Aim: Human protein disulfide isomerase (hPDI) is a key enzyme and a redox-regulated chaperone responsible for oxidative protein folding in the endoplasmic reticulum. This work aims to reveal the molecular mechanism underlying the redox-regulated functions of hPDI by determining the crystal structures of hPDI in different redox states. Results: The structures of hPDI (abb′xa′) in both the reduced and oxidized states showed that the four thioredoxin domains of a, b, b′, and a′ are arranged as a horseshoe shape with two CGHC active sites, respectively, in domains a and a′ facing each other at the two ends. In reduced hPDI, domains a, b, and b′ line up in the same plane, whereas domain a′ twists ∼45° out. The two active sites are 27.6 Å apart. In oxidized hPDI, the four domains are differently organized to stay in the same plane, and the distance between the active sites increases to 40.3 Å. In contrast to the closed conformation of reduced hPDI, oxidized hPDI exists in an open state with more exposed hydrophobic areas and a larger cleft with potential for substrate binding. Innovation: This is the first report of the high-resolution structures of hPDI containing all four domains in both the reduced and the oxidized states. It reveals the redox-regulated structural dynamic properties of the protein. Conclusion: The redox-regulated open/closed conformational switch of hPDI endows the protein with versatile target-binding capacities for its enzymatic and chaperone functions. Antioxid. Redox Signal. 19, 36–45.
Introduction
Formation of the correct disulfide bonds is vital for proper folding of membrane and secretory proteins. Unformed or mismatched disulfide bonds can lead to misfolding and aggregation of proteins, which can ultimately cause abnormal or pathological conditions, such as deficiency in antibody production, infertility, and neurodegeneration (4). Protein disulfide isomerase (PDI) is a key enzyme (15) and a chaperone (37) abundantly located in the endoplasmic reticulum for catalyzing oxidation, reduction, and isomerization of disulfide bonds and assisting the proper folding of proteins.
PDI was discovered about a half century ago as the first folding catalyst (14). It is composed of four thioredoxin domains a, b, b′, and a′, an x-linker between domains b′ and a′, and a C-terminal acidic tail (2, 12, 29) (Fig. 1A). Both domains a and a′ contain an active site CGHC that is responsible for the oxidoreductase activity (6), and domain b′ contains a large hydrophobic pocket that provides the major site for substrate binding (22, 27). All the four thioredoxin domains have been found to work synergically so as to promote a complex oxidative protein-folding process, including thiol–disulfide exchanges as well as significant conformational changes of the substrates (13).
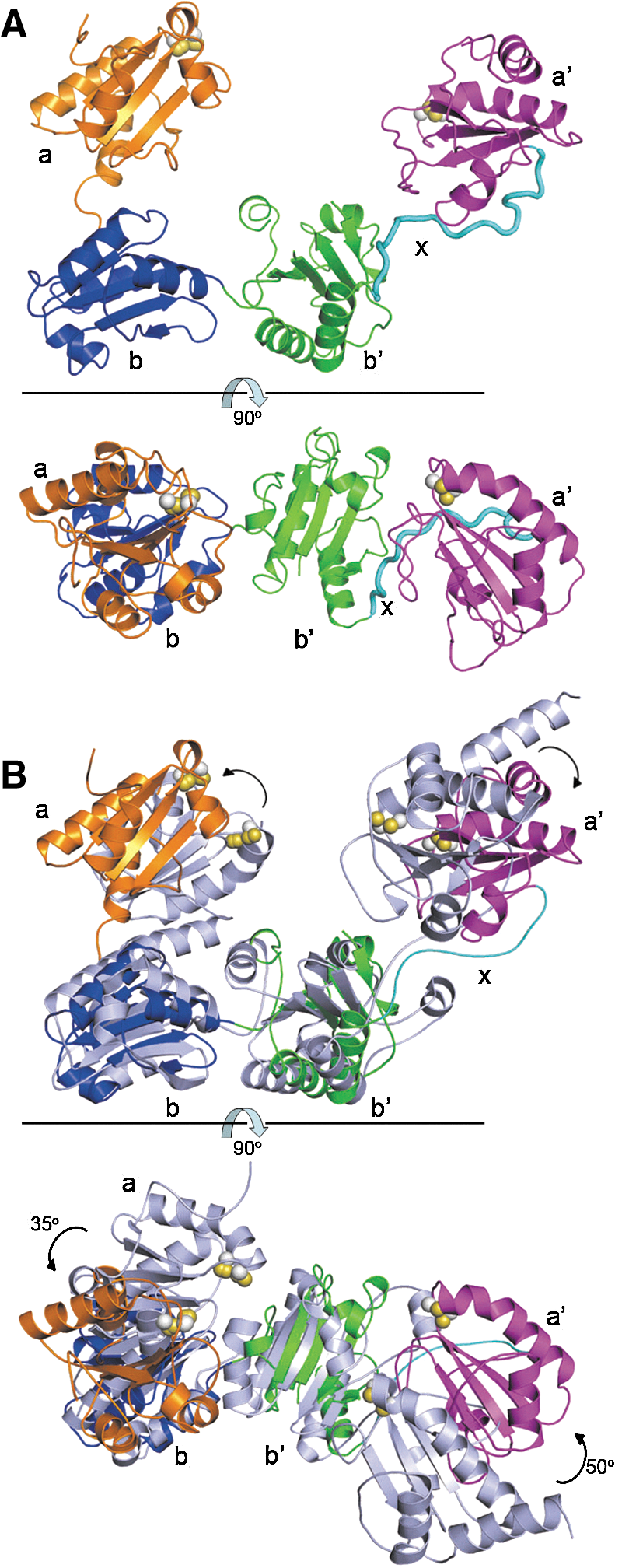
The overall structure of reduced hPDI. (A) Domain organization of the hPDI molecule. The residue numbering is for hPDI with signal sequence. The fragment (residues −19 to 0) is the MH6SSGLEVLFQGPGS tag derived from the vector. The CGHC motifs in the a and a′ domains are active sites. (B) Ribbon diagrams of the crystal structure of reduced hPDI from the front view (upper panel) and the top view (lower panel). Each domain is colored as (A). The side chains of the cysteine residues of the active sites in a and a′ are shown in space-filling representation with the beta carbon atoms in white and the sulfur atoms in yellow. (C) Superimposition of the structures of reduced hPDI (colored as panel A) and reduced bb′xa′ (35) (colored in light blue). (D) Superimposition of the structures of reduced hPDI (colored as panel A) and yPDI (32) (colored in light blue) based on the bb′ domains. The rotation of domains a and a′ in hPDI are indicated by arrows. hPDI, human protein disulfide isomerase; yPDI, yeast protein disulfide isomerase.
Decades of trials to obtain the high-resolution structure of full-length PDI failed until 6 years ago, and the crystal structures of yeast protein disulfide isomerase (yPDI) at 4°C (32) and 22°C (31) were determined. However, for human protein disulfide isomerase (hPDI), only the structures of each single thioredoxin domain (18, 19, 27; PDB 1X5C) and the domain combinations bb′ (8) and bb′xa′ (35) have been determined to date. The structure of full-length yPDI cannot represent the structure of hPDI, because hPDI is not functionally equivalent to yPDI (21) and is dissimilar to it in several aspects (15). For example, the sequence identity of the bb′ fragment, which determines the substrate specificity, is only 17.7% (15). This sequence identity is even lower than that (22%) between hPDI and another PDI family member, human ERp57 (15), yet hPDI and human ERp57 have been demonstrated to be associated with different substrates (25). Furthermore, we recently demonstrated that the major flexibility of hPDI is determined by the intrinsic dynamic property of the b′xa′ region (34), but not by that of the ab region as indicated for yPDI (31). Our recent structural work on bb′xa′ of hPDI showed that the b′xa′ region undergoes a large redox-regulated interdomain conformational change, which can directly modulate the chaperone activity of hPDI (35). However, the exact structural arrangement of the four thioredoxin domains of hPDI under reduced or oxidized conditions has yet to be determined.
Innovation
Protein disulfide isomerase (PDI) has been known for a half century; however, no three-dimensional structure of full-length vertebrate PDI has been reported to date. Here, we determined the crystal structures of human protein disulfide isomerase (hPDI) containing all four thioredoxin domains (abb′xa′) in different redox states, which revealed a closed conformation of reduced hPDI and an open conformation of oxidized hPDI. This study provided structural insights into the redox-regulated dynamic properties of hPDI and revealed the diverse target-binding modes of hPDI in different redox states.
Although hPDI has already been a subject to intense biochemical studies, the molecular mechanism underlying its versatile biological functions still remains largely unclear (33). For example, how does hPDI distinguish unfolded or misfolded substrates to catalyze different reactions (oxidative folding or reductive unfolding)? How does hPDI undergo various conformational changes during its catalytic cycle? What is the effect of the potential conformational changes of hPDI on the binding and folding of substrates, and how are the conformational changes regulated? Currently, a major obstacle for us to answer these questions is the lack of a high-resolution structure of full-length hPDI and a detailed description of the dynamic properties of the protein.
In this work, we determined for the first time the crystal structures of hPDI containing all the four thioredoxin domains (abb′xa′) in both the reduced and oxidized states to 2.5 and 2.9 Å, respectively. The closed conformation of reduced hPDI and the open conformation of oxidized hPDI not only reveal the redox-regulated structural dynamic properties of the protein but also provide a mechanistic explanation for the diverse target-binding capacities of hPDI.
Results
The overall structure of reduced hPDI
The short C-terminal tail (composed of 29 amino acid residues) of hPDI was deleted for crystallization, because this flexible tail has been determined to be not essential for chaperone and enzymatic activities (23), and it may hinder protein crystallization. The crystals of reduced hPDI were achieved in the presence of 50 mM DTT and diffracted to 2.5 Å for structural determination (Table 1).
Data for the outermost shell are given in parentheses.
Rmerge=100 ΣhΣi|Ih,I−<Ih>|/ΣhΣiIh,i, where the outer sum (h) is over the unique reflections, and the inner sum (i) is over the set of independent observations of each unique reflection.
As defined by the validation suite MolProbity (7).
Residues from −19 to 0 are the N-terminal (MH6SSGLEVLFQGPGS) tag derived from the vector.
hPDI, human protein disulfide isomerase.
From the front view of the overall structure of reduced hPDI (Fig. 1B, upper panel), the four thioredoxin domains abb′a′ are arranged as a horseshoe shape with domains a and a′ at the two ends and domains bb′ as the base. Rotation of the structure ∼90° with respect to the horizontal line (Fig. 1B, lower panel) shows that domains a, b, and b′ are located in the same plane, whereas the domain a′ is twisted ∼45° out of the abb′ plane. The two active sites in domains a and a′ exist in the reduced state as expected, and the distance between the N-terminal cysteine (Cys 53 in domain a and Cys 397 in domain a′) sulfur atoms of the active sites is 27.6 Å.
Compared with the reduced bb′xa′ structure we solved recently (35), the bb′xa′ fragment in the reduced hPDI structure shows a high similarity, especially the b′xa′ region (Fig. 1C). There are limited structural variations in the b domain probably due to the contacts between domains a and b in the hPDI structure. The structures of reduced hPDI and yPDI were also compared on the basis of the bb′ domains. Since the structure of yPDI at 22°C (31) is at a lower resolution (3.7 Å) than that at 4°C (2.4 Å) (32), and the active sites in the 22°C structure no longer face each other (31), here after, the 4°C structure was used to represent the yPDI structure. Domains a and a′ in reduced hPDI exhibit anticlockwise rotations of ∼30° and ∼20°, respectively (Fig. 1D, lower panel), and the active site of domain a′ moves ∼10 Å toward the hydrophobic pocket in the b′ domain (Fig. 1D, upper panel). Nevertheless, the distances between the two active sites in reduced hPDI (27.6 Å) and in yPDI (26.7 Å) are similar.
The overall structure of oxidized hPDI
Our previous work showed that hPDI undergoes redox-regulated conformational changes, which can directly modulate its chaperone activity (35). To unveil the details of the redox-dependent conformational changes and the related regulation mechanism, the structure of oxidized hPDI is undoubtedly required. The crystals of oxidized hPDI were achieved in the same conditions as those for reduced hPDI, except that DTT was omitted; they diffracted to 2.9 Å for structural determination (Table 1).
In this structure, the active site of domain a′ exists in the oxidized form, whereas the active site of the domain a was best modeled (16) as a mixture of oxidized (70%) and reduced (30%) forms. Similarly to reduced hPDI, the overall oxidized hPDI structure also adopts a horseshoe shape (Fig. 2A, upper panel), but is distinct from that of reduced hPDI in that the four thioredoxin domains abb′a′ are arranged in the same plane (Fig. 2A, lower panel). Furthermore, the distance between the two active sites increases to 40.3 Å from the 27.6 Å found in reduced hPDI. We next compared the oxidized hPDI structure with the yPDI structure by superimposition of the two structures on the basis of the bb′ domains, and found that there are also significant rotations of both domain a (∼35°) and domain a′ (∼50°) of the oxidized hPDI structure with respect to the corresponding domains in the yPDI structure (Fig. 2B, lower panel). Taken together, the overall structure of oxidized hPDI is fairly different from that of reduced hPDI and from yPDI, which suggests that the dynamic properties and versatile binding capacities of hPDI may be controlled by its redox switch (see below for details).
The overall structure of oxidized hPDI. (A) Ribbon diagrams of the crystal structure of oxidized hPDI from the front view (upper panel) and the top view (lower panel). Each domain is colored as Fig. 1A. The side chains of the cysteine residues of the active sites are shown in space-filling representation with the beta carbon atoms in white and the sulfur atoms in yellow. (B) Superimposition of the structures of hPDI (colored as Fig. 1A) and yPDI (colored in light blue) based on the bb′ domains from the front view (upper panel) and the top view (lower panel). The rotation of domains a and a′ in hPDI are indicated by arrows.
Redox-regulated domain rotations of hPDI
With the determined structures of the reduced and oxidized forms of hPDI, we next analyzed the conformational changes of hPDI induced by its redox switch. Since the bb′ domains have been suggested to form a rigid base, and the a and a′ domains represent two flexible arms, we superimposed the reduced and oxidized structures based on the bb′ domains. Unexpectedly, none of α-helixes and β-sheets of bb′ domains can be fitted well, resulting in a large rmsd value of ∼3.4 Å (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/ars). Considering that, as revealed in our previous work, the ab region in hPDI showed less flexibility than the b′xa′ region (34), we then superimposed the two structures on the basis of the ab domains. The result showed that the ab domains in the two structures are very similar and can be well superimposed with each other with an rmsd value of ∼1.9 Å. As expected, significant structural changes between the reduced and oxidized hPDIs are located in the b′xa′ region (Fig. 3A). Compared with reduced hPDI, the a′ domain in oxidized hPDI rotates ∼45° around the x linker outward (away from the b′ domain from the front view of the overall structures), which leads to an increase in the distance between the two active sites in domains a and a′ (Fig. 3A, upper panel). After an ∼90° rotation of the superimposed structures, it is obvious that the b′xa′ region rotates ∼45° anticlockwise with respect to the ab domains, causing the four thioredoxin domains abb′a′ to lineup in a same plane (Fig. 3A, lower panel).
Superimposition of the structures of reduced hPDI (green) and oxidized hPDI (magenta). (A) Superimposition of the structures of reduced hPDI and oxidized hPDI based on the ab domains from the front view (upper panel) and the top view (lower panel). The side chains of the cysteine residues of the active sites are shown in space-filling representation with the beta carbon atoms in green and magenta for reduced and oxidized hPDI, respectively, and the sulfur atoms in yellow. (B) Superimposition of domains ab of reduced hPDI and oxidized hPDI based on the b domain from the front view (upper panel) and the top view (lower panel). (C) Superimposition of domains b′xa′ of reduced hPDI and oxidized hPDI based on the b′ domain from the front view (upper panel) and the top view (lower panel). (D) Superimposition of domains bb′ based on the b domain. (E) Superimposition of domains b′x based on the b′ domain.
To further analyze the details of the conformational changes, we compared the structures of the reduced and the oxidized forms of hPDI by superimposing the two structures on the basis of single b or b′ domains. When the structures are superimposed on domain b, domain a shows a small rotation of ∼10° with respect to domain b (Fig. 3B), and domain b′ shows an ∼20° rotation (Fig. 3D). When the structures are superimposed on domain b′, domain a′ in oxidized hPDI shows a rotation of ∼45° clockwise with respect to domain b′ (Fig. 3C, upper panel) and another rotation of ∼25° from the top view of the structure (Fig. 3C, lower panel). Interestingly, the C-terminal half of the x linker also undergoes a rotation of ∼45° in relation to the b′ domain (Fig. 3E). All the above structural analyses demonstrate that the redox-regulated conformational changes in hPDI are mainly located in the b′xa′ region, which is consistent with our previous biochemical studies (35). Given that the site of the redox switch in the b′xa′ region is exactly the active site of domain a′ (35), the latter thus plays a direct role in mediating the interdomain rotations in the b′xa′ region, which will ultimately result in the different architectures, that is, the closed or open conformations of the reduced or oxidized forms of hPDI, respectively (Fig. 3A).
Diverse target-binding capacities of hPDI in different redox states
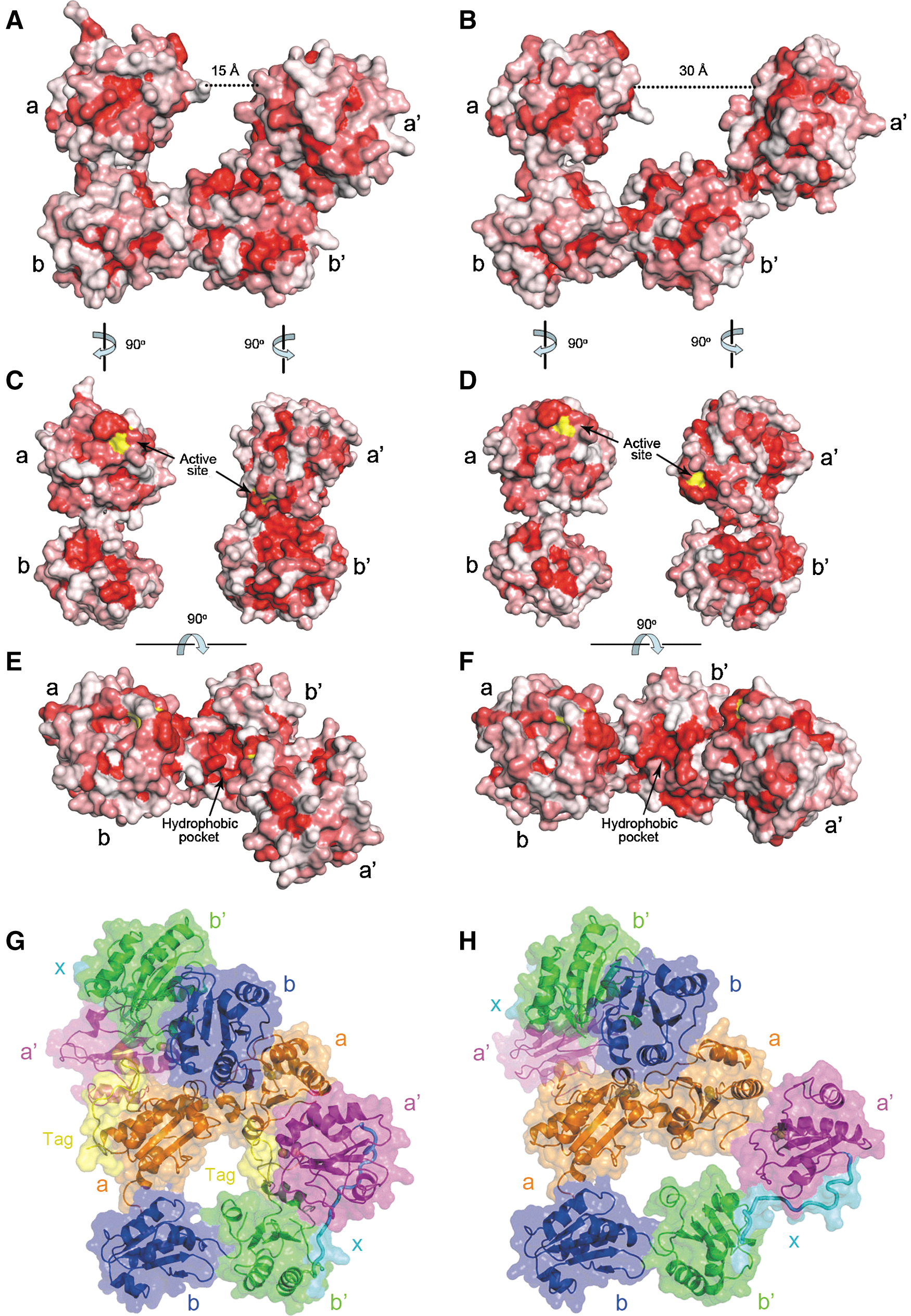
hPDI binds to its substrates largely by hydrophobic interactions and by forming mixed disulfide bonds between the cysteine residues in the active sites and those in the substrate. Surface property analysis revealed a continuous hydrophobic surface inside the horseshoe shape of the reduced and the oxidized structures, which is composed of a hydrophobic pocket in domain b′, the hydrophobic patches around the active sites in domain a and a′, and a hydrophobic patch in domain b (Fig. 4C, D). However, the substrate accessibility of the active site in domain a′ is markedly different in the different redox states. In reduced hPDI, the hydrophobic patch in domain a′ together with the hydrophobic pocket in domain b′ forms a narrow hydrophobic tunnel, and the active site of domain a′ is largely shielded in this tunnel (Fig. 4C). In contrast, the active site of domain a′ and its surrounded hydrophobic patch are fully exposed in oxidized hPDI (Fig. 4D), thus resulting in greater accessibility of the active site and a more expanded hydrophobic surface for substrate binding.
The structures of reduced hPDI and oxidized hPDI reveal different target-binding properties. (A, B) Surface representation of reduced hPDI (A) and oxidized hPDI (B). The color is coded ranging from hydrophobic (red) to hydrophilic (white) according to the normalized consensus hydrophobicity scale of the surface-exposed residues (10). The diameters of the open side of the cleft formed by four thioredoxin domains are indicated. (C, D) Surface representation of the ab and b′xa′ domains in reduced hPDI (C) and oxidized hPDI (D) in an open-book view from the middle of the structures. The surfaces of the active sites are colored in yellow. (E, F) A top view of the surface representation of reduced hPDI (E) and oxidized hPDI (F) by rotating ∼90° from (A, B), respectively. (G, H) In the crystal-packing profile, the N-terminal tag and the a domain of one hPDI molecule insert into the potential substrate-binding cleft formed by the abb′a′ domains of another hPDI molecule in the reduced (G) or the oxidized (H) form.
Furthermore, the cleft of the horseshoe shape, which is enveloped by the hydrophobic areas of all four domains for interactions with substrates, is also quite different in the reduced and oxidized hPDI structures. The volume of the cleft in oxidized hPDI is approximately twofold larger than that in reduced hPDI (Fig. 4A, B, and Supplementary Fig. S2). In particular, the diameter of the open side of the cleft in oxidized hPDI is about 30 Å (Fig. 4B), significantly longer than that in reduced hPDI (∼15 Å) (Fig. 4A), indicating that oxidized hPDI may accommodate relatively large substrates, but reduced hPDI can merely interact with small targets. More intriguingly, in the crystal packing, the a domain of one hPDI molecule was found to be inserted into the cleft of the horseshoe shape of another molecule, which may mimic the binding mode between hPDI and substrates (Fig. 4G, H). In the reduced hPDI structure, only two α-helices of the a domain of one hPDI molecule and the small N-terminal tag are inserted into the cleft of another molecule (Fig. 4G). Here, the hydrophobic patch around the active site in domain a′ and the hydrophobic pocket in domain b′ directly clamp the hydrophobic residues from the N-terminal tag (Supplementary Fig. S3), thus resembling the interaction of reduced hPDI with a small target. In contrast, in the oxidized hPDI structure, more than a half of the a domain of one PDI molecule is inserted into the cleft (Fig. 4H), which probably represents the binding between oxidized hPDI and a large target. However, the electronic density of the N-terminal tag is undefined (reflecting its flexible conformation), possibly due to the oxidized b′xa′ region adopting an open form that cannot tightly clamp the N-terminal tag. Taken together, the crystal-packing profiles clearly show that oxidized hPDI is able to accommodate larger targets than the reduced form does, which also strongly suggests that the versatile target-binding properties of hPDI are regulated by its different redox states.
The molecular mechanism of the redox-regulated conformational changes of hPDI
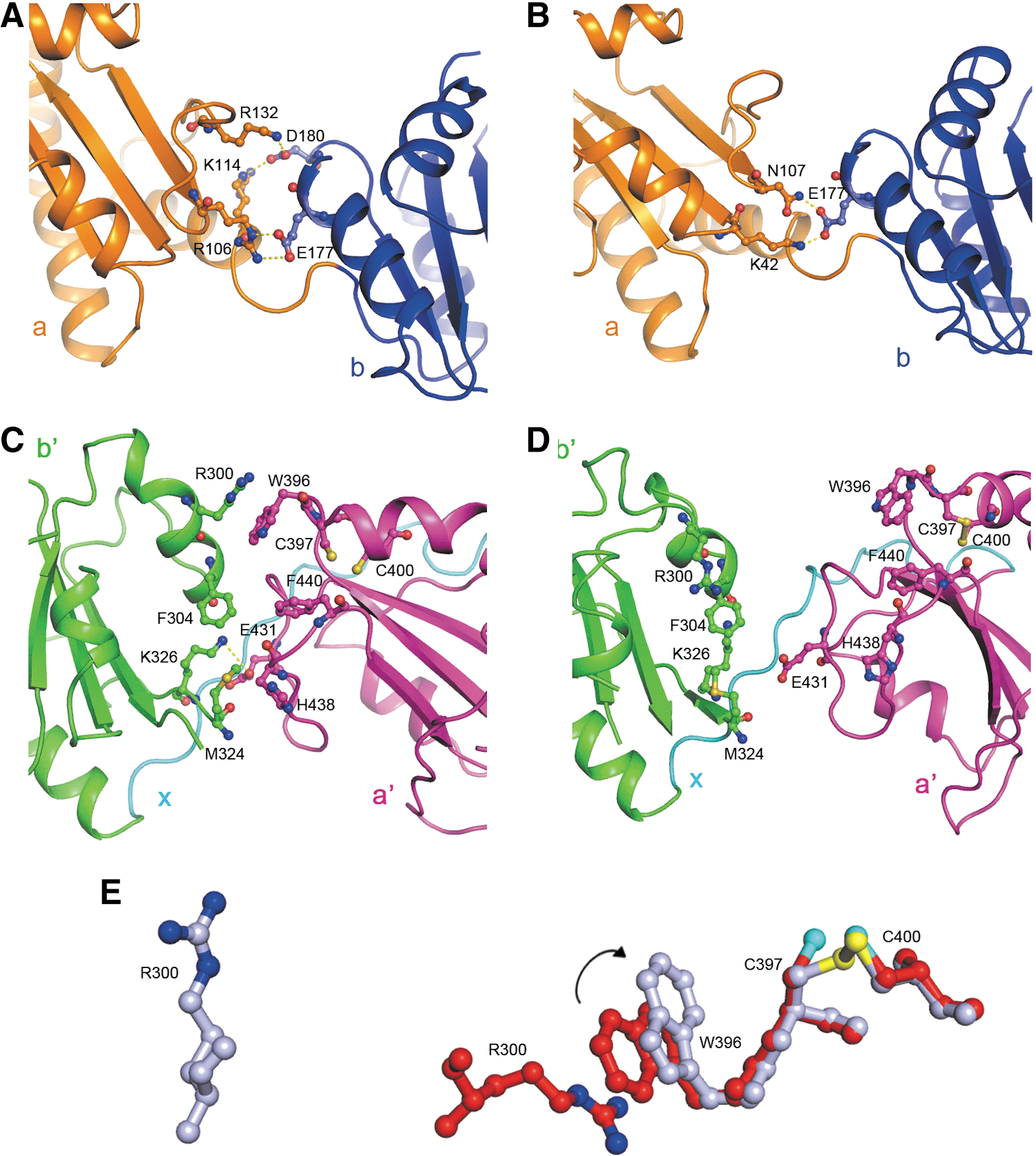
To understand how the redox switch of the active sites of hPDI induces the domain rearrangements demonstrated above, we next analyzed in detail the contacts between the redox-active domains (domains a and a′) and their adjacent redox-inactive domains (domains b and b′). Although the position of domain a is somewhat fixed relative to domain b as demonstrated above, the contacts between domains a and b are different in reduced and oxidized hPDIs. In reduced hPDI, residues E177 and D180 in domain b form a number of salt bridges with residues R106, K114, and R132 in domain a (Fig. 5A), whereas in oxidized hPDI, only residue E177 interacts with residues K42 and N107 in domain a while residue D180 no longer makes contacts with domain a (Fig. 5B). All the above interactions are not close to the active site.
Interactions between the redox-active domains and redox-inactive domains of hPDI at different redox states. (A, B) Interactions between domains a and b in reduced (A) and oxidized (B) hPDI. Residues involved in the interactions are shown in stick representation. The salt bridges and hydrogen bonds are indicated in yellow dotted lines. (C) Interactions between domains b′ and a′ in reduced hPDI. The residues involved in the interactions and the active site cysteines are shown in stick representation. (D) The architecture of the b′ and a′ domains in oxidized hPDI. The corresponding residues shown in (C) are also shown in stick representation, but no interactions are observed. (E) The key residues triggering the redox-regulated interdomain conformational changes of hPDI. The active site cysteines (C397 and C400) and W396 in domain a′, and R300 in domain b′ are all shown in stick representation in red for reduced hPDI and in light blue for oxidized hPDI, which are superimposed on the basis of the main chain of C397 and C400. The sulfur atoms are shown in cyan for the reduced form and in yellow for the oxidized form, and the nitrogen atoms of the guanidinium group are shown in blue. The rotation of the side chain of W396 is indicated by arrow, and the cation-π interaction between residues R300 and W396 is disrupted in oxidized hPDI.
On the other hand, for the b′xa′ region, multiple contacts between the b′ domain and the a′ domain were found in reduced hPDI, including a cation-π interaction between residues R300 and W396, a salt bridge between residues K326 and E431, and the hydrophobic interactions formed by residues F304, M324, W396, F440, and H438 in domains b′ and a′ (Fig. 5C). All these interactions are disrupted in oxidized hPDI (Fig. 5D). On the basis of the structural comparison, one explanation for this redox-regulated large interdomain conformational change in the b′xa′ region is that the formation of the disulfide bond of the active site in domain a′ (Cys 397 and Cys 400) causes the rotation of the side chain of W396, which breaks the cation-π interaction between residue R300 in domain b′ and residue W396 in domain a′ (Fig. 5E). The effect of the breakage of the cation-π interaction may be further amplified and may induce the disruption of other contacts between the domains a′ and b′, thus leading to the separation of the two domains. The mutation of W396 or R300 did significantly impair the redox-regulated conformational changes of hPDI (35), supporting the hypothesis that these two residues play a key role in these conformational changes.
Discussion
Oxidative protein folding is a synergistic process that includes both the proper folding and the formation of native disulfide bonds of nascent polypeptides, which are both essentially catalyzed by hPDI. A half century after its discovery, the crystal structures of hPDI with all four thioredoxin domains (abb′xa′) are now presented in this article in both the reduced and oxidized forms, which provide great mechanistic insights into the redox-regulated conformational changes and the related target-recognition mode of hPDI.
The structures of hPDI are different from the structure of yPDI published earlier (32). First, the positions of the a and a′ domains in relation to the central bb′ base are obviously different in yPDI and hPDI (Figs. 1D and 2B). Furthermore, the contacts between domains a and b and those between domains b′ and a′ are also distinct (Fig. 5vs. Supplementary Fig. S4). These differences may contribute to the opposite internal flexibility of hPDI (34) and yPDI (31). Moreover, hPDI has a larger and deeper hydrophobic pocket embedded in domain b′ and a more extensive hydrophobic patch in domain b than yPDI (Supplementary Fig. S5), which may provide an explanation for the observation that hPDI shows intrinsic chaperone activity in assisting the folding of denatured proteins without disulfide bonds (5), whereas yPDI does not (17).
Superimposition of hPDI structures in different redox states revealed a large redox-dependent interdomain rotation, which happens basically in the b′xa′ region probably due to two reasons. First, we observed extensive interactions between domain b′ and the regions around the active site in domain a′ in reduced hPDI. Thus, oxidation of this active site can directly modulate the interactions between the two domains. In contrast, almost no interaction exists between domain b and the regions around the active site in domain a. Second, the x linker significantly improves the conformational plasticity of the b′xa′ region. Previous work showed that this linker can adopt alternative conformations (27, 34), which are mediated by the a′ domain (34). Superimposition of the structures of reduced and oxidized hPDI revealed an ∼45° rotation of the C-terminal half of the x linker, which underpins the large interdomain conformational change in the b′xa′ region.
Significant alterations between the hPDI structures in different redox states further revealed the redox-regulated versatile target-binding capacities of hPDI. In reduced hPDI, the open side of the cleft formed by the four thioredoxin domains for accommodating the substrate is narrow (∼15 Å), and the volume of the cleft is ∼6816 Å3 (Supplementary Fig. S2A), which is estimated to be sufficient to accommodate a small folded protein of ∼50 residues. However, in oxidized hPDI, the open side of the cleft becomes wider (∼30 Å), and the volume of the cleft is also much larger, ∼14453 Å3 (Supplementary Fig. S2B), which may accommodate a larger protein of ∼100 residues. Furthermore, in reduced hPDI, the active site and the surrounding hydrophobic patch in domain a′ pack with the hydrophobic pocket in domain b′ to form a narrow tunnel, which is capable of tightly clamping small targets (Supplementary Fig. S3). In contrast, in oxidized hPDI, all these structural segments are fully exposed, providing the potential for binding to large targets (Fig. 4D). In bacteria, the pathways for oxidation and for reduction/isomerization are separated and catalyzed by DsbA and DsbC, respectively (9). In eukaryotes, the two pathways converge on PDI; therefore, not only oxidized but also reduced PDI is necessary for oxidative protein folding (15). Moreover, a balance of oxidized and reduced PDI determines ER redox homeostasis by directly modulating the activity of Ero1, a flavoprotein oxidase (3, 20). The diverse target-recognition models of hPDI in different redox states presented in this article shed light on the different functions of oxidized and reduced hPDI.
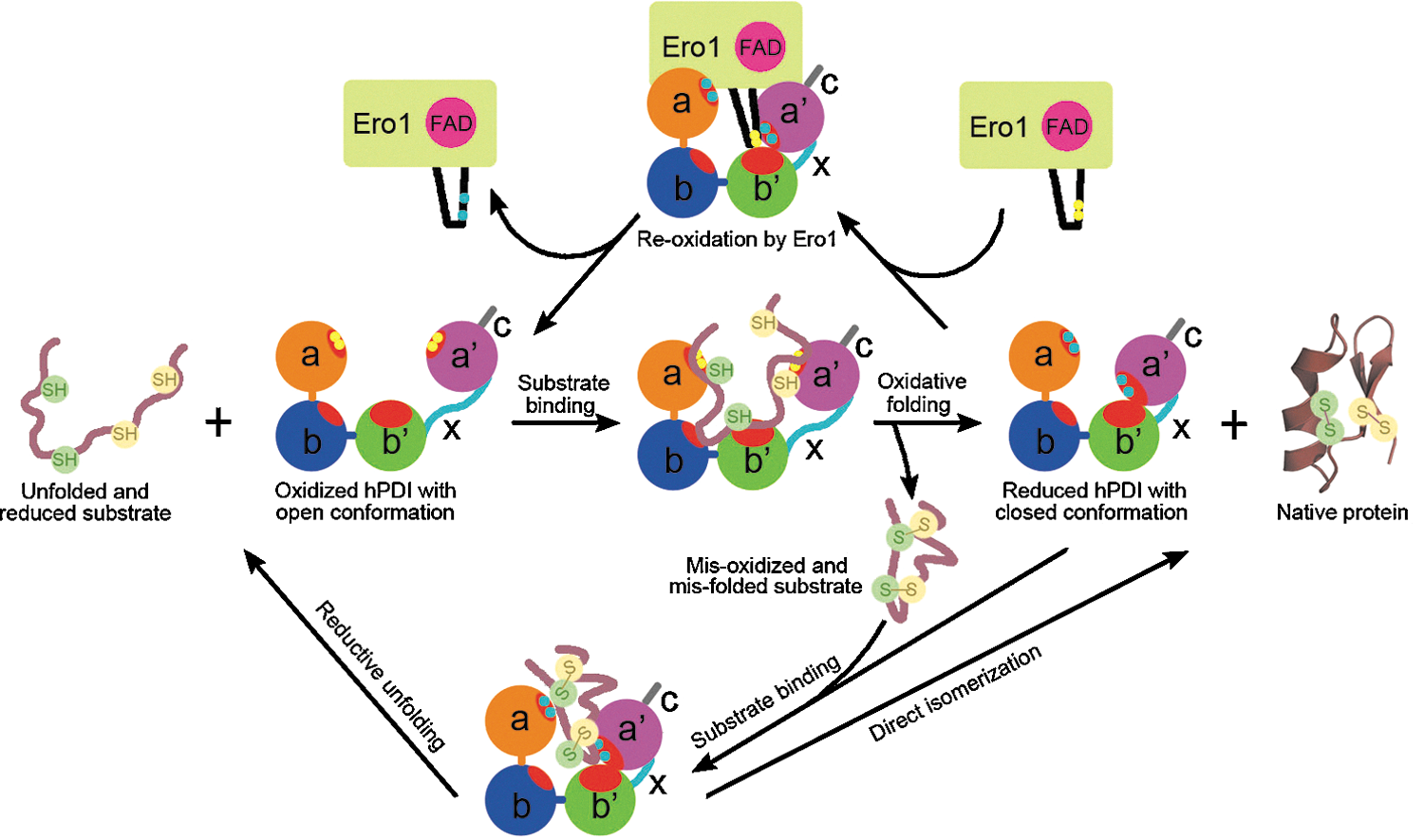
A general mechanistic model for hPDI-catalyzed reactions is proposed in Fig. 6. The oxidized hPDI with open conformation and extensively exposed hydrophobic binding sites binds with reduced/unfolded substrates, transfers oxidizing equivalents to make disulfide bonds in the substrate, and thereby switches to being reduced, which induces conformational changes, limits the binding sites, and expels the oxidized/folded protein product. The reduced hPDI with closed conformation may be reoxidized by interaction with the extruded active site of Ero1 (30, 36). It may also capture specific exposed hydrophobic regions of misoxidized/misfolded substrates, catalyzing a direct isomerization to produce a native protein or a reductive unfolding reaction to make the trapped folding intermediate re-enter the oxidative folding cycle.
Schematic model of hPDI functional cycle. The hydrophobic areas of hPDI are colored in red. The cysteine residues of the active sites in hPDI and the outer active site (Cys 94 and Cys 99) in Ero1 (30) are indicated by yellow circles for the oxidized form and cyan circles for the reduced form. The oxidized hPDI adopts an open conformation with a large substrate-accommodating cleft and extensive hydrophobic areas, which preferentially bind with the unfolded/reduced substrate to initiate oxidative protein folding. When transferring the oxidative equivalents to make disulfide bonds in the substrate, hPDI is concomitantly reduced and switched to a closed conformation, which induces the release of a native protein or a misoxidized folding intermediate in some cases. The latter may expose some hydrophobic regions, which can be captured by reduced hPDI with a closed conformation to start a direct isomerization or a reductive unfolding reaction. The reduced substrate can re-enter the oxidative folding cycle to reach its native structure, and the resulting reduced hPDI can be reoxidized by oxidase Ero1 through the interactions between the active site in the a′ domain (36) and the extruded active site of Ero1 (30) to regain the open conformation.
Materials and Methods
Plasmid construction, protein purification, crystallization, and data collection
The gene encoding hPDI without the C-terminal tail and signal sequence (residues from 18–479) was cloned into modified pET32a, and the resulting protein contains an N-terminal MH6SSGLEVLFQGPGS tag. The hPDI protein purified as described previously (24) in 50 mM Tris-HCl buffer (pH 8.0) containing 0.1 M NaCl and 1 mM EDTA was concentrated to 10 mg/ml for crystallization at 16°C using the hanging-drop vapor diffusion method. Drops in a ratio of 1 μl protein—in the presence or absence of 50 mM DTT (for reduced or oxidized hPDI, respectively)—to 1 μl well solution were equilibrated against 200 μl of 25% (w/v) PEG 3350 in 0.1 M Bis–Tris–HCl buffer (pH 9.8) containing 0.2 M ammonium acetate. Crystals were flash-cooled in liquid nitrogen. The diffraction data were collected at the Shanghai Synchrotron Radiation Facility beamline BL17U at a wavelength of 0.979 at 100 K. Data were indexed, integrated, and scaled using the HKL-2000 program package (28). Data collection statistics are listed in Table 1.
Structure determination and refinement
Structures of reduced and oxidized hPDI were solved by the molecular replacement method with the program Phaser (26). For reduced hPDI, the structure of hPDI bb′xa′ (35) was used as the search model. For oxidized hPDI, the structures of domains ab, domain b′, and domain a′ of reduced hPDI were used as the search models. Additional residues were manually modeled in the program coot (11). Refinement was performed using the program Phenix (1). The structural coordinates have been submitted to PDB with code 4EKZ for reduced hPDI and code 4EL1 for oxidized hPDI.
Footnotes
Acknowledgments
We thank the BL17U beamline of the Shanghai Synchrotron Radiation Facility for the X-ray beamline time, Mr. Feng Gao for the diffraction data collection, and Dr. Jiang Yu for helpful discussion. This work was supported by the National Major Basic Research Program of China (2011CB910303, 2012CB911002, and 2011CB910503), the National Natural Science Foundation of China (31070657 and 31190062), and the Knowledge Innovation Program of the Chinese Academy of Sciences (KSCX2-YW-R-154 and KSCX2-EW-J-3).
Author Disclosure Statement
The authors declare that they have no competing financial interests.
Abbreviations Used
References
1.
AdamsPD, Grosse-KunstleveRW, HungLW, IoergerTR, McCoyAJ, MoriartyNW, ReadRJ, SacchettiniJC, SauterNK, TerwilligerTC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr, 58:1948–1954. 2002.
2.
AlanenHI, SaloKE, PekkalaM, SiekkinenHM, PirneskoskiA, RuddockLW. Defining the domain boundaries of the human protein disulfide isomerases. Antioxid Redox Signal, 5:367–374. 2003.
3.
Appenzeller-HerzogC, RiemerJ, ZitoE, ChinKT, RonD, SpiessM, EllgaardL. Disulphide production by Ero1alpha-PDI relay is rapid and effectively regulated. EMBO J, 29:3318–3329. 2010.
4.
BenhamAM. The protein disulfide isomerase family: key players in health and disease. Antioxid Redox Signal, 16:781–789. 2012.
5.
CaiH, WangCC, TsouCL. Chaperone-like activity of protein disulfide isomerase in the refolding of a protein with no disulfide bonds. J Biol Chem, 269:24550–24552. 1994.
6.
DarbyNJ, CreightonTE. Functional properties of the individual thioredoxin-like domains of protein disulfide isomerase. Biochemistry, 34:11725–11735. 1995.
7.
DavisIW, Leaver-FayA, ChenVB, BlockJN, KapralGJ, WangX, MurrayLW, ArendallWB, SnoeyinkJ, RichardsonJS, RichardsonDC. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res, 35:W375–W383. 2007.
8.
DenisovAY, MaattanenP, DabrowskiC, KozlovG, ThomasDY, GehringK. Solution structure of the bb′ domains of human protein disulfide isomerase. FEBS J, 276:1440–1449. 2009.
9.
DepuydtM, MessensJ, ColletJF. How proteins form disulfide bonds. Antioxid Redox Signal, 15:49–66. 2011.
10.
EisenbergD, SchwarzE, KomaromyM, WallR. Analysis of membrane and surface protein sequences with the hydrophobic moment plot. J Mol Biol, 179:125–142. 1984.
11.
EmsleyP, CowtanK. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr, 60:2126–2132. 2004.
12.
FreedmanRB, GanePJ, HawkinsHC, HlodanR, McLaughlinSH, ParryJW. Experimental and theoretical analyses of the domain architecture of mammalian protein disulphide-isomerase. Biol Chem, 379:321–328. 1998.
13.
FreedmanRB, KlappaP, RuddockLW. Protein disulfide isomerases exploit synergy between catalytic and specific binding domains. EMBO Rep, 3:136–140. 2002.
14.
GoldbergerRF, EpsteinCJ, AnfinsenCB. Acceleration of reactivation of reduced bovine pancreatic ribonuclease by a microsomal system from rat liver. J Biol Chem, 238:628–635. 1963.
15.
HatahetF, RuddockLW. Protein disulfide isomerase: a critical evaluation of its function in disulfide bond formation. Antioxid Redox Signal, 11:2807–2850. 2009.
16.
HerasB, EdelingMA, SchirraHJ, RainaS, MartinJL. Crystal structures of the DsbG disulfide isomerase reveal an unstable disulfide. Proc Natl Acad Sci U S A, 101:8876–8881. 2004.
17.
KatiyarS, TillEA, LennarzWJ. Studies on the function of yeast protein disulfide isomerase in renaturation of proteins. Biochim Biophys Acta, 1548:47–56. 2001.
18.
KemminkJ, DarbyNJ, DijkstraK, NilgesM, CreightonTE. Structure determination of the N-terminal thioredoxin-like domain of protein disulfide isomerase using multidimensional heteronuclear 13C/15N NMR spectroscopy. Biochemistry, 35:7684–7691. 1996.
19.
KemminkJ, DijkstraK, MarianiM, ScheekRM, PenkaE, NilgesM, DarbyNJ. The structure in solution of the b domain of protein disulfide isomerase. J Biomol NMR, 13:357–368. 1999.
20.
KimS, SiderisDP, SevierCS, KaiserCA. Balanced Ero1 activation and inactivation establishes ER redox homeostasis. J Cell Biol, 196:713–725. 2012.
21.
KimuraT, HosodaY, KitamuraY, NakamuraH, HoribeT, KikuchiM. Functional differences between human and yeast protein disulfide isomerase family proteins. Biochem Biophys Res Commun, 320:359–365. 2004.
22.
KlappaP, RuddockLW, DarbyNJ, FreedmanRB. The b′ domain provides the principal peptide-binding site of protein disulfide isomerase but all domains contribute to binding of misfolded proteins. EMBO J, 17:927–935. 1998.
23.
KoivunenP, PirneskoskiA, KarvonenP, LjungJ, HelaakoskiT, NotbohmH, KivirikkoKI. The acidic C-terminal domain of protein disulfide isomerase is not critical for the enzyme subunit function or for the chaperone or disulfide isomerase activities of the polypeptide. EMBO J, 18:65–74. 1999.
24.
LiSJ, HongXG, ShiYY, LiH, WangCC. Annular arrangement and collaborative actions of four domains of protein-disulfide isomerase: a small angle X-ray scattering study in solution. J Biol Chem, 281:6581–6588. 2006.
25.
MaattanenP, KozlovG, GehringK, ThomasDY. ERp57 and PDI: multifunctional protein disulfide isomerases with similar domain architectures but differing substrate-partner associations. Biochem Cell Biol, 84:881–889. 2006.
26.
MurshudovGN, VaginAA, DodsonEJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr, 53:240–255. 1997.
27.
NguyenVD, WallisK, HowardMJ, HaapalainenAM, SaloKE, SaaranenMJ, SidhuA, WierengaRK, FreedmanRB, RuddockLW, WilliamsonRA. Alternative conformations of the x region of human protein disulphide-isomerase modulate exposure of the substrate binding b′ domain. J Mol Biol, 383:1144–1155. 2008.
28.
OtwinowskiZ, MinorW. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol, 276:307–326. 1997.
29.
PirneskoskiA, KlappaP, LobellM, WilliamsonRA, ByrneL, AlanenHI, SaloKE, KivirikkoKI, FreedmanRB, RuddockLW. Molecular characterization of the principal substrate binding site of the ubiquitous folding catalyst protein disulfide isomerase. J Biol Chem, 279:10374–10381. 2004.
30.
SevierCS, KaiserCA. Ero1 and redox homeostasis in the endoplasmic reticulum. Biochim Biophys Acta, 1783:549–556. 2008.
31.
TianG, KoberFX, LewandrowskiU, SickmannA, LennarzWJ, SchindelinH. The catalytic activity of protein disulfide isomerase requires a conformationally flexible molecule. J Biol Chem, 238:33630–33640. 2008.
32.
TianG, XiangS, NoivaR, LennarzWJ, SchindelinH. The crystal structure of yeast protein disulfide isomerase suggests cooperativity between its active sites. Cell, 124:61–73. 2006.
33.
WallisAK, FreedmanRB. Assisting oxidative protein folding: how do protein disulphide-isomerases couple conformational and chemical processes in protein folding?Top Curr Chem, 2012[Epub ahead of print]DOI:10.1007/128_2011_171.
34.
WangC, ChenS, WangX, WangL, WallisAK, FreedmanRB, WangCC. Plasticity of human protein disulfide isomerase: evidence for mobility around the x-linker region and its functional significance. J Biol Chem, 285:26788–26797. 2010.
35.
WangC, YuJ, HuoL, WangL, FengW, WangCC. Human Protein-disulfide Isomerase Is a Redox-regulated Chaperone Activated by Oxidation of Domain a′J Biol Chem, 287:1139–1149. 2012.
36.
WangL, LiSJ, SidhuA, ZhuL, LiangY, FreedmanRB, WangCC. Reconstitution of human Ero1-Lalpha/protein-disulfide isomerase oxidative folding pathway in vitro. Position-dependent differences in role between the a and a′ domains of protein-disulfide isomerase. J Biol Chem, 284:199–206. 2009.
37.
YaoY, ZhouY, WangC. Both the isomerase and chaperone activities of protein disulfide isomerase are required for the reactivation of reduced and denatured acidic phospholipase A2. EMBO J, 16:651–658. 1997.
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.