
Abstract
Introduction
Cell death occurs mainly by apoptosis and necrosis, pathways that differ functionally and mechanistically. The critical role of mitochondria in the intrinsic apoptotic pathway is well documented (89, 172, 184) and entails changes in respiratory capacity and mitochondrial membrane potential, as well as increased mitochondrial permeability transition (95). Necrosis, on the other hand, is usually triggered by infection, trauma, or toxins (114), and is associated with major ultrastructural abnormalities of mitochondria (95). Generally, apoptosis may occur with low or moderate, but lethal oxidative stimuli, whereas necrosis would result from severe oxidative challenges that overcome the cellular antioxidant defenses and energy-transducing pathways (100). The intracellular ATP levels constitute a critical signal directing the cells towards either type of cell death (203), because apoptosis requires energy in the form of ATP to assemble the apoptotic machinery (104, 151), which is dissipated during necrosis due to depletion of energy stores and damage of energy-transducing capacity in mitochondria.
Mitochondrial thiols that maintain redox reactions mainly include GSH and thioredoxin-2 (Trx2) and the associated enzymes—glutathione reductase (GR) and thioredoxin reductase (TrxR)—supporting the activities of glutathione peroxidase, glutaredoxin-2 (Grx2), and peroxiredoxin-3 (Prx3) and −5 (Prx5) (34, 70, 148). These mitochondrial thiols have been shown to influence the cellular death pathway (49, 175). Under physiological conditions, the mitochondrial thiol-based antioxidant systems maintain steady-state levels of H2O2 and an adequate cell's redox status, thereby preventing cell death by the pathways mentioned above. Hence, the mitochondrial thiol state is a critical mediator of metabolic-, signaling-, and cell death-related processes. Thiol groups in proteins play an important role in redox signaling by shuffling between oxidized and reduced states (41).
The Mitochondrial GSH Pool and Redox Status
GSH, synthesized by two ATP-dependent steps involving γ glutamylcysteine synthetase and GSH synthase (128), is found in two major pools in cytosol and mitochondria: the latter is the most abundant thiol in mitochondria and acts as a cofactor for glutathione peroxidase, glutathione-S-transferases, and sulfiredoxins (110, 150). Cellular viability and redox status are controlled in part by GSH (45), which plays a dual role by participating in the reduction of peroxides and acting as an nucleophile upon conversion of electrophilic centers to thioether bonds (147). GSH is imported from the cytosol via transporters in the outer and inner mitochondrial membranes (54): dicarboxylate- and 2-oxoglutarate carriers in the inner mitochondrial membrane were identified first in kidney (25, 26, 97, 113) and then in liver (204). The involvement of GSH in redox pathways results in GSSG formation. However, at variance with cytosolic GSSG, mitochondrial GSSG cannot be exported, thus increasing mitochondria susceptibility to protein thiol oxidation and increasing the significance of systems involved in the interconversion between GSH and GSSG to maintain the redox status and provide an environment appropriate for disulfide bond formation during folding of nascent proteins (71).
The mitochondrial GSH:GSSG ratio is greater than 100:1 and is widely used as an indicator of the redox status, calculated by the Nernst equation (E hc=E 0+30 log ([GSSG]/GSH]2) (80). The redox potential of mitochondrial GSH/GSSG couple was calculated as approximately −300 mV and that of the Trx2reduced/Trx2oxidized couple as −340 mV (87). Although these redox couples are maintained independently in nonequilibrium steady state across different subcellular compartments, these values indicate a more reducing environment in mitochondria than in cytosol (−280 mV for Trx1 and −260 to −200 mV for GSH/GSSG) and in endoplasmic reticulum (−185 mV for GSH/GSSG) (58, 82).
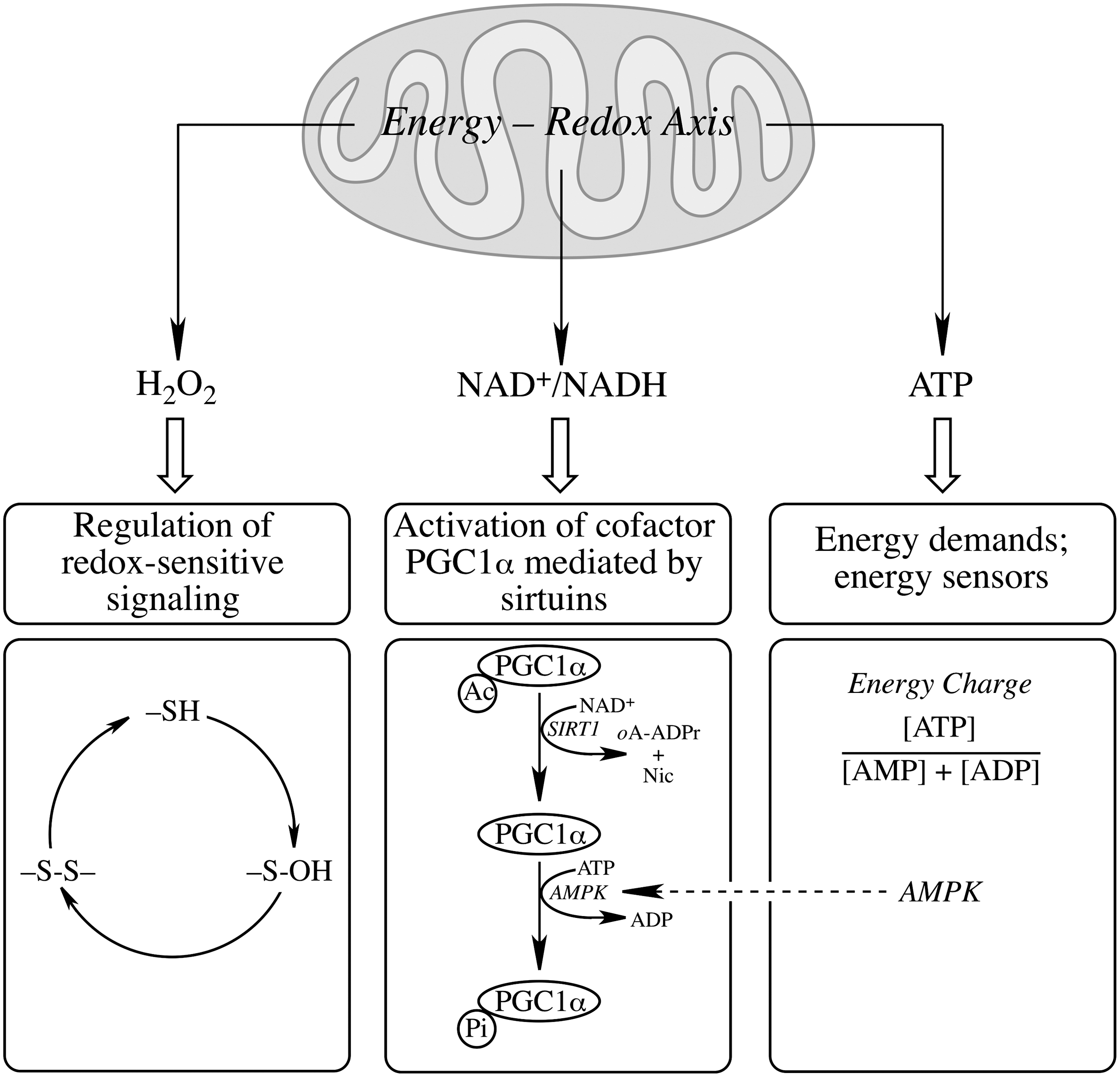
The mitochondrial redox status cannot be viewed independent of its energy-transducing capacity but integrated in a mitochondrial energy–redox axis (Fig. 2). The energy component of this axis is encompassed by the generation of reducing equivalents (NADH and FP2H2) by the tricarboxylic acid cycle (TCA) and their flow through the respiratory chain with concomitant generation of
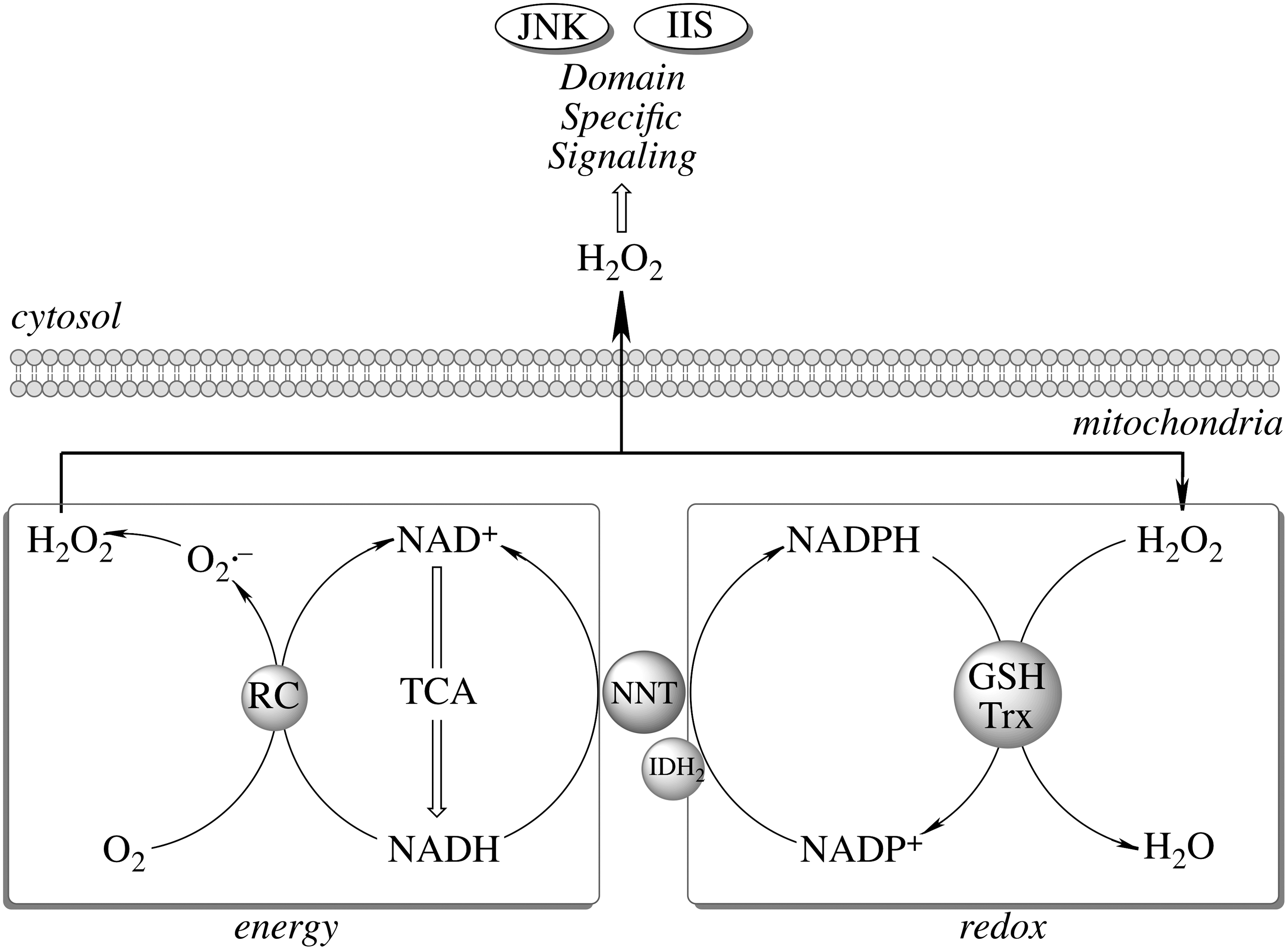
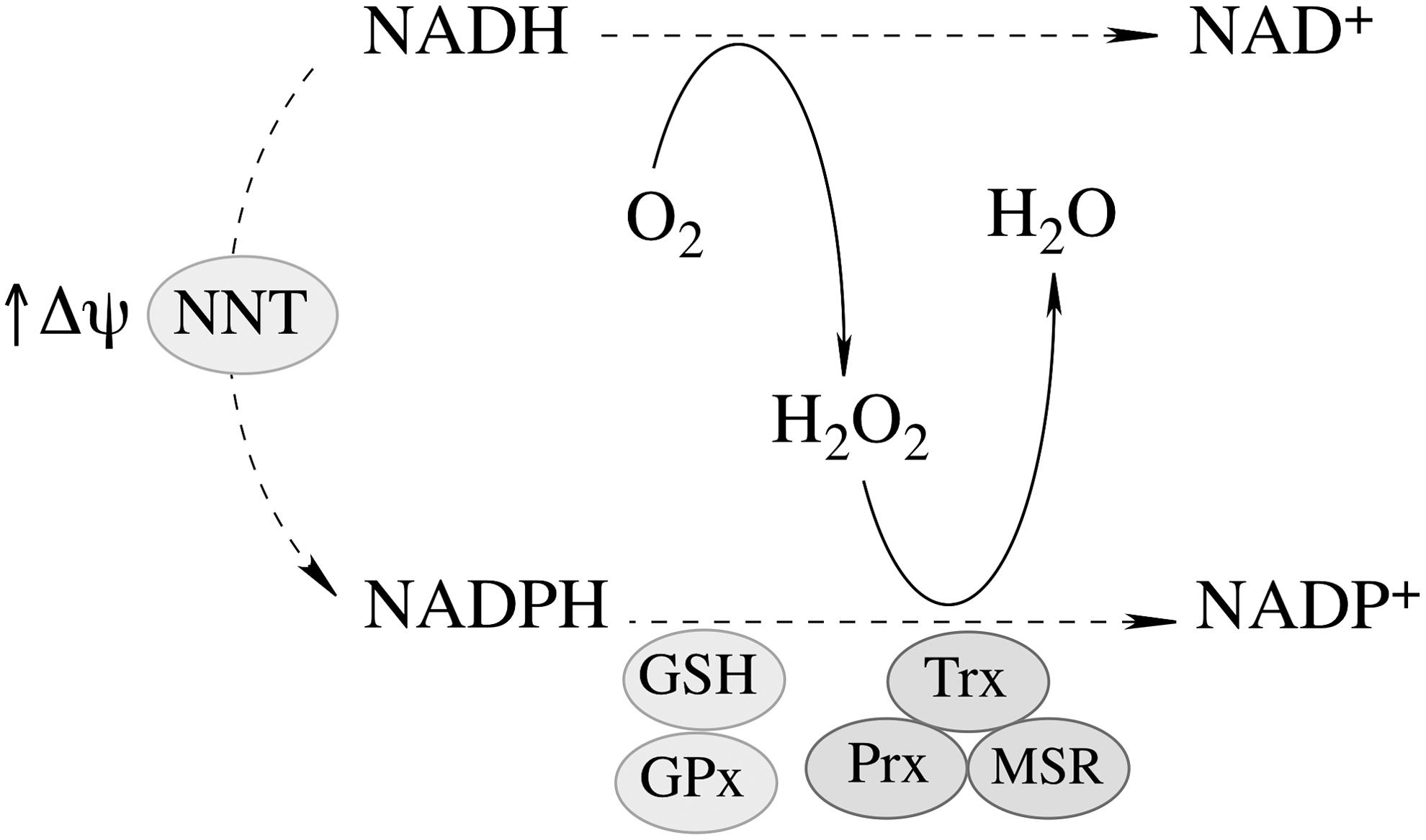
The proton gradient across the mitochondrial inner membrane strongly stimulates the forward reaction (Fig. 3), [i.e., the generation of NADPH and the subsequent H2O2 reduction (197)]. NNT plays an important role in regulating cellular redox homeostasis, energy metabolism, and apoptotic pathways (196). Knockdown of NNT in PC12 cells results in an altered redox status encompassed by decreased cellular NADPH levels and GSH/GSSG ratios and increased H2O2 levels, as well as an impaired mitochondrial energy-transducing capacity. The activation of redox-sensitive signaling (JNK) by H2O2 after NNT suppression induces mitochondrion-dependent intrinsic apoptosis and results in decreased cell viability (196). The oxidized cellular redox state and decline in bioenergetics, as a consequence of NNT knockdown, cannot be viewed as independent events, but rather as interdependent relationships coordinated by the mitochondrial energy–redox axis. Disruption of electron flux from fuel substrates to redox components due to NNT suppression induces not only mitochondrial dysfunction but also cellular disorders or cell death through redox-sensitive signaling.
Mitochondrial Protein S-Glutathionylation
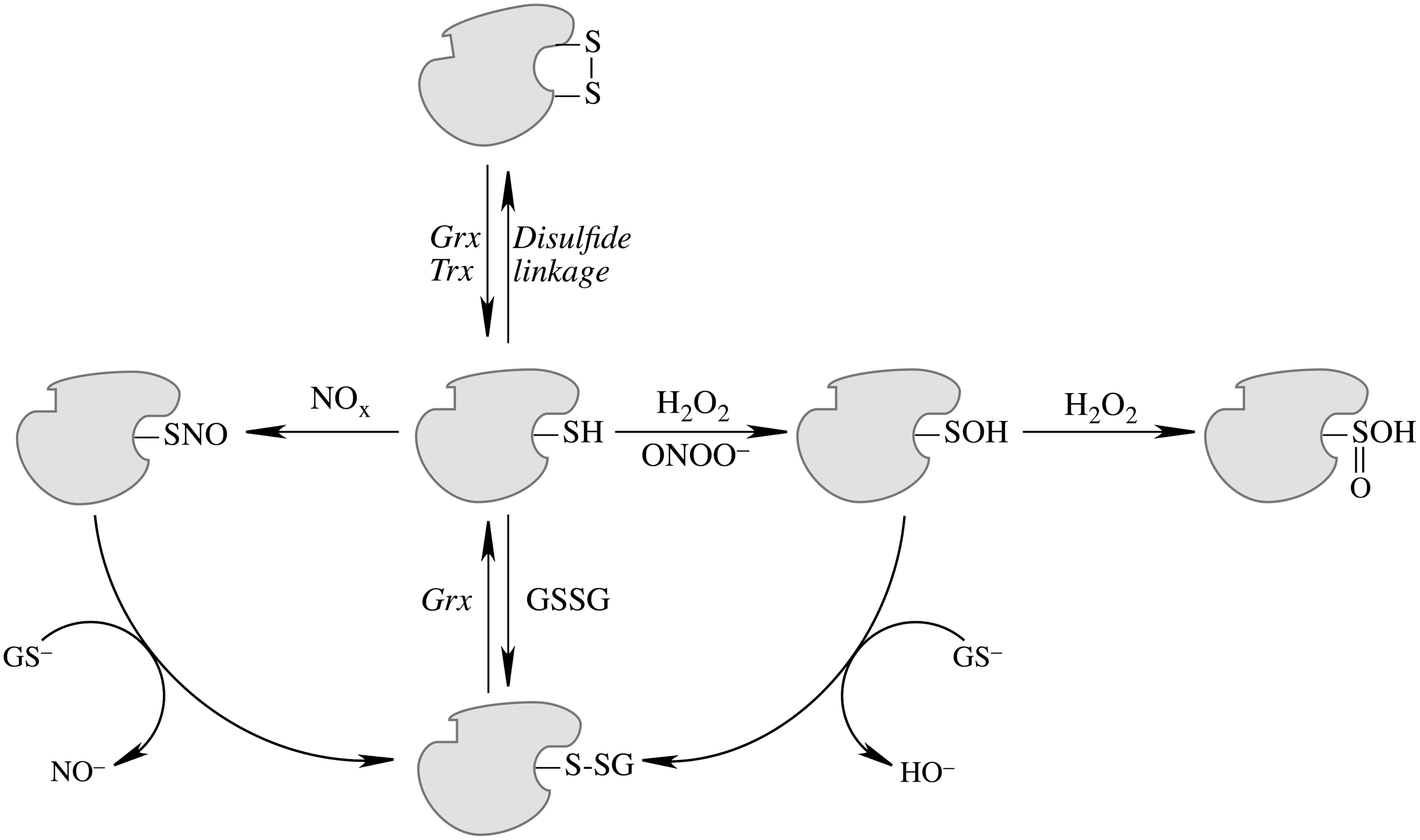
Mitochondrial generation of oxidants and free radicals is associated with reversible and irreversible modifications of target proteins (Fig. 4), mainly involving S-nitrosylation and S-glutathionylation of redox-sensitive cysteinyl residues (Fig. 5) and nitration of tyrosyl residues. The increased GSSG formation and the lack of export of GSSG from mitochondria renders these organelles more susceptible to oxidative conditions and S-glutathionylation reactions through thiol-disulfide exchange (Equation 2) that may be associated with impairment or protection of protein function. Protein mixed disulfides are also formed upon the reaction of GSH with S-nitrosylated proteins by a S-thiolation mechanism (Equation 3) and sulfenic acid intermediates (Equation 4). Protein mixed disulfides can be formed by one- or two-electron pathways: the former yields a protein thiyl radical (Equation 5) that upon conjugation with a thiol (e.g., GSH) forms a protein disulfide anion radical (Equation 6); the disulfide anion radical reduces O2 to

Regardless of the molecular mechanisms, S-glutathionylation is one of the most important protein post-translational modifications and is viewed as a regulatory device for proteins involved in energy metabolism, redox signaling, and apoptosis (40, 41, 63, 92, 116). In mitochondria, aconitase (57), α-ketoglutarate dehydrogenase (133), isocitrate dehydrogenase (90), succinyl-CoA transferase (51), and aldehyde dehydrogenase (185) can be inhibited upon glutathionylation. Electron transport chain complexes I (170), II (24), and V (51,186) are also sensitive to glutathionylation. S-glutathionylation of succinyl-CoA transferase and ATP synthase (F1 complex, α-subunit) in brain mitochondria resulted in a decrease of activity and a substantially low reduction potential (−171 mV); supplementation of mitochondria with respiratory substrates to complex I or complex II increased NADH and NADPH levels, restored GSH levels through reduction of GSSG and deglutathionylation of mitochondrial proteins, and resulted in a more reducing mitochondrial environment (−291 mV) (51). Excessive protein glutathionylation upon treatment with diamide at high concentrations resulted in bioenergetics failure and cell death; however, low diamide concentrations lead to an apparently adaptive response [i.e., increased glycolytic flux and cell viability remained unchanged (63)]. Treatment of mitochondria from human dopaminergic neuroblastoma cells with neuromelanin increased GSH and free thiol levels by releasing GSH from glutathionylated mitochondrial complex I, thereby exposing critical thiols to detrimental oxidation and subsequent mitochondrial permeability transition and apoptosis (125). These results support the notion that reversible formation of mixed disulfides could serve as a mechanism that protects critical sulfhydryls in mitochondria from further oxidation (e.g., protein sulfinic and sulfonic acids) (62, 145).
The deglutathionylation of protein mixed disulfides is the domain of Grx (116) by a monothiol mechanism (67). The oxidized form of Grx is reduced by GSH, regenerated from GSSG by NADPH-supported glutathione reductase (GR). Cytosolic Grx1 is involved in multiple cellular processes (30, 68). Mitochondrial Grx2 (52) is about 1.5–3-fold more efficient than cytosolic Grx1 in protein de-glutathionylation (102) and is strongly implicated in mitochondrial redox control. Oxidized Grx1 is exclusively reduced by GSH, whereas oxidized Grx2 can also be a substrate for TrxR (79), which enables Grx2 catalysis in a wide range of GSH/GSSG values and conditions of oxidative stress (15).
The role of Grx2 function in the maintenance of the mitochondrial redox status gains further significance when considering that Grx2 knockdown led to increased sensitivity to cell death (103), whereas overexpression of Grx2 decreased the susceptibility of cells to oxidants and inhibited cytochrome c release and caspase activation (48); moreover, inhibition of Grx1 by cadmium did not sensitize to oxidative damage (103). The protein level of Grx2 is less than 1/20 of that of Grx1: this emphasizes the regulatory role of Grx2 upon specific mitochondrial protein targets rather than an antioxidant itself. The cytoprotective role of Grx2 may be related to the activation of Akt signaling and involves the redox-sensitive transcription factor NF-κB and anti-apoptotic Bcl-2 (123). Human Grx2 has been characterized as an iron–sulfur center-containing component of the thioredoxin family that may serve as a redox sensor that controls the activation of Grx2 during conditions of oxidative stress (68); this expands the interaction between oxidants, mitochondrial redox status, and protein glutathionylation. Grx1 activity in the mitochondrial intermembrane space (47, 137) is involved in the regulation of complex I and VDAC activity (88), mitochondrial membrane potential, and apoptosis, and implicated in neurodegenerative diseases (155).
Mitochondrial Thiols, H2O2, and Domain-Specific Signaling
Imbalanced H2O2 regulation can shift the cell from a reduced state to an oxidized state and further induce apoptosis and/or necrosis (6). Moreover, mitochondria provide a setting for relatively high
Maintenance of mitochondrial H2O2 homeostasis is the domain of glutathione peroxidase and Prx: the latter are a family of thiol peroxidases involved in peroxide reduction. Mitochondrial Prx3 and Prx5 are involved in the enzymatic degradation of H2O2, organic hydroperoxides, and ONOO− (46, 139). Prx3 belongs to the typical 2-cysteine class of Prx (38) and is the target of up to 90% of H2O2 generated in the mitochondrial matrix with a high reaction rate (2×107 M −1·s−1) especially at low levels of H2O2 (36, 38, 148). Accordingly, overexpression of Prx-3 reduces H2O2 production and lipid peroxidation and protects cells from different inducers of apoptosis such as hypoxia, TNFα, cadmium, and oxidant-generating drugs (19, 21, 131, 188). Prx3 is overexpressed in human breast cancers (129) and, hence, it prevents apoptosis induced either by radiation therapy or cisplatin (31). Conversely, Prx3 knockdown leads to increased mitochondrial oxidant production and protein carbonyl content, altered mitochondrial morphology, and renders cells susceptible to apoptosis (19, 44, 60, 101, 120). Prx3 levels are found significantly lower in brains of Alzheimer's disease patients (91) and deficiency in Prx3 is also associated with amyotrophic lateral sclerosis (ALS), Parkinson's disease, and Down syndrome (94, 192), thus emphasizing the significance of mitochondrial Prx in neurodegenerative disorders. Mitochondrial Prx5, a 17 kDa atypical 2-Cys Prx (157), is less effective than Prx3 in reducing H2O2 but has a higher reactivity towards ONOO− (173). Overexpression of the Prx5 inhibits H2O2 accumulation, TNFα-induced JNK activation, H2O2-induced DNA damage, and p53-induced apoptosis (11, 12, 205), whereas Prx5-deficient cells show higher levels of protein and DNA oxidative damage and are more susceptible to apoptosis (44, 96, 146).
Regulation of mitochondrial Prx activity is performed at the level of gene expression and by its oxidation (73, 194). A disrupted mitochondrial redox status activates transcription of Prx3 by FOXO3a, nuclear factor erythroid 2-related factor (Nrf2), and PGC1α, and adaptively strengthens antioxidant defenses (7, 28, 134). Post-translationally, Prx3 oxidation is found as an early event during receptor-mediated apoptosis, which leads to increased mitochondrial H2O2 levels and further affects assembly of the apoptotic machinery (37).
Prxs are considered important regulators of the cellular H2O2 steady-state levels: for the typical 2-Cys Prx (Prx1-3), the peroxidatic cysteine, CysP-SH, at the redox-sensitive N-terminal, is oxidized by H2O2 to CysP-SOH, followed by reaction with a nearby resolving cysteine (CysR-SH) at the N-terminal of the other subunit to form an intermolecular disulfide and release H2O. This disulfide is then reduced by Trxs. At high concentrations of H2O2, oxidation of the intermediate Prx-Cys-SOH to Prx-Cys-SO2H (sulfinic acid form) results in loss of peroxidase activity (35). Sulfiredoxin (Srx) can reduce the sulfinic acid form of Prx back to Prx-Cys-SOH (189). Mammalian Srx translocates to mitochondria under oxidative conditions to reduce over-oxidized mitochondrial Prx3 (130, 190) in an ATP-driven reaction followed by binding of Srx to 2-Cys Prx enzymes and release of the γ-phosphate from ATP to the sulfinic moiety and reduction of the resulting sulfinic phosphoryl ester by either mitochondrial GSH or thioredoxin (20, 74, 83, 149, 153) (Fig. 6).
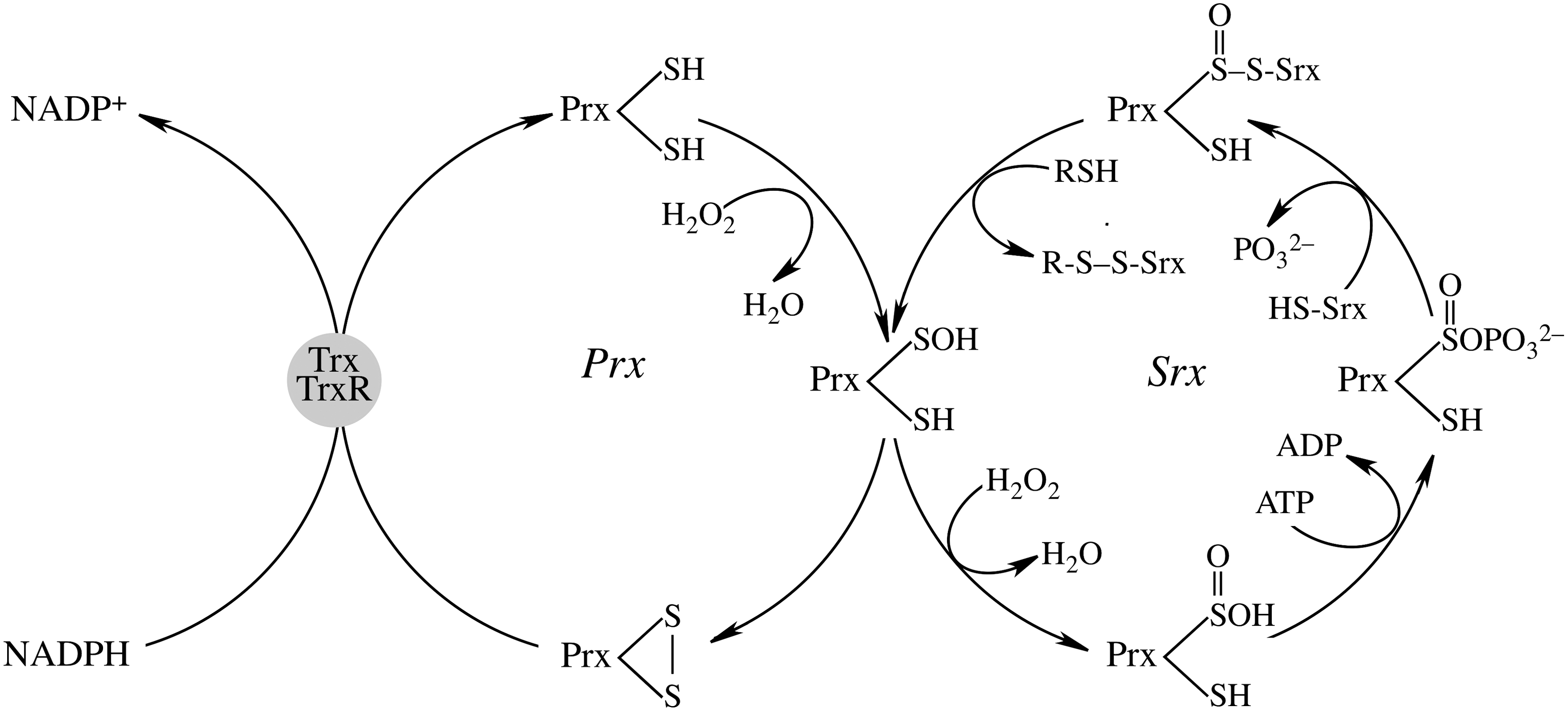
Expression of Srx is primarily regulated by the Nrf2 (10, 138, 164) along with other phase II enzymes: Trx, Prx, GPx, and MnSOD. In addition to Nrf2, the expression of Srx is also regulated by AP-1 in pancreatic β-cells (53) and in rat neurons (183). Hence, Srx expression is induced in various oxidative and nitrosative stress conditions and is seen as an adaptive and protective mechanism to avoid sustained excessive oxidant production due to hyperoxidation of Prx and its inactivation (1, 9, 10, 164). Currently, the major role of Srx is to catalyze the reversible sulfinic modification of 2-Cys Prxs but not the sulfinic acid form of other over-oxidized proteins such as GAPDH and DJ-1 (149). The biological significance of this reversible hyperoxidation of Prx enzymes is still unclear: it has been proposed that inactivation of Prx by over-oxidation results in higher levels of H2O2, which may be engaged in regulation of distinct signaling pathways (193); albeit attractive, this proposal needs to be viewed in light of the spatial considerations for H2O2 signaling (82, 178). Srx−/− mice show normal viability but an increased mortality during endotoxic shock; this may suggest a protective role of Srx through regulation of Prx function and cellular H2O2 levels (141).
The reducing power for Prx is transmitted through thiols of the Trx system: NADPH→TrxR→Trx→Prx (65, 199). Trx is highly efficient in redox reactions via thiol-disulfide exchanges (108, 118, 143), thus impacting cellular functions such as antioxidant defenses and redox control of transcription and signal transduction (8, 66). Trx is also involved in the reduction of methionine sulfoxides via methionine sulfoxide reductases (MSR). Mitochondrial Trx2 is found at its highest levels in metabolically active tissues (18, 167) and its oxidation after exposure to peroxides and diamide is an early event in oxidative stress; overexpression of Trx2 increases mitochondrial membrane potential, inhibits cytochrome c release from mitochondria (42), and protects the cells against TNFα-, diamide-, and tert-butylhydroperoxide-induced oxidation, cytotoxicity, and cell death (22, 23, 59). Trx-2-deficient cells show accumulation of intracellular oxidants, cytochrome c release, and activation of the intrinsic apoptotic pathway (169) and Trx2−/− mice show increased apoptosis in early embryos leading to embryonic lethality (132) that coincides with mitochondria maturation. This strengthens the association of mitochondrial metabolic function and oxidant regulation (i.e., the mitochondrial energy–redox axis). This is also supported by studies showing that Trx2+/− mice show reduced ATP production and electron-transport chain complexes activities (140). Knocking down TrxR leads to Trx2 oxidation and this sensitizes cells to oxidant-induced cell death (152). The mitochondrial generation of oxidants and the reducing power of TrxR2 determine the redox status of Trx2, which can be viewed as a marker of mitochondrial dysfunction and oxidant-induced cell death (81, 82, 87, 124).
Trx2 may be involved in the regulation of apoptosis through its interaction with Apoptosis Signal-regulating Kinase 1 (ASK1) (200). Upon pro-inflammatory cytokine (TNFα) or oxidative stress (H2O2) stimulation, mitochondrion-localized ASK1 disassociates from Trx2 and mediates a JNK-independent caspase-mediated apoptotic pathway (200). Overexpression of Trx2 inhibits ASK1-induced apoptosis, while knockdown of Trx2 increases TNFα/ASK1-induced cytochrome c release (156, 200). The mitochondrial permeability transition (MPT) plays significant roles in activation of apoptosis and necrosis (72, 144, 174, 198): Trx2 protects isolated mitochondria from MPT induced by peroxide or Ca++ (61) and Trx2-deficient cells show lower mitochondrial membrane potential (182). Whether Trx2 regulates the MPT by either interacting with MPT pore or modulating the mitochondrial oxidant levels is still unclear (61). Mitochondrial GSH also plays an important role in maintaining mitochondrial inner membrane permeability (111). The association of Trx2 with cytochrome c both in vivo and in vitro could provide a mechanism for the inhibitory role of Trx2 in apoptotic signaling (169). Taken together, the interactions involving Trx2, ASK1, cytochrome c, and MPT seem to be critical in the regulation of mitochondria-mediated apoptotic pathway.
Mitochondrial GSH in Cell Viability and Function
The properties and roles of mitochondrial GSH were first studied during the mid-1960s to mid-1970s (77, 78) and then advanced in the 1980s (115), showing the role of mitochondrial GSH in maintaining cell viability. Depletion of mitochondrial GSH decreased the cellular viability (158), whereas increasing mitochondrial GSH protected against oxidative and nitrosative stress (121). Increased mitochondrial GSH oxidation and decreased GSH/GSSG ratios as a function of age were accompanied by oxidative damage of the mtDNA (43, 165, 166, 181).
In the central nervous system, mitochondrial GSH depletion in astrocytes led to cell death via necrosis rather than apoptosis and decreases in mitochondrial GSH below 50% resulted in neuronal degeneration (69, 122). Expectedly, loss of mitochondrial GSH in neurons was accompanied by increase in oxidant levels, collapse of mitochondrial membrane potential, and cell death (195). The chemoprotectant 3H-1,2-dithiole-3-thione protected against oxidative and electrophilic neurotoxicity in neuroblastoma cells and primary neurons due to its ability to increase mitochondrial GSH (76). Interestingly, dopamine at nontoxic concentrations strongly increased mitochondrial GSH and afforded a greater protection against cytotoxicity (75). GSH was substantially decreased in cerebral cortex and striatum mitochondria in a model of brain focal ischemia, in which the loss in mitochondrial GSH did not correlate with minimal total GSH losses in the tissue (4). In this model, bilateral injections of GSH monoethylester—prior to induction of unilateral focal ischemia—increased mitochondrial GSH in the striatum of ischemic and nonischemic hemispheres, albeit with no reduction of infarct volume. This could be potentially used to study the effects of modulating brain mitochondrial glutathione in a range of brain disorders and warrants further research (3). The above studies establish the importance of the role of mitochondrial GSH in maintaining brain function.
In liver, TNF-α increased the susceptibility of hepatocytes after mitochondrial GSH depletion and restoration of mitochondrial GSH levels had protective effects against TNF-α (32). Decreased intracellular GSH levels markedly enhance the cytotoxicity of alkylating agents; however, it shifts the mode of cell death to necrosis rather than apoptosis. This study poses an important question as to whether raising GSH levels enables the switch from necrosis to apoptosis, thus viewing apoptosis as a more desirable cell death pathway that circumvents the destructive inflammatory response associated with necrosis (49).
Mitochondria have also been shown to undergo morphological and functional changes in chronic experimental models of alcoholism in which ethanol is oxidized to acetaldehyde in liver (168). In chronic models of alcoholism, there is a distinct mitochondrial damage characterized by abnormalities like its swelling, disruption, disorganization of the normal cristae organization, all of which finally translates into a lower energy-transducing capacity (i.e., ATP levels) (17, 39, 168). These effects stem partly from a low mitochondrial GSH pool, as a consequence of dysfunctional GSH transport into the mitochondria, which weakens binding of cytochrome c to cardiolipin in the inner mitochondrial membrane and affects membrane permeabilization (84, 110, 135). Decreased mitochondrial GSH is also linked to disrupted Ca++ homeostasis via disturbances in the pyridine nucleotide pool mainly caused by decreased mitochondrial GSH (99, 106).
The transport of GSH into mitochondria was found to be closely associated with the apoptotic machinery due to the interaction of GSH with the BH3 groove of Bcl-2; pro-apoptotic Bax and BH3-only proteins suppressed GSH transport into the mitochondria upon inhibition of GSH-Bcl-2 binding (206). Bcl-2 binding to GSH enhanced its affinity for the 2-oxoglutarate carrier on the inner mitochondrial membrane (187).
Conclusions and Perspectives
The interlaced networks of mitochondrial thiols constitute a regulatory device to maintain mitochondrial redox status and modulate cytosolic redox signaling in normal and stress conditions. Disturbances in this regulatory device can affect transcription, growth, and ultimately influences cell survival/death. Modification of sulhydryl groups on signal proteins by oxidants and their control exerted by thiol-containing molecules such as glutathione, Grx, Trx, and Prx, forms the core of redox signaling. Each of them plays a distinct role in the overall process. GSH/GSSG determines the mitochondrial redox status due to its high molecular concentration and can be seen as a “redox buffer”; Prx3 acts more in H2O2 removal and therefore affects the H2O2 signal pathway as a “redox sensor”; Trx2 acts more as a “redox transmitter” to transfer the reducing equivalents from NADPH to other thiol-molecules such as Prx3. The primary role of Grx2 in mitochondria is to control protein glutathionylation/deglutathionylation and thereby regulate functions of important mitochondrial enzymes in response to change in mitochondrial redox status. Mitochondrial thiols thereby form an intricate network that constitutes complex crosstalk involved in oxidants detoxification and maintenance of cellular and mitochondrial redox homeostasis, as well as the modulation of cytosolic redox-sensitive signaling and cell death. Emerging evidence suggests that the mitochondrial thiol/disulfide systems are critical for the progression of several pathologies (Table 1). Thus, the modulation of key mitochondrial thiol proteins, which participate in oxidative stress responses, redox signaling, maintenance of the bioenergetic machinery, and cell death programming, provides a pivotal direction in developing new therapies towards the prevention and treatment of these diseases.
The table summarizes the involvement of selective mitochondrial thiols in different disease models addressed in this review. *Glutaredoxin-1 in the inter-membrane space.
Footnotes
Acknowledgments
This study is supported by National Institutes of Health Grants R01AG016718 and P01AG026572 (to Roberta Díaz Brinton; Project 1 to EC) and Grant 17RT-0171.
Author Disclosure Statement
No competing financial interests exist.