails to eliminate parasites from tissues, and the pathogen is able to chronically persist.
Host-Derived Nitroxidative Stress During T. cruzi Infection
In the acute infection, resident macrophages at the site of parasite invasion are among the first professional phagocytes that are invaded by T. cruzi (29, 39). On invasion, infective insect-borne metacyclic trypomastigotes should survive and evade the highly oxidative environment found inside the phagosome in order to establish the infection (1, 46, 50). We have recently shown that during the phagocytosis of T. cruzi metacyclic trypomastigotes, macrophage membrane-associated NADPH oxidase is activated, resulting in a sustained (60–90 min) superoxide radical (O2 •−) production toward the internalized parasite (1). O2 •− either dismutates to H2O2 or reacts with nitric oxide (·NO) derived from the inducible nitric oxide synthase (iNOS) in a diffusion-controlled reaction, to yield peroxynitrite (ONOO−; k=1×1010 M −1 s−1), a strong oxidant and potent cytotoxic effector molecule against T. cruzi (1, 2, 16). The control of intraphagosomal parasite survival, before the replicative amastigotes reach the “safe” cytoplasmic environment, largely depends on the macrophage production of ONOO− (1, 46). In this scenario, the levels of parasite antioxidant defenses at the onset of macrophage invasion may tilt the balance toward pathogen survival (44, 46, 48). Some parasites manage to evade the initial assault of the immune system and infect other organs such as the heart and digestive tract, disseminating the infection and favoring the progression to the chronic stage of CD. Chronic chagasic cardiomyopathy is characterized by the presence of pseudo cysts of amastigote nests in the cardiac fiber. T. cruzi invasion to cardiomyocytes triggers the production of inflammatory mediators (TNF-α, IL-1β) and the induction of cardiomyocyte iNOS with the subsequent generation of sustained amounts of ·NO (13, 32). T. cruzi invasion and proinflammatory cytokines along with ·NO generation lead to cardiomyocyte mitochondrial dysfunction with an increase in reactive oxygen species that may contribute to cardiomyocyte death and chronic heart failure in chagasic patients (7, 24).
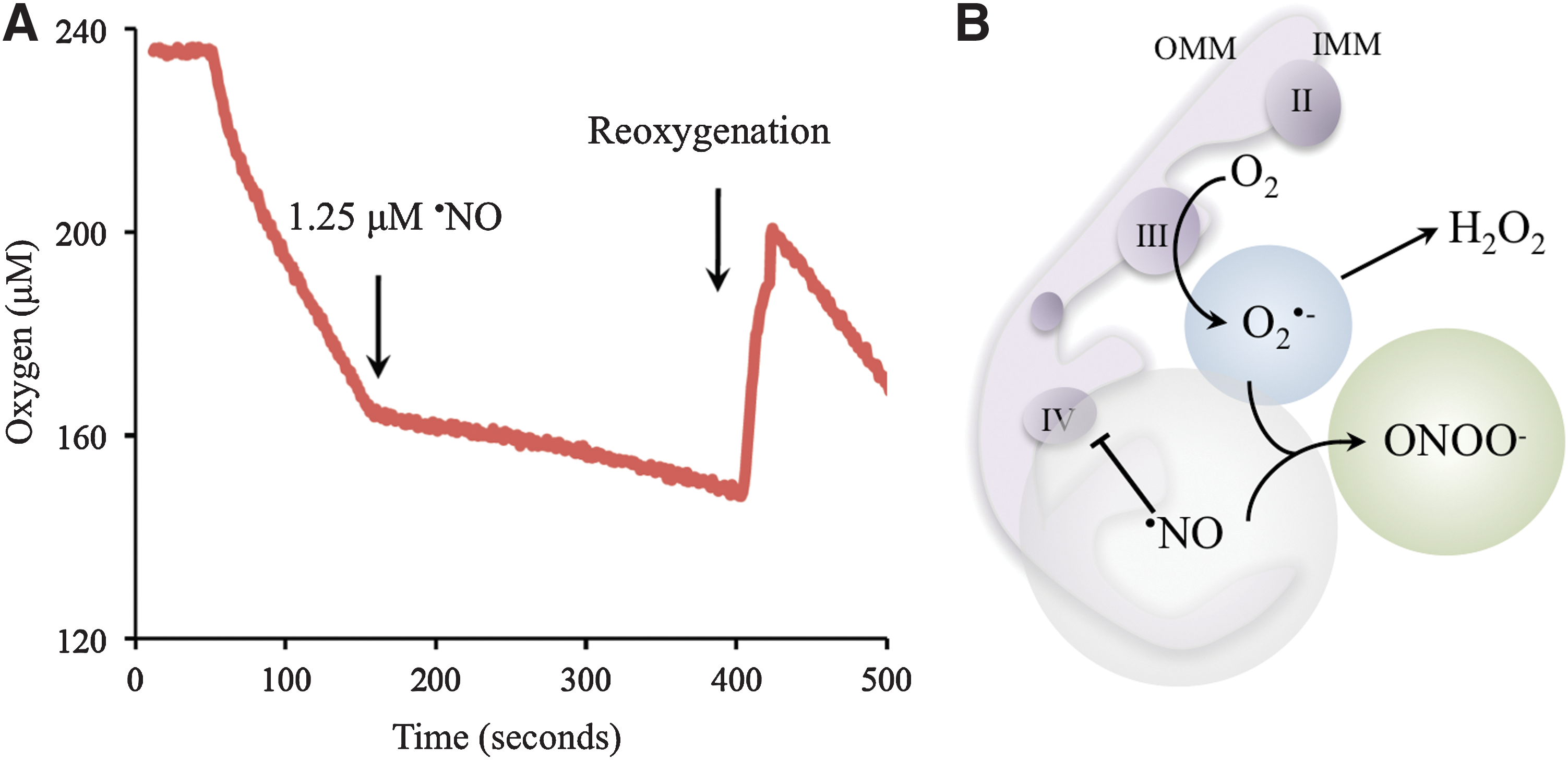
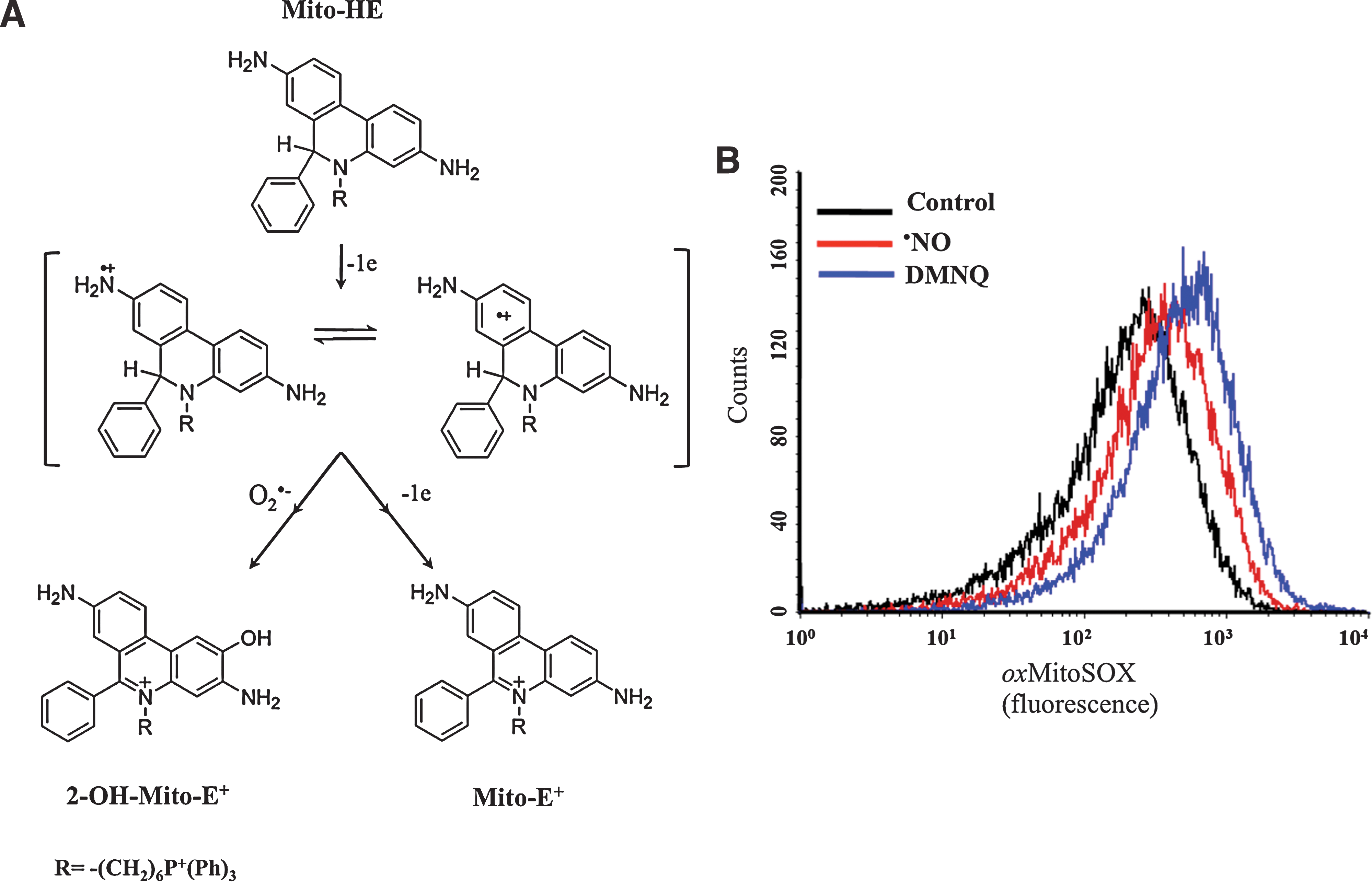
The ·NO pathway is indicated as a parasite control mechanism, as revealed by experiments performed on infected cardiomyocytes cultures. In addition, the role of the IL-12/INF-γ/iNOS axis in the control of T. cruzi infection and its implication for the outcome in CD has been well documented in key experiments using knockout mice for INF-γ, IL-12, and NOS isoforms (20, 37). ·NO oxidative damage largely depends on its reaction with superoxide (Fig. 1). Peroxynitrite can cause damage by direct reactions via one or two electron oxidations mechanisms to several molecules such as thiols and metal centers (21), also yielding secondary reactive species, including hydroxyl radical (·OH), nitrogen dioxide (·NO2), and carbonate (CO3·−) radicals that can oxidize lipids, DNA, and participate in protein oxidation and nitration, stable indicators of nitroxidative stress (9, 21, 40) (Fig. 1). Host-tyrosine nitrated proteins have been identified in heart lesions in the mouse model of CD (17, 40). Moreover, nitrated and oxidized plasma proteins as well as circulating myeloperoxidase [a heme-peroxidase released by PMNs that also participates in protein tyrosine nitration (54)] have been found in the plasma of patients with CD (17, 18). ·NO is not a strong oxidant or reactive species per se and by itself is unlikely to account for direct damage to the parasite (21). The inhibition of the respiratory chain by ·NO in mammalian mitochondria via interactions with cytochrome c oxidase is accompanied by a larger steady-state level of reduced respiratory complexes, which, in turn, favors intramitochondrial O2 •− formation (11, 55). After the acute infection, host-derived ·NO may diffuse and reach intracellular amastigotes, leading to the generation of intramitochondrial O2 •− and ONOO− formation. Results from our laboratory showed that T. cruzi mitochondrial oxygen consumption was reversibly inhibited by ·NO (Fig. 2A, B), and mitochondrial O2 •− production under these conditions was detected using the mitochondria-targeted ethidium-derived probe [MitoSOX™, live-cell permeable probe selectively targeted to mitochondria (59)] (Fig. 3A, B). This result makes feasible the concept of ·NO-mediated control of parasite proliferation in CD via intra-parasitic ONOO− formation. In this scenario where host-derived oxidant mediators are actively generated, the antioxidant armamentarium of T. cruzi becomes crucial for parasite survival and persistence.

T. cruzi Antioxidant Network
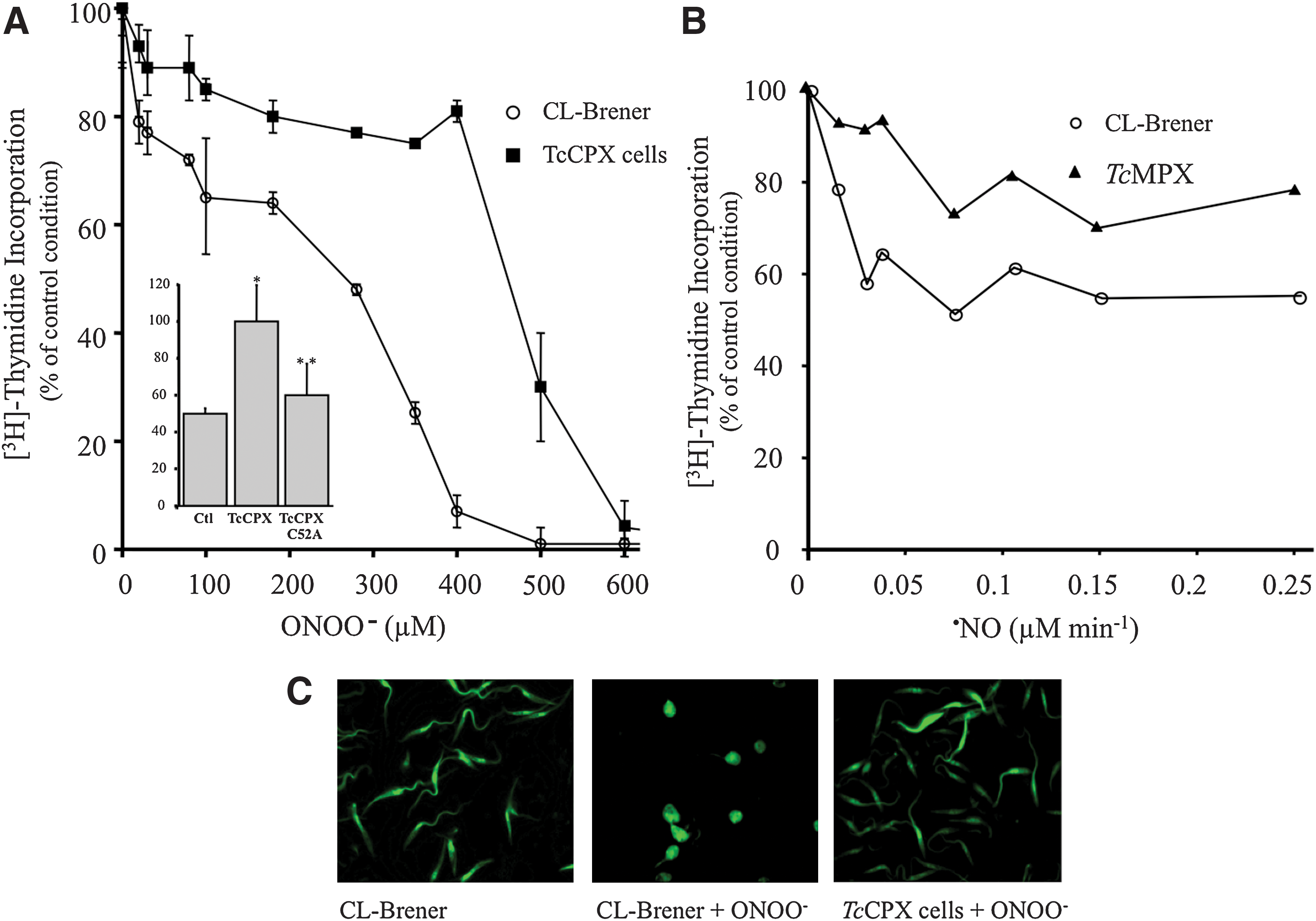
Antioxidant defenses in T. cruzi rely on a sophisticated system of linked pathways in which reducing equivalents from NADPH (derived from the pentose phosphate pathway) are delivered to a variety of enzymatic detoxification systems through the dithiol trypanothione [T(SH)2, N 1,N 8-bisglutathionylspermidine] and the thioredoxin homologue tryparedoxin (TXN; Fig. 3) (26). Trypanothione is synthesized in two sequential steps in which two molecules of glutathione (GSH) are covalently bound to the terminal NH2 groups of spermidine by a single cytosolic enzyme called T. cruzi trypanothione synthetase (TcTS) (42). Five distinct peroxidases have been identified in T. cruzi, differing in their subcellular location and substrate specificity. Glutathione peroxidase-I (TcGPXI, located at the cytosol and glycosome) and TcGPXII (located at the endoplasmic reticulum) confer resistance against hydro- and lipid-hydroperoxides, respectively, and use GSH and/or TXN as reducing substrates (72, 76). The T. cruzi cytosolic tryparedoxin peroxidase (TcCPX) and T. cruzi mitochondrial tryparedoxin peroxidase (TcMPX, typical two-cysteine peroxiredoxins belonging to the AhpC/Prx subfamily) have the capacity to detoxify ONOO−, H2O2 and small-chain organic hydroperoxides using TXN (Fig. 4) (48, 51, 66, 77). Parasite resistance against ONOO− toxicity is afforded by CPX overexpression (Fig. 4A), while the mutation in the peroxidatic Cys52 (Cys52Ala) of CPX fails to confer resistance, indicating the catalytic nature of the process (Fig. 5A, inset). Moreover, TcMPX overexpressers are more resistant to ·NO fluxes, which is in line with the predicted ·NO-mediated intramitochondrial ONOO− formation and detoxification by this peroxiredoxin (Fig. 5B). Whether the complete system of TXN, T(SH)2, and TR is found in the mitochondria is still a matter of debate. Finally, an ascorbate-dependent heme-peroxidase (TcAPX), located at the endoplasmic reticulum, confers resistance against H2O2 challenge using ascorbate as the reducing substrate (48, 73). The complete detoxification capacities of the different peroxidases of T. cruzi are still under investigation. In particular, further studies are required to find out whether thiol-dependent GPX-I and GPX-II can catalyze ONOO− reduction and detoxification in T. cruzi.

T. cruzi contains four iron superoxide dismutases (FeSODs) that detoxify O2 •− generated in the cytosol (TcSODB1), glycosomes (TcSODB1-2), and mitochondria (TcSODA and C) (33). The biochemical and cell biology characterization of the T. cruzi FeSODs repertoire in terms of free radical events has been minimally explored (28, 47, 64, 74). The role of mitochondrial TcFeSODA could be particularly relevant, as this organelle is a key source of O2 •− and presumably ONOO−. The preliminary data of our laboratory indicate that, although fluxes of O2 •− and ·NO (SIN-1, 0–10 mM) cause a very modest enzyme inactivation, TcFeSODA activity is inhibited by biologically relevant ONOO− concentrations, which is accompanied by nitration on tyrosine residues (Martinez, Piacenza, et al., unpublished results). The nitration of a critical tyrosine near the active site can account for the observed inhibition, as previously shown for Tyr-34 in MnSOD (38, 53, 78). These data support the concept that TcFeSODA play an important role in parasite oxidant detoxification pathways by preventing ONOO− formation in this organelle trough O2 •− dismutation, all of which contribute to neutralize cytotoxicity. Due to its unique characteristics when compared with the mammalian counterparts, components of the T. cruzi antioxidant system have been considered good targets for chemotherapy (23, 26, 30).
Since the end of the 1960s and the beginning of the 1970s, two drugs have been used for the treatment of CD: a 5-nitrofuran, nifurtimox [NFX, (4[5-nitrofurfurylidene)amino]-3-methylthiomorpholine-1,1-dioxide] and a 2- nitroimidazole, benznidazole (BZ, N-benzyl-2-nitroimidazole-1-acetamide) (62). Both are prodrugs that undergo intracellular activation by nitroreductases (NTRs), leading to cytotoxicity within the parasite, although the precise mechanism of action and parasite targets remain to be established (70, 71, 75). Since a long time, it has been proposed that NFX trypanocidal activity involves the generation of nitroanion radicals through NTR type II activity (one-electron reduction), which, in turn, generates O2 •− radicals (19, 34). In addition, NFX-derived metabolites can lead to adduct formation with GSH and TSH, contributing to parasite oxidative stress (58). However, the direct evidence between drug-induced oxidative stress and trypanocidal activity is limited and arises from functional studies where (i) T. brucei parasites lacking cytosolic SODB are more sensitive to NFX and BZ (52); and (ii) the reported overexpression of mitochondrial TcFeSODA in an in vitro-derived BZ-resistant T. cruzi strain (41). It is now known that BZ and NFX trypanocidal activity depends on NTR type I activity, which is absent in mammalian cells and is the base for parasite selectivity (70). NTR type I activity catalyzes the two-electron reduction of nitroheterocyclic compounds producing toxic hydroxylamine derivatives and other metabolites that can induce biomolecule modifications and DNA strand breaks, leading to parasite death (8, 36, 75).
Moreover, it was recently found that in the BZ-resistant T. cruzi population, the parasite phenotype is associated with the loss of a NTR gene copy with changes in neither parasite SOD expression nor activity (36). The unambiguous determination of BZ-NFX toxicity mechanisms as well as the participation of antioxidant enzymes, and especially that of FeSOD, in resistant T. cruzi phenotypes needs further investigation. SODs readily eliminates O2 •− and may contribute to T. cruzi survival in the vertebrate host by immune evasion mechanisms, such as (i) protection from direct cytotoxic effects of O2 •−, mainly generated by parasite mitochondria; (ii) inhibiting the formation of ONOO− in ·NO-challenged parasites; and (iii) participating in the modulation of redox signaling processes. While the protein fold and structure of FeSOD are considered similar to mammalian MnSOD, efforts are being made to unravel the structural characteristics to allow the generation of specific inhibitors (6).
Redox Signaling of T. cruzi Programmed Cell Death
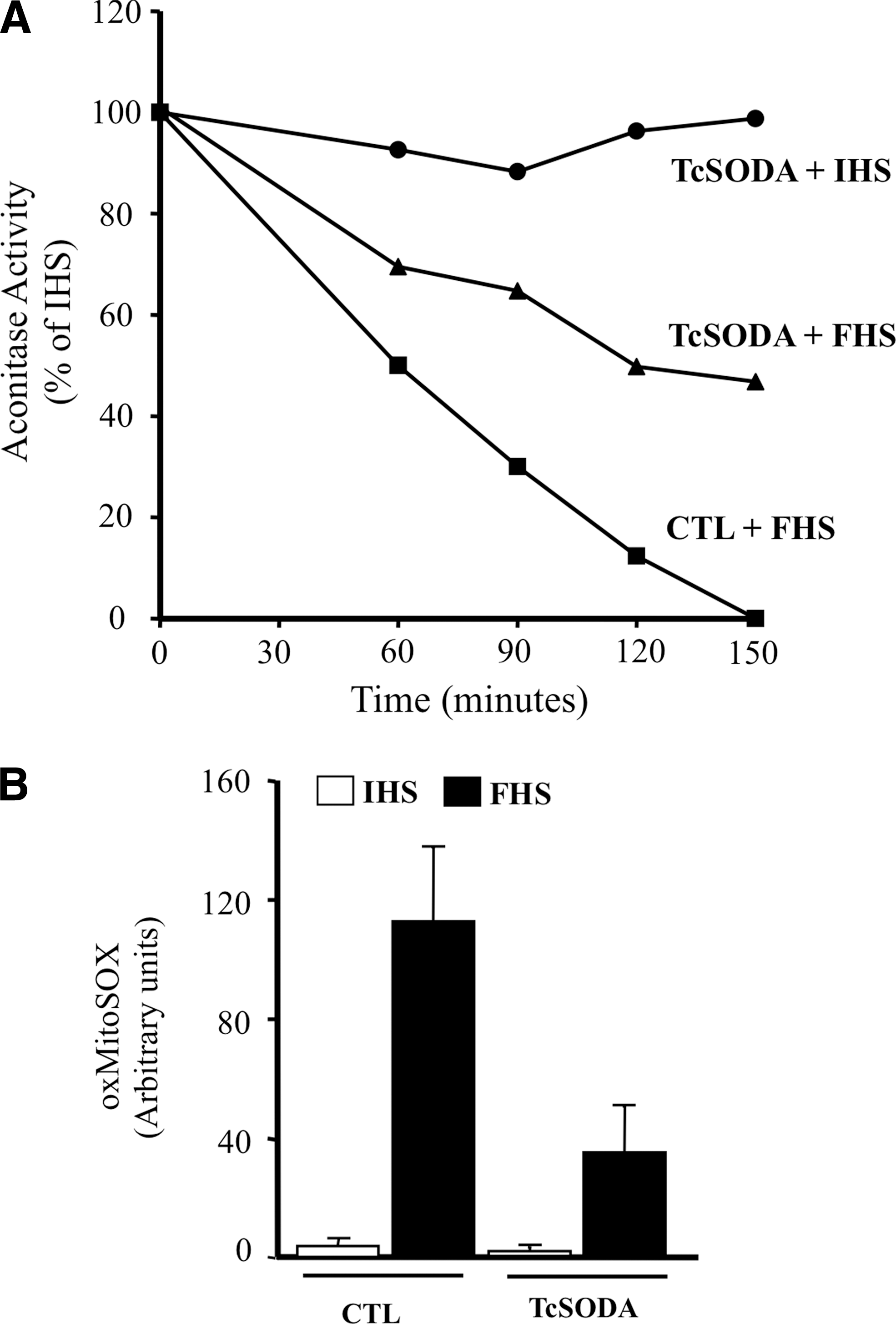
T. cruzi can undergo a programmed cell death process that is triggered by a variety of stimuli and involving alterations of cellular homeostasis (3, 27, 49). The intracellular misbalance of the parasite redox status can disengage programmed cell death in epimastigotes of T. cruzi (27, 44, 47). After appropriate challenges (fresh human serum [FHS]), a rapid loss of low-molecular thiols (i.e., T(SH)2, glutathionyl-spermidine, and GSH) occurs accompanied by mitochondrial dysfunction; disruption of mitochondrial homeostasis in the parasite is characterized by mitochondrial Ca2+ influx, an increase in mitochondrial O2 •− production, impaired oxidative phosphorylation, oxygen consumption, and the release of cytochrome c to the cytosol (47). FHS-dependent mitochondrial O2 •− generation is evidenced by aconitase inhibition due to the disruption of the 4Fe4S cluster (Fig. 6A). The T. cruzi cell death phenotype is accompanied by phosphatydylserine exposure to the outer leaflet of the plasma membrane and by nuclear DNA condensation and fragmentation, all common morphological features of metazoan apoptosis (27, 47, 49). Notably, the overexpression of FeSODA prevented FHS-triggered processes, including aconitase inhibition, MitoSOX oxidation, and programmed cell death, underscoring the role of mitochondrial O2 •− in the redox signaling of this process (Fig. 6A, B).
Intracellular apoptotic amastigotes have been found in experimentally infected cardiomyocytes as well as in vivo in the heart tissue of infected mice (14, 15). Interactions of apoptotic parasites with immune cells may locally down regulate the host-inflammatory response with the concomitant production of anti-inflammatory cytokines (TGF-β), thus favoring parasite proliferation and transition to the chronic stage (22, 44). It has also been proposed that apoptosis enables the regulation of parasite densities in distinct host compartments, facilitating a sustained infection in the vertebrate host (67). Thus, interfering with the parasite apoptotic machinery may reveal new targets for drug design. The presence in T. cruzi of four FeSODs localized in different subcellular compartments argues in favor of a pivotal role of O2 •− in parasite redox signaling.
Antioxidant Enzymes As Virulence Factors
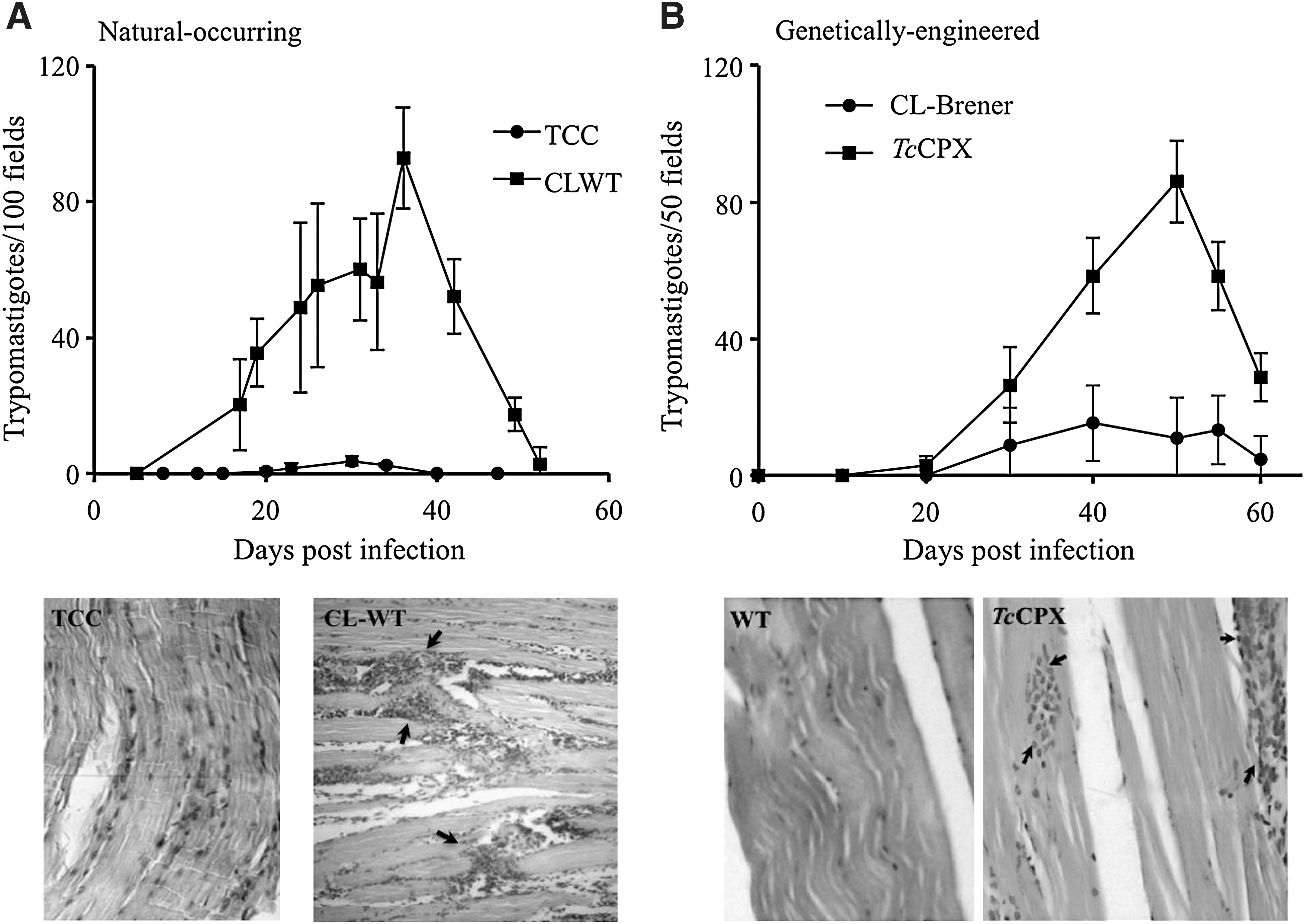
T. cruzi consists of a mixed population of strains classified in two discrete typing units recently designated as T. cruzi I-VI (81), which circulate in the sylvatic and domestic cycles, respectively (63). The biochemical and genetic heterogeneity between strains is, in part, responsible for the diverse clinical manifestations of the disease, ranging from asymptomatic to severe cardiac and digestive presentations (31). The pathogenesis of CD depends on parasite and host factors that control virulence, tissue tropism, tissue damage, and ability to maintain long-term infections in the vertebrate host. A number of virulence factors have been identified for T. cruzi and include complement C2 receptor inhibitor trispanning (CRIT) protein (12), calreticulin (TcCRT) (56), gp35/50 (79), gp82 (61), cruzipain (35), oligopeptidase B (10), and transialidases (45), among others. Taking into account the establishment of a nitroxidative stress during CD (acute or chronic stages), the antioxidant armamentarium of T. cruzi becomes decisive. Several proteomic analyses have suggested the up-regulation of members of the T. cruzi antioxidant network (TcTS; TcMPX; TXN; FeSODA and TcAPX) in the infective metacyclic trypomastigote compared with the noninfective epimastigote stage (5, 43). Due to the fact that T. cruzi is not a clonal population, we search for the up-regulation of different components of the antioxidant network in several strains belonging to the major phylogenetic groups during the differentiation process to the infective metacyclic trypomastigotes. Our results show for all the analyzed strains an up-regulation of TcCPX, TcMPX, and TcTS protein content during metacyclogenesis, making this a general preadaptation process that allows T. cruzi to deal with the nitroxidative environment found in the vertebrate host (50). Most important, a positive association was found between virulence and the protein levels of antioxidant enzymes (TcCPX, TcMPX, and TcTS) in several T. cruzi strains, highlighting the interplay between the antioxidant enzyme network and host oxidative defense mechanisms (Table 1) (50). In accordance, strains of higher virulence showed an important heart inflammatory infiltrate with high parasitemias in the acute phase of CD. (Fig. 7A). At the cellular level, parasites that overexpressed TcCPX were resistant to macrophage killing due to peroxynitrite detoxification (Table 2) (1). Moreover, the overexpression of TcCPX in trypomastigotes augments virulence, as evidenced by a threefold increase in parasitemia with the presence of higher inflammatory infiltrates in the heart and skeletal muscle when compared with the wild strain (Fig. 7B). In this line, the experiments performed with genetically modified parasites fully recapitulated the association observed between the virulence and antioxidant enzyme content of T. cruzi wild strains (compare Fig. 7A and B) (1). These results reinforce the concept that the success of infection in the acute phase depends on parasite antioxidant enzyme levels. Whether FeSOD constitute a virulence factor for T. cruzi infection remains to be established and is currently under investigation in our laboratory. T. cruzi cytosolic FeSODB is particularly resistant to peroxinitrite inactivation, suggesting its participation mainly as an antioxidant defense enzyme, while mitochondrial FeSODA may act as an oxidative stress sensor participating in O2 •—-mediated redox processes of cell signaling (Martinez, Piacenza, et al., unpublished results). Interestingly, highly virulent strains of T. cruzi showed less amastigote apoptotic death and higher proliferation rates than less virulent strains in cardiomyocyte infections (14). Taking into account that mitochondrial O2 •− mediates programmed cell death in T. cruzi, we propose that FeSOD may also play a role in the pathogenesis of the disease.
Correlation is significant at the 0.001 level (two tailed).
TcMPX, T. cruzi mitochondrial tryparedoxin peroxidase; TcCPX, T. cruzi cytosolic tryparedoxin peroxidase; TcTS, T. cruzi trypanothione synthetase.
Adapted from Piacenza et al. (46)
The load of T. cruzi amastigotes in the macrophage (J-774 line) culture was evaluated 24 h after infection with metacyclic trypomastigotes (parasite:macrophage ratio for infection 5:1). Data are mean±standard error of three independent experiments.
INF-γ/LPS was added 5 h before the infection.
p<0.01.
LPS, lipopolysaccharide.
Adapted from Alvarez et al. (1)
Concluding Remarks
T. cruzi antioxidant enzymes acting synergistically represent the first line of defense to cope with O2 •−, H2O2, and ONOO− generated while infecting mammalian cells either in the acute and presumably in the chronic stage of the disease. Particularly interesting is the emerging evidence that the single T. cruzi mitochondrion can play a central role as a “powerhouse” of the reactive oxygen and nitrogen species, including O2 •− and ONOO− (the latter arising from the combination of O2 •− with mammalian host cell-derived •NO), when infecting non-phagocytic cells such as cardiomyocytes; these alterations in T. cruzi mitochondrion redox homeostasis and its modulation by mitochondrial antioxidant systems may facilitate the progression of a “low grade” infection to a chronic phase. Deciphering the relative role of different arms of the enzymatic antioxidant network in the various cellular compartments should be a part of future investigations. In summary, the assessment and definition of the contribution of the parasite antioxidant systems toward virulence and persistence could further define them as relevant targets for the development of new pharmacological strategies that treat CD, especially in the chronic stage.
Footnotes
Acknowledgments
This work was support by grants from the National Institutes of Health, 1R01AI095173-01 (NIH, USA), and Universidad de la República (CSIC, Uruguay) to R.R. A.M. is supported by a fellowship of Agencia Nacional de Investigación e Innovación (ANII, Uruguay).