
Abstract
Introduction
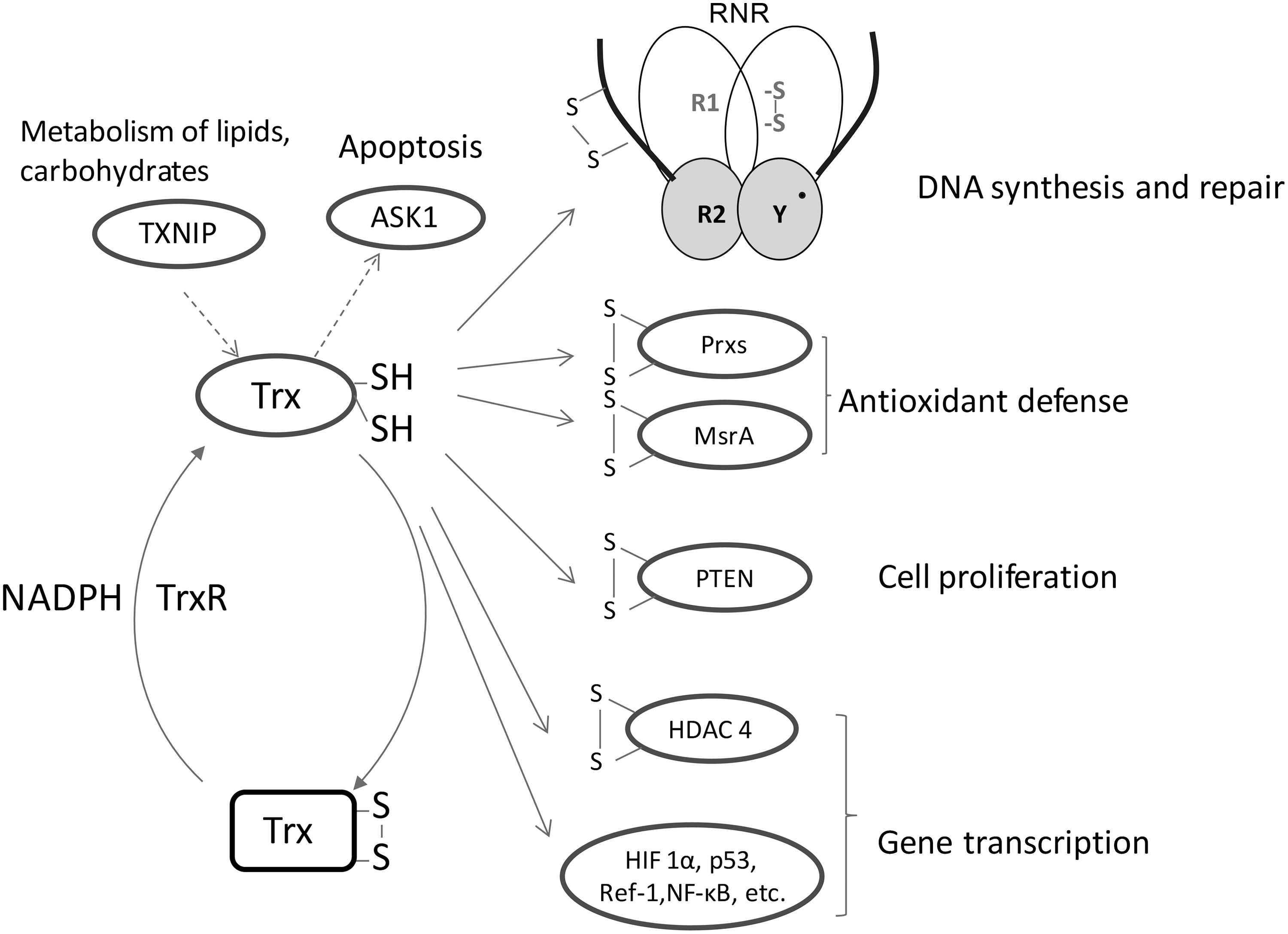
Role of Trxs in Mammalian Cell Death Progression
Mammalian Trx systems
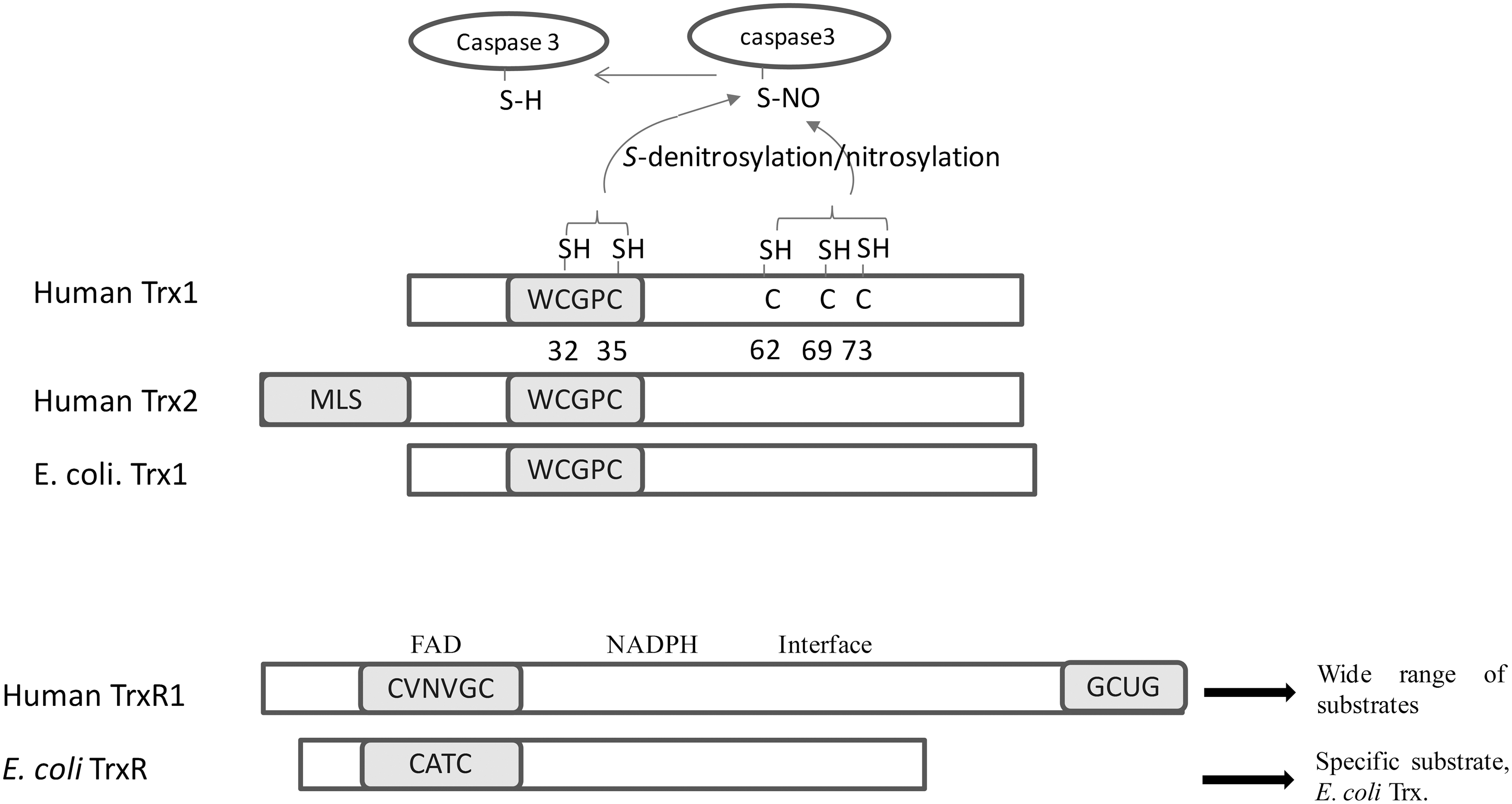
Both cytosolic Trx1 and mitochondrial Trx2 in mammalian cells contain an active site Trp-Cys-Gly-Pro-Cys- in a typic Trx-fold structure (Fig. 2) (32). Human Trx1 with 105 amino-acid residues has three structural Cys residues at positions 62, 69, and 73, apart from Cys32 and Cys35 in the active site. Cys62 and Cys69 in Trx-S2 can form a second disulfide bond under oxidative stress conditions (Fig. 2). This two-disulfide bond form of Trx-S2 is poorly characterized but cannot be reduced by TrxR directly, and its formation leads to the inactivation of Trx1 (20, 23, 40, 67). In addition, these extra structural Cys residues have been suggested to be involved in many post-translational modifications of human Trx1, such as S-nitrosylation, glutathionylation, and dimerization (70).
Regulation of cell death by Trx1
Trx1 is a central redox regulator that mediates the activation of many transcription factors which are involved in cell growth, apoptosis, and inflammation, such as NF-κB, activator protein 1 (AP-1), p53, hypoxia-inducible factor 1, and redox factor 1 (Ref-1) (25, 32). The reduction status of some Cys residues in the DNA binding site of the transcription factors, for example, Cys62 of NF-κB p50, is critical for the DNA binding. Under oxidative stress conditions, Trx1 is found to be translocated from the cytosol to the nucleus (22). The presence of Trx can maintain the cysteine in its reduced state and promote the DNA binding activity of NF-κB and AP-1 (22). Moreover, Ref-1 can also translocate from the cytosol to the nucleus, and it interacts with Trx1 physically. The association of Ref-1 with Trx1 can increase the DNA binding activity of transcription factors such as AP-1 (21).
Trx1 can prevent the cell apoptosis process by a direct association with apoptosis signal regulating kinase 1 (ASK1), a mitogen-activated protein kinase kinase kinase (MAP3K) (53). This MAP3K activates the c-Jun N-terminal kinase and p38 MAP kinase pathways and is required for tumor necrosis factor (TNF)-α-induced apoptosis. Trx1 can bind with the N-terminal noncatalytic region of ASK1. The interaction between Trx1 and ASK1 is highly dependent on the redox status of Trx1 and only reduced Trx binds. Reactive oxygen species (ROS) such as H2O2 caused by stress or cytokine treatment will lead to the oxidation of Trx either directly or via Prxs, the dissociation of Trx1 from ASK1, and the subsequent activation of ASK1 and subsequent apoptosis (Fig. 3) (53). In addition, Trx1 can induce ASK1 ubiquitination and degradation to inhibit ASK1 activity in endothelial cells. In the process, the Trx1 active site Cys32 or Cys35 is necessary and sufficient for the association of Trx1 with Cys250 in ASK1 (33, 76).
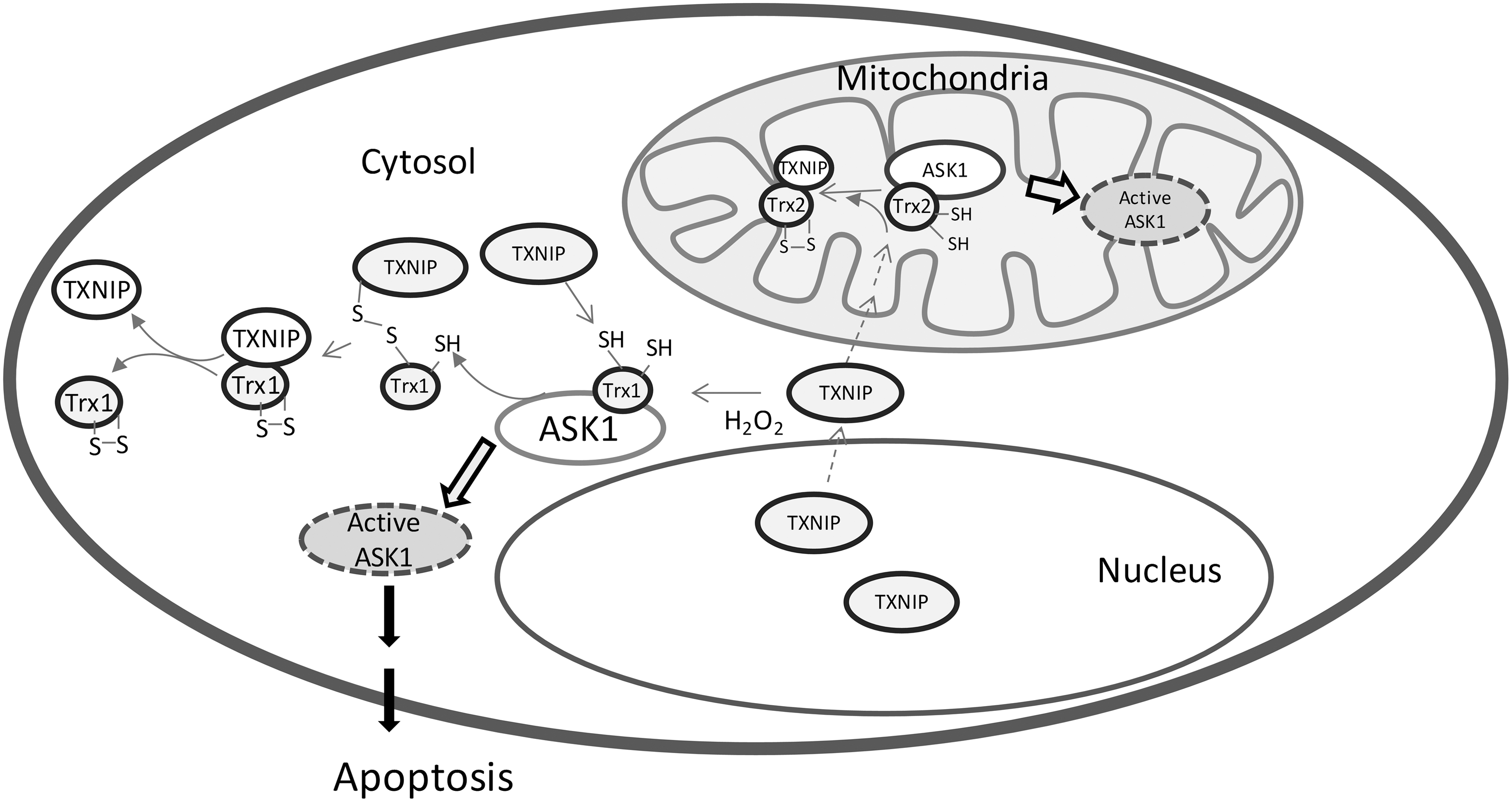
Since Trx1 exerts an anti-apoptotic function, it is not surprising that the inhibition of Trx1 induces cell death. Trx interacting protein (TXNIP, TBP2, or vitamin D3 up regulated protein 1) is an endogenous inhibitor of Trx1 that was identified in a yeast two-hybrid system (44). TXNIP forms a complex with reduced Trx1, but not oxidized Trx1. This process may involve a disulfide exchange interaction between the disulfide bond of Cys63 and Cys247 in TXNIP and reduced Trx active site dithiols (Fig. 3) (48). TXNIP, thus, binds and inhibits Trx1, and this binding results in cellular oxidative stress. [For a review on the role of Trx-TXNIP in apoptosis, see the reference (72)]. Indeed, the overexpression of TXNIP made fibroblasts, cardiomyocytes, and pancreatic β-cells more susceptible to apoptosis. Glucose-caused β-cell death has been shown to be due to the stimulation of the overexpression of TXNIP by glucose (10). However, it should be pointed out that TXNIP is a member of the α-arrestin protein superfamily, and it does not only act as the inhibitor of Trx, but also emerges as a metabolic regulatory protein (74). TXNIP plays a dual role in apoptosis in redox-dependent (29, 51) and -independent regulatory (10, 12, 73) manners, which is reviewed by Spindel et al. (58). More recently, it is reported that the binding of Trx with TXNIP can prevent the TXNIP degradation and adipocyte differentiation (14, 75).
In agreement with an unknown role of Trx1 in embryogenesis, probably involving oxidative stress defense and transcription regulation, homozygotes with a targeted disruption of the mouse TXN gene died shortly after implantation, indicating that Trx1 is essential for early differentiation and morphogenesis of the mouse embryo (41).
Regulation of cell death by Trx2
Mitochondria are considered a major site for ROS production. ROS level is a determining factor for cell death. The low level of ROS promotes the cells into apoptosis, while the high level of ROS causes cell necrosis. Along with Prx3, the Trx2 system is a major player that controls ROS levels in mitochondria and, therefore, plays a key role in regulating cell apoptosis. The deficiency of Trx2 resulted in the elevation of cellular ROS level, cytochrome c release from mitochondria, and the activation of caspase 3 and 9 in chicken DT40 (61). Very interestingly, the transfection of hTrx2 or redox-inactive hTrx2CS can rescue the Trx2-deficeint DT40 from cell death. This may be due to the fact that Trx2 maintains the Bcl-xL protein level and controls the mitochondrial outer membrane permeabilization in a redox-active, site-independent mechanism (64). Trx2 can also bind and inhibit mitochondrial-located ASK1 apoptotic activity. Cys30 in the N-terminal domain of TXNIP is critical for the binding with Trx2 (76). Recently, it has been proposed that the Trx2-ASK1 signaling pathway also involves an intracellular shuttling of TXNIP. Under normal conditions, TXNIP is located in the cytosol and also primarily in the nucleus. Mitochondrial Trx2 is bound with ASK1 and inhibits its protein kinase activity. On oxidative stress, TXNIP translocates from the nucleus to the mitochondria, competes for the binding to Trx2, and results in the apoptotic process (Fig. 3) (55). Another possible mechanism of Trx2 protection against cell death is via the regulation of p66Shc, a lifespan regulator (42). p66Shc has been revealed to be a disulfide substrate of Trxs (18). Thus, the p66Shc tetramer that is able to cause cell death can be reduced by the mitochondrial Trx system into the inactive form p66Shc dimer (18).
Mitochondrial Trx2 is essential for the normal development of the mouse embryo. Homozymous mutants embryos with the silencing TXN2 gene showed massive increased apoptosis at 10.5 days postcoitus and died in the Theiler stage 15/16 (45). The timing of the embryonic lethality coincides with the maturation of the mitochondria. The embryonic fibroblasts from Trx2-null embryos were not viable. The heterozygous mice are fertile, but theTrx2 decrease mice showed increased oxidative damage to nuclear DNA, lipid, and proteins in the liver (49).
The role of Trx2 in the regulation of apoptosis has been demonstrated by the protection of Trx2 against oxidant-induced cell death. The liver from the Trx2+/− mice has an increased apoptosis compared with wild type (WT) mice on diquat treatment (49). WT Trx2 overexpression, but not a C93S Trx2 mutant protein, can significantly inhibit TNF-α-induced apoptosis in HeLa cells (19). The redox statue of Trx2 is a key factor for many oxidant-induced cell death processes, and Trx2 get oxidized after the treatment of peroxides and diamide (11). Most recently, we found that some cationic triphenylmethanes such as brilliant green and gentian violet, the long time-used antifungal and antibacterial agents, accumulate in the mitochondria and cause the oxidation and degradation of Trx2. The disruption of the mitochondrial Trx system resulted in a subsequent release of cytochrome c and apoptosis inducing factor (AIF) from mitochondria into the cytosol. Very interestingly, HeLa cells were more sensitive to brilliant green than to fibroblasts. In HeLa cells, Trx2 down-regulation by small interfering RNA (siRNA) resulted in an increased sensitivity, whereas for normal fibroblasts, the Trx2 or Trx1 down-regulation had no effects (77).
Besides Trx1 and Trx2, the Trx family contains many other thiol-disulfide oxidoreductases with a CXXC active site such as Grxs, an endoplasmic reticulum-localized transmembrane Trx-related protein, and a macrophage migration inhibitory factor. These proteins have also been indicated as playing critical roles in the regulation of apoptosis, which have been summarized in reviews (32, 62, 72).
Role of TrxRs in Mammalian Cell Death Progression
Structure and reaction mechanism of mammalian TrxR
Three TrxRs, cytosolic TrxR1, mitochondrial TrxR2, and testis-specific Trx glutathione reductase (GR) have been found in mammalian cells, corresponding to the localization of three types of Trxs (31, 40, 59). All the three mammalian TrxRs are selenoenzymes, with a selenocysteine (Sec, U) residue in their C-terminus (4, 37). Sec is known to be the 21st genetically translated amino acid that is encoded by a UGA codon, which, in most circumstances, acts as stop codon. The Sec incorporation into the selenoprotein polypeptide chain requires a complex machinery containing the Sec insertion sequence (SECIS) element, tRNA[Ser]Sec, SECIS binding protein 2 (SBP2), and other components in mammalian cells and the availability of selenium (46). The Sec incorporation efficiency affects the activity of selenoprotein TrxR (56). The deficiency of Sec synthesis machinery can also change the existing form of TrxR (39). For example, selenium deficiency causes the Sec residue to be replaced by Cys in the rat liver TrxR (39). On the other hand, the Trx system can reduce disulfide bonds and/or glutathione (GSH) mixed disulfides in oxidized SBP2, and participate in the regulation of SBP2 compartmental location and selenoprotein synthesis efficiency (47).
TrxRs from mammalian cells possess different properties as compared with the TrxR from E. coli, yeast, or plants (Fig. 2) (69). The TrxR from mammalian cells and higher eukaryotes are large homodimmeric flavoproteins with 55 kDa or larger subunits, instead of the bacterial TrxRs, which are smaller dimeric flavoproteins with 33 kDa subunits (69). Mammalian TrxRs have a very broad range of substrates, including selenite, which could be reduced to selenide for selenoprotein synthesis (34, 36). In contrast, TrxRs in prokaryotes, yeast, and plants have a narrow substrate specificity (36).
In contrast to the single active site with a CXXC motif in the dimeric bacterial TrxR, mammalian TrxRs are a head to tail dimer and have an N-terminal active site disulfide CVNVGC as well as a conserved C-terminal sequence Gly-Cys-Sec-Gly (78). The structures of the mammalian TrxRs have been solved by X-ray crystallography (7, 13, 17, 54). The overall structure of TrxR is similar to that of GR with an identical N-terminal active site, NADPH and flavin adenine dinucleotide (FAD) binding domains (Fig. 2). However, mammalian TrxR1 uniquely contains a 16-residue C-terminal tail. Mechanisms of the action of mammalian TrxR have been proposed that electrons from NADPH are transferred to the N-terminal redox-active dithiols via FAD, then to the selenenylsulfide of the other subunit, and, subsequently, to the substrates (Fig. 4) (13, 78, 79). Crystal structures of oxidized and NADPH-reduced mouse TrxR2 have shown a similar overall structure as that of rat TrxR1 (7).

Role of TrxRs in mammalian cell death progression
To elucidate the roles of TrxR in vivo, several TrxR knock-out models have been studied (8, 15, 27). Mice with a ubiquitous inactivation of TrxR2 were dead at embryonic day 13 (15). TrxR2-deficient embryologic fibroblasts grew slower than their WT counterparts and were highly sensitive to buthionine sulfoximine treatment to block GSH synthesis (15). The inactivation of TrxR1 also led to early embryonic lethality around E9.5 (8) or E10.5 (27). Surprisingly, mice with a heart-specific inactivation of TrxR1 developed normally and appeared healthy (27), and the mice with TrxR-null hepatocyte also survived for more than 1 year (52, 60). These results indicate that TrxR1 and TrxR2 are critical in embryo development, but they are not essential for cell proliferations. The nonessential role of TrxR1 in cell proliferation is consistent with the observation that the down-regulation of TrxR1 by siRNA did not change Trx1 redox status and caused cell toxicity (66, 77). The mechanistic details are unknown as yet.
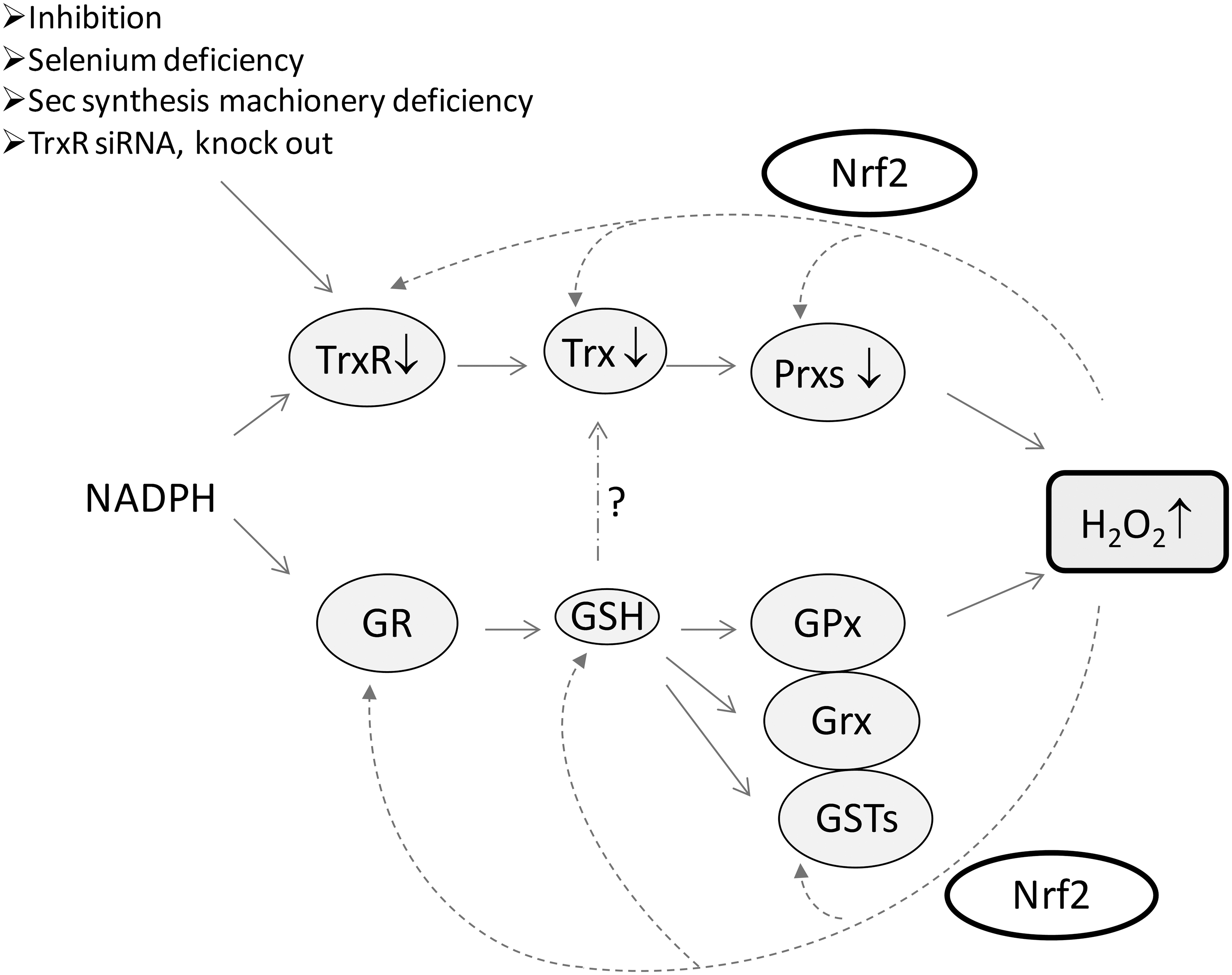
One reason that TrxR is not essential for cell proliferation may be due to the compensatory effects of another disulfide reductase system, GSH and Grx system, and this cellular response process may be regulated by the Nrf2 pathway (60). The decrease in TrxR1 activity resulting in Nrf2 activation was also observed under other conditions such as selenium deficiency (Fig. 5) (9, 39). Grxs are Trx-fold proteins with a CPYC or CGFS active site motif. In the GSH-Grx system, electrons transfer from NADPH to GR, then to GSH, and, subsequently, to Grx. The functions of Trx and GSH-Grx systems are partially overlapping (35). In particular, when TrxR is absent, Trx may get the electrons from the GSH-Grx system to maintain its reduced status (Y. Du, J. Lu, A. Holmgren, unpublished results).
Regulation of Activity in Trx System and Its Medical Applications
Mammalian TrxR as an anticancer target
Though TrxR1 is not essential for cell proliferation, TrxR emerges as a new target for anticancer drug development, because many malignant cells appear to be more dependent on an efficient Trx system. TrxR and Trx are overexpressed in aggressive cancers (5). The correlation between the Trx system and cancer hallmarks has been reviewed (5). Moreover, mouse lung carcinoma cells with TrxR stable-knockdown exhibited a similar cell morphology and anchorage-independent growth property as normal cells (71). The cells transfected with the siTrxR1 construct grew in a monolayer, whereas the control cancer cells grew in a multilayer and loosely attached to the culture dish. Growth of the TrxR1-knockdown cells in soft agar was inhibited compared with control cells (71). Growing unanchored in soft agar is a characteristic of many malignant cells (71). When these TrxR-knockdown cells were injected into mice, the tumor progression and metastasis were dramatically reduced (71). The requirement of protein disulfide reductase for tumor growth was originally reported by Apffel et al. (2), even before the identification of the enzymes to be Trx and TrxR by Holmgren (23). These results strongly suggest that TrxR is critical for cancer cell growth in vivo. In addition, a main potential substrate of the Trx system, RNR, is also a well-known anticancer target. The blockage of the Trx system activity will decrease RNR activity (43). More importantly, the inhibition of TrxR does not only decrease the activity of TrxR but also results in the elevation of ROS production and the oxidation of Trx (Fig. 5).
As a unique Sec-containing flavoprotein, TrxR is very suitable as a target for drug development. The C-terminal active site Sec makes the enzyme highly reactive with electrophilic agents due to its low pKa value and easily accessible location. Indeed, many TrxR inhibitors selectively exhibit the inhibition of TrxR against GR (3). This is also verified in vivo (65). Moreover, the inhibition of TrxR may result in a modified TrxR, which induces rapid cell death (1). Indeed, many clinically used anticancer compounds (3, 63), including alkylating and platinum-containing drugs, arsenic trioxide (35), and chemoprevention agents such as flavonoids (38) and curcumin (16), have been found to possess TrxR inhibitory effects. However, since TrxR is involved in a wide range of cell activities, the inhibition of TrxR may also cause some side effects in normal cells. Thus, further investigation that elucidates the different roles of TrxR along with Trx in normal and cancer cells and the reaction mechanism of TrxR inhibitors may facilitate and enlarge the therapeutic windows and selectivity in treatment.
Principles for some TrxR inhibition reactions
We have classified TrxR inhibitors as four types (36) (Fig. 6). The first type is metal- or metalloid-containing compounds, including gold, platinum-, mercury-, arsenic-containing compounds, some other organometallic complexes, and so on. “Hard and soft acids and bases” theory provides a useful principle for understanding the reaction mechanism between these compounds with TrxR (28). Since Se2− is a softer base than S2−, it reacts preferentially with soft bases such as gold(I), platinum(II), mercury(II), and arsenic(III). This principle may also explain why Sec-containing TrxR shows a priority to react with arsenic compounds than Cys-containing proteins. Though the metal- or metalloid-containing compounds have a high priority to attack the C-terminal Sec, the N-terminal CVNVGC active site in TrxR is also involved in the reaction (35). Zinc or lead compounds only show their inhibitory effects in the absence of EDTA in vitro. In cells or in vivo, they do not display the selective inhibition of TrxR as arsenic and mercury compounds do. The reason may be that arsenic and mercury compounds are softer acids and, thus, have a higher affinity with TrxR. For this type of inhibitors, one interesting observation is that some inhibitor has the activity to cross-link TrxR with Trx or other small proteins (50).
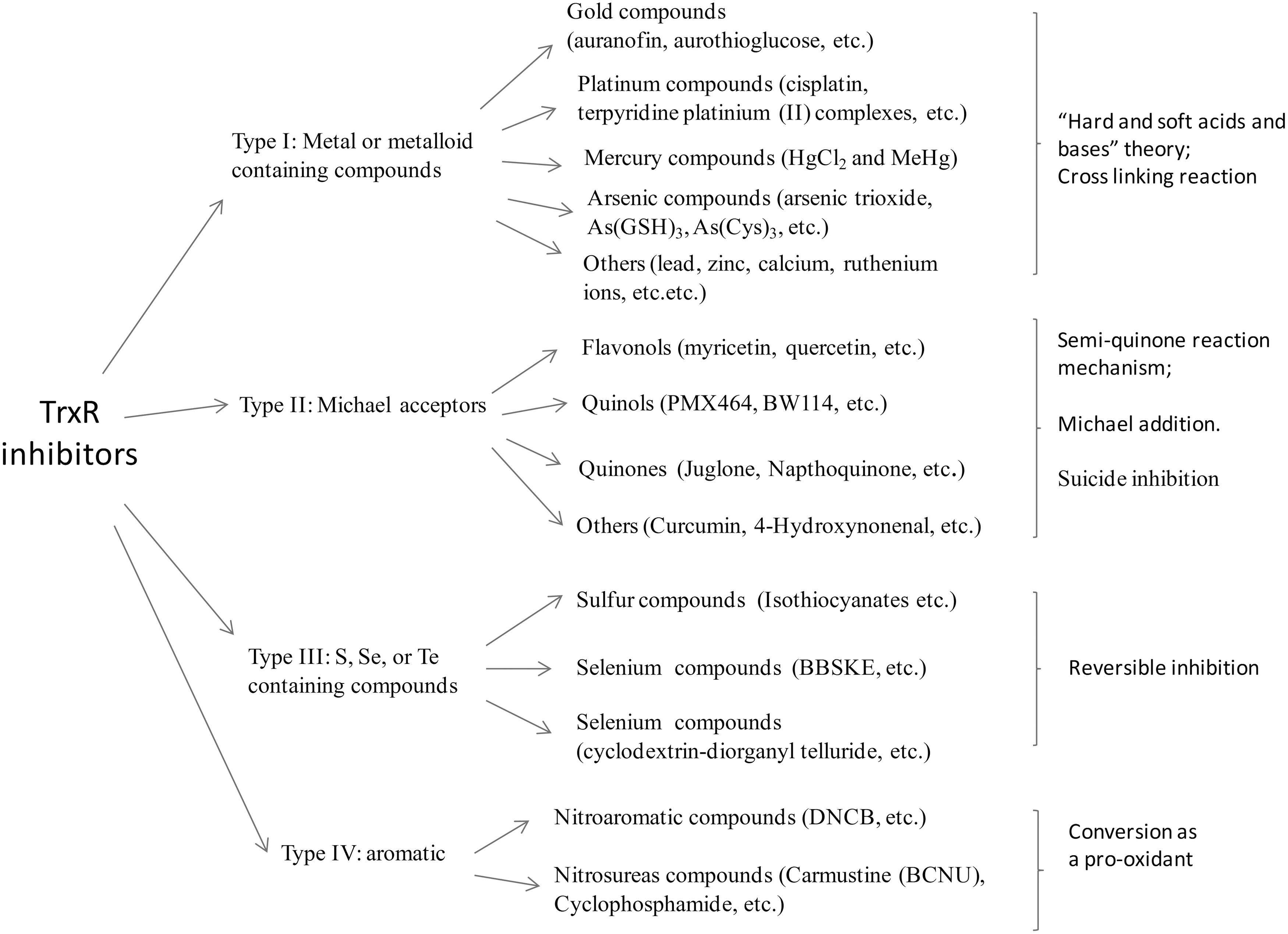
The second type of TrxR inhibitors are Michael acceptors (α, β-unsaturated carbonyl compounds), including flavonoids, quinols, quinines, and so on. The Michael acceptor curcumin has an α, β-unsaturated ketone structure that is equilibrated with its enol form. The latter is a highly active electrophilic agent that can have a Michael conjugation with the selenide of Sec in TrxR (16). The inhibition of TrxR by the flavonoids myricetin, quercetin, quinols, and quinones may involve a direct reaction between the TrxR and semiquinone (38). This may also indicate that many quinones act as “suicide inhibitors.” They can be reduced by TrxR, and, at the same time, the reduced product semiquinones inhibit TrxR.
The third type is sulfur, selenium, or telluride-containing compounds. The inhibition of TrxR by 1,2-[bis(1,2-benzisoselenazolone-3(2H)-ketone)]ethane (BBSKE) is reversible, which is different from most of the other inhibitors. The fourth type is alkylation agents. Dinitrochlorobenzene is an aromatic alkylation agent. This compound can have a covalent bonding with C-terminal Sec and also converts the antioxidant enzyme into a pro-oxidant (Fig. 4).
Concluding Remarks
In summary, the mammalian Trx system is obviously a key player in cell death for a large number of cellular activities they are involved in. The redox status of Trx is a determining factor for the cell death, which is controlled by TrxR activity and the cellular ROS level. Mammalian TrxR is a suitable anticancer target due to its unique structural properties. Various types of compounds can exert inhibitory effects on TrxR. The challenges are how to design and synthesize the compounds to possess higher selectivity related to the inhibitory efficiency and ROS production capacity via TrxR.
Footnotes
Acknowledgments
The authors acknowledge the support from the Swedish Research Council Medicine (3529), the Swedish Cancer Society (961), the K.A. Wallenberg Foundation, Åke Wiberg Stiftelse, and the Karolinska Institutet.