
Abstract
Reactive oxygen and nitrogen species are currently considered not only harmful byproducts of aerobic respiration but also critical mediators of redox signaling. The molecules and the chemical principles sustaining the network of cellular redox regulated processes are described. Special emphasis is placed on hydrogen peroxide (H2O2), now considered as acting as a second messenger, and on sulfhydryl groups, which are the direct targets of the oxidant signal. Cysteine residues of some proteins, therefore, act as sensors of redox conditions and are oxidized in a reversible reaction. In particular, the formation of sulfenic acid and disulfide, the initial steps of thiol oxidation, are described in detail. The many cell pathways involved in reactive oxygen species formation are reported. Central to redox signaling processes are the glutathione and thioredoxin systems controlling H2O2 levels and, hence, the thiol/disulfide balance. Lastly, some of the most important redox-regulated processes involving specific enzymes and organelles are described. The redox signaling area of research is rapidly expanding, and future work will examine new pathways and clarify their importance in cellular pathophysiology. Antioxid. Redox Signal. 18, 1557–1593.
III. Protein Targets Sensitive to Oxidant Species
A. Chemistry and biochemistry of thiol oxidation
IV. Control of Cellular Levels of Peroxides and the Thiol Redox Balance
V. Redox-Regulated Systems and Their Role in Signaling Pathways
A. Transcription factor OxyR: a classic example of redox signaling
B. The Gpx3/Orp1-Yap1 system in yeast. Peroxidases may act as sensors of peroxides.
D. NF-κB: the first mammalian transcription factor shown to undergo redox regulation
E. The Keap1-Nrf2-ARE pathway: redox signaling by nutritional phytochemicals
F. Hypoxia-inducible factor-1: a critical modulator of oxygen homeostasis
G. Ero1: Disulfide bond formation and maintenance of redox homeostasis in ER
I. Control of mitochondrial functions and redox regulation of permeability transition
I. Introduction
A. Cell signaling and redox signaling
For all the above reasons, at appropriately low concentrations, ROS are currently viewed as signaling molecules that are able to communicate to cells or to the subcellular micro-environment any alterations in redox conditions in order to transmit specific messages (54, 88, 103, 108, 111, 286, 287, 298, 314, 332, 372). Hormones, growth factors, neurotransmitters, and cytokines have been shown to be capable of inducing in mammalian cells transient increases in hydrogen peroxide (H2O2) acting as a second messenger and enabling the signaling event to take place. Well-recognized cases are platelet-derived growth factor (334), epidermal growth factor (EGF) (13), vascular endothelial growth factor (VEGF) (301), tumor necrosis factor (TNF)-α (239), and insulin (224). Identification of signaling processes that depend on changes in local redox conditions has led to a redefinition of the concept of oxidative stress, from a “disturbance in the prooxidant-antioxidant balance in favor of the former” (319) to a “disruption of redox signaling and control” (183). Redox signaling is, thus, a condition in which oxidant species cause a post-translational modification of proteins that transmits and amplifies the signal (308) involving mainly cysteine residues forming sulfenic acid, disulfides, nitrosothiols, or mixed disulfides with glutathione (101). This type of signaling is, thus, characterized by a reversible redox switch involving different types of proteins such as receptors, channels and transporters, transcription factors, kinases, and phosphatases, all of which are able to change their conformations and functions.
B. ROS involved in redox signaling
The main ROS produced by cells are superoxide (O2 •−), H2O2, hydroxyl radical (•OH), and singlet oxygen (1O2) (Fig. 1). Many other radicals and oxidants are formed as a consequence of the generation of the above primary species, such as lipid peroxides and other oxidized forms involving proteins and nucleic acids (146). The enzymatic formation of nitric oxide and its diverse biological functions (328) gave great support to the idea that free radicals or oxidants may act as second messengers. Nitric oxide can also give rise to several derivatives (328) that markedly contribute to redox changes in the cell. Various oxidant species have been proposed to be involved in redox signaling. Considering the different chemical features and reactivity of the above-mentioned ROS, it is essential first to examine critically which are the most probable candidates for potential signaling roles.

1. Superoxide anion
Superoxide anion is the product of the univalent reduction of dioxygen and, at physiological pH, is a negatively charged species, as the pKa of the weak acid HO2• is 4.8 (121). It, therefore, requires an anion channel to cross membranes, as occurs for the superoxide produced by NADPH oxidase in endothelial cells (153). However, in mitochondria, O2 •− is protonated by protons extruded by active respiration, and the resulting uncharged species may diffuse back into the matrix (142). In the cell, O2 •− is very rapidly dismuted to H2O2 by superoxide dismutase (SOD) at a rate constant of about 109 M −1 s−1(109). H2O2 should, therefore, be considered the true signaling molecule (113). In addition, the signaling action of O2 •− may depend on its interaction with nitric oxide and the consequent formation of peroxynitrite (251).
Superoxide anion has been shown to play a prominent role in bacteria as an activator of the transcription factor SoxR, resulting in the transcription of several enzymes against oxidative stress (270). Superoxide anion interacts with the reduced iron sulfur clusters of SoxR, which are rapidly oxidized, giving rise to a conformational change and transcription activation (270).
2. H2O2 and hydroperoxides
H2O2 is the most likely candidate to serve as a second messenger in signaling processes (128, 285). In comparison with other ROS, H2O2 is more abundant, as its concentration in tissues is estimated to be in the nanomolar to low micromolar range (60). It is also relatively stable and can cross biological membranes (29, 128, 285). Although a few studies have reported that H2O2 gradients across membranes can form and that some membranes exhibit a limited permeability to H2O2 (7, 312, 324), note should be taken of the fact that specific aquaporins can facilitate the diffusion of H2O2 through membranes, thus enabling the signaling molecule to move easily from one compartment to another (28). When acting as a second messenger, H2O2 readily interacts with a “sensor” thiol, with the production of sulfenic acid (see section III.A.1.a.).
Similar to H2O2, lipid hydroperoxides emerge to play a role in redox signaling events acting on specific sensor proteins, favored by their potential capacity for trans-bilayer, intermembrane, and intercellular movements (129). The enzymes cyclooxygenase (COX) and lipoxygenase (LOX) play an important role in the formation of hydroperoxides, which, in turn, stimulate these enzymes, thus leading to an amplification of the process (129). In particular, enhanced 12/15 LOX activity appears to be involved in cell death signaling pathways due to oxidative stress (69), and the hydroperoxides produced by this enzyme can also inactivate protein tyrosine phosphatase (PTP) (71). During these processes, a critical preventive action is exerted by reduced glutathione (GSH) and GPx4, also called phospholipid hydroperoxide glutathione peroxidase (GPx) (69) (see also section IV.A.3.).
3. Hydroxyl radical and singlet oxygen
When it interacts with biological molecules, hydroxyl radical exhibits very high rate constants, near the diffusion limit (146). In other words, since it reacts with almost any molecule it encounters, its action is highly nonspecific, and there are thus no valid reasons for including these species among the second messengers of redox signaling processes (113).
Singlet oxygen is an electronically excited form of oxygen with no spin restriction regarding its reactivity, and, hence, it can easily oxidize organic molecules, although not selectively. In cells, it may be produced in a nonenzymatic photochemical process in the presence of chemicals or drugs. It may also be produced in dark reactions, for example, the spontaneous dismutation of O2 •−, or during inflammatory processes in the reaction between H2O2 and the hypochlorite generated by myeloperoxidase (195). Singlet oxygen can attack and modify several amino acid residues such as tyrosine, methionine, histidine, and tryptophan. Cysteine residues are also oxidized beyond the disulfide or sulfenic acid state to higher oxidation states, with the formation of sulfinic, sulfonic, and thiolsulfinate residues (195), some of which are irreversible in a biochemical context (see section III). The high reactivity of singlet oxygen associated with a short lifetime may, therefore, limit its action as a signaling agent. Nevertheless, in plants, singlet oxygen, a major ROS produced in the photosystem II of chloroplasts, is one of the pathways of signaling to the nucleus (19). Lipid hydroperoxides deriving from singlet oxygen attack on unsaturated lipids can also mediate a photooxidative type of signaling (130).
C. RNS involved in signaling processes
Nitric oxide (•NO) generated by the enzyme nitric oxide synthase (NOS) is a molecule with distinct physiological functions (328). The cyclic guanosine-3′-5′-monophosphate (cGMP)-dependent pathway of •NO signaling relies on the activation of the soluble guanylate cyclase (sGC), which catalyzes the formation of the second messenger cGMP, which in turn is able to regulate vascular smooth muscle relaxation, platelet aggregation, and neurotransmission (110, 162). Other pathways, distinct from the •NO-sGC-cGMP cascade, should be considered in order to set •NO among the participants in redox signaling processes. A c-GMP-independent signaling function of •NO involves post-translational modification of protein thiols, which depends on the formation of nitrosothiol residues, largely mediating the functions of •NO on redox signal transduction pathways (110, 156, 315).
The direct reaction of nitric oxide with thiols results in the formation of a radical intermediate (RS-N•-OH), which in turn reacts with molecular oxygen in a one-electron oxidation process, yielding O2 •− and a nitrosothiol (RS-NO) (137). Catalysis by metalloproteins is another pathway of RS-NO formation. Nitric oxide can coordinate to a ferric porphyrin, forming a ferric nitrosyl (or ferrous nitrosonium) species, which, after a reaction with a nucleophilic thiol, generates a nitrosothiol in a reductive nitrosylation process (110, 212). Ferric cytochrome c was recently shown to catalyze the formation of S-nitrosoglutathione (GSNO) in the presence of •NO and GSH (49). Protein S-nitrosylation also takes place by transnitrosylation between protein cysteine residues and GSNO (49).
RS-NO may also be indirectly generated by nitrogen dioxide (•NO2) after the interaction of •NO with oxygen. As a one-electron oxidant, •NO2 may lead to the formation of a thiyl radical, which can easily combine with a •NO molecule (110) according to reaction 1:
In general, any process that is able to catalyze the formation of a thiyl radical can potentially give rise to the formation of nitrosothiols (48). Nitrogen dioxide, after combination with •NO, can generate the electrophilic species N2O3, which can then react with thiols, again forming RS-NO (110):
A very large number of proteins have been shown to undergo nitrosylation (315) (and references therein), a modification which, according to the specific enzyme protein, can lead either to activation or inhibition of activity (249, 315 and references therein). In particular, protein–protein transnitrosylation, in which the •NO residue is transferred in a process involving two or more proteins giving rise to a transnitrosylation cascade, is important for regulating cellular signaling functions (249). However, according to the redox potential of the cysteines, and other features such as low pKa of the cysteine residue and a hydrophobic environment, only specific proteins can undergo S-nitrosylation, resulting in selective activation or inhibition of well-defined signaling pathways (156, 249).
Both GSH and thioredoxin systems play a significant role in promoting denitrosylation of proteins (315). GSNO, formed by transnitrosylation between GSH and S-nitrosylated proteins (268), can be reduced by a highly specific GSNO reductase (GSNOR) that uses the reducing equivalents of NADH and produces NH3 and glutathione disulfide (GSSG) (217). The latter, in turn, can be reduced by glutathione reductase (GR) setting up a cyclic process (268). GSNOR, therefore, acts as a modulator of the cellular S-nitrosylation status based on an equilibrium involving GSNO and S-nitrosylated proteins (315). The thioredoxin system has been shown to act as a denitrosylase on a large number of substrates, including caspase-3 (23, 315). However, in specific conditions, oxidized Trx can undergo nitrosylation, acting as a transnitrosylating factor on specific proteins (315).
II. Cellular Sources of ROS
A. NADPH oxidase
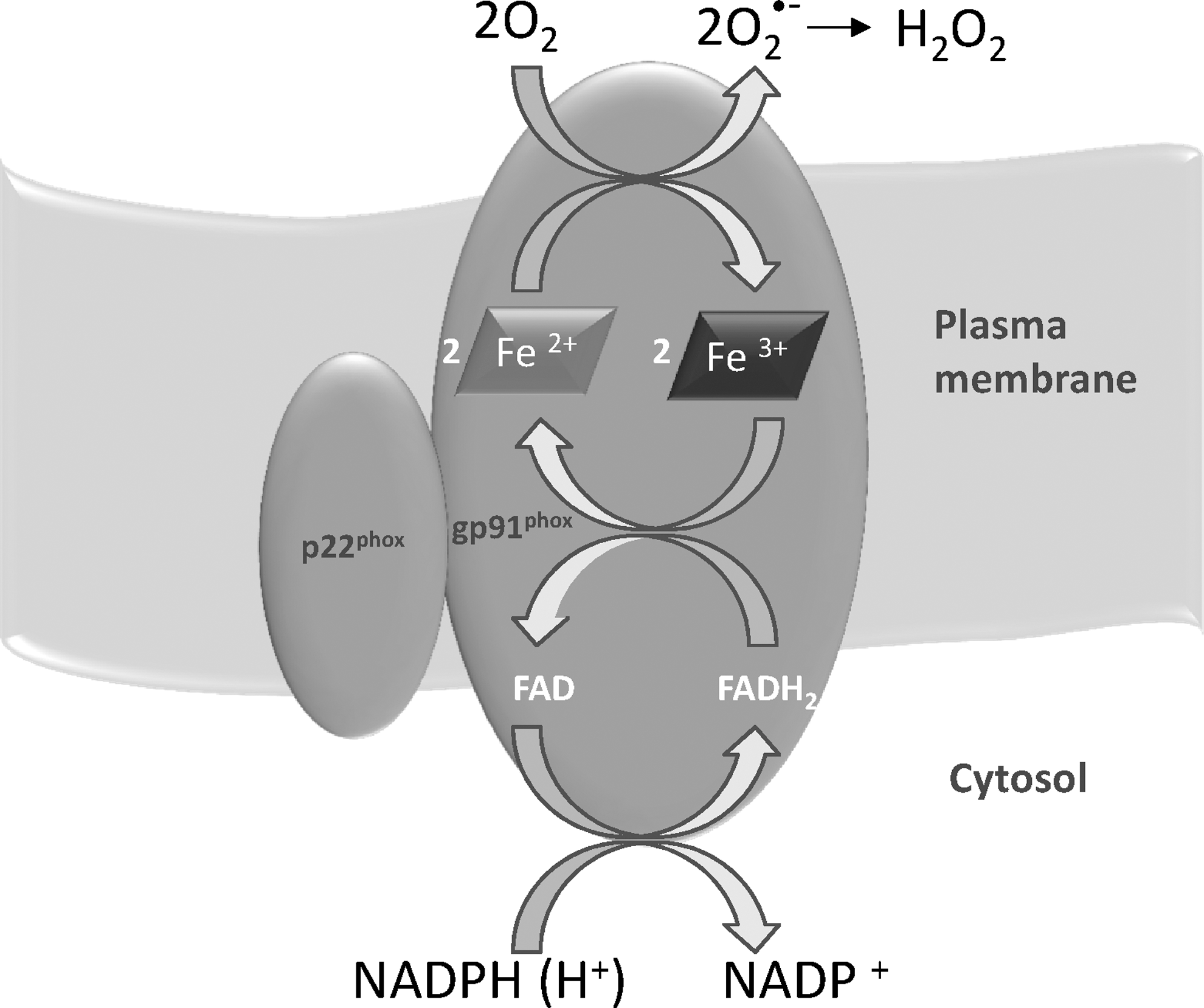
Although many different sources of ROS have been clearly identified in cells, unlike most of the biological ROS generators, NADPH oxidase has the unique property of forming superoxide and H2O2 in a highly regulated mode, as required in signaling processes (Fig. 2). This characteristic places it in a privileged position with regard to the other pathways of ROS formation. NADPH oxidase was first observed in phagocytic cells such as neutrophils and macrophages in the process called “respiratory burst” that is a sudden and considerable consumption of oxygen leading to the production of ROS for cell defence against pathogens (201). NADPH oxidase is a multienzyme complex that comprises both membrane-bound (Nox) and cytosol proteins. However, the presence of NADPH oxidase is not restricted to cells engaged in the innate immune response to invading microorganisms, as distinct homologs of Nox2 (Nox1, Nox3, Nox4, Nox5, Duox1, and Duox2) have been found in several tissues (1, 124, 202), indicating the specific role of this enzyme in redox signaling. Of note, while Nox1–4 requires association with p22phox, the latter is not necessary for the activity of Nox5, Duox1, and Duox2. In addition, the regulatory proteins recruited to the membrane for the formation of an active complex are not required for Nox4 or Nox5 (1 and references therein). Nox proteins are widely distributed in several tissues, and their location is not restricted to the plasma membrane, as they are also found in subcellular organelles such as endoplasmic reticulum (ER), nucleus, and mitochondria (1 and references therein), showing that this ROS-producing enzyme can influence various signaling pathways according to the type of cell and its intracellular location. Duox1 and Duox2 (ThOX1 and 2) are mainly expressed in thyroid tissue and are regulated by calcium ions (84), such as Nox5, expressed in testis, spleen, and lymph nodes (16) and thus suggesting a link between Ca2+ levels and redox signaling. Notably, Duox2-transfected HEK293 cells can produce ROS in a calcium-dependent manner (4). Nox4, originally located to the kidney, is now known to be widely distributed in several types of cells (316 and references therein). Of note, Nox 4 essentially produces H2O2, instead of O2 •− and ROS also form spontaneously without stimulation (316). Nox4 has recently been found to be also located in mitochondria (39, 223), and, therefore, its activity is added to the many enzymes producing ROS in mitochondria (section II.B.), suggesting reciprocal regulation of ROS production involving Nox4 and mitochondria (39). The presence of Nox4 in mitochondria influences the thiol redox state of many mitochondrial proteins such as adenine nucleotide translocase (ANT), the components of the electron transport chain, and the tricarboxylic acid cycle (223). Nox4 has also been found over-expressed in breast and ovarian tumors (138), suggesting that H2O2 produced by Nox4 plays a role in the signaling processes of tumorigenesis. The wide distribution of homologs of the phagocytic enzyme in various types of cells and in subcellular organelles indicates the variety of functions involving NADPH oxidases such as cell proliferation, differentiation, and survival (20, 201). Notably, while Nox2 and Duox1 and 2 are functionally linked to their specific heme peroxidases, other targets such as protein thiols are to be taken into account for the other homologs (355). NADPH oxidase, activated by several distinct stimuli such as cytokines and growth factors, thus generates H2O2, acting as a second messenger influencing discrete thiol-sensitive pathways. The effects of NADPH oxidase also include sustained cross-talk involving it and other cellular ROS generators, in particular mitochondria (1).
B. Mitochondria
The capability of mitochondria to generate and release H2O2 and O2 •− was discovered about 40 years earlier (112, 219). Superoxide is first formed from the autoxidation of some components of the respiratory chain and is rapidly dismuted to H2O2 by SODs, present in both the matrix (Mn-SOD) and the intermembrane space (Cu, Zn-SOD) (258) (Fig. 3). Once formed, H2O2 is removed by mitochondrial peroxidases, although some of it may easily diffuse to the cytosol through aquaporins in the inner mitochondrial membrane (207). The quantitatively most important sites of superoxide production are Complex I and Complex III (42). Complex I produces O2 •− that is essentially directed toward the matrix, whereas Complex III also releases O2 •− to the cytoplasmic side (325). Other enzymatic sites of superoxide production in mitochondria are α-glycerophosphate dehydrogenase, which, similar to Complex III, generates electrons toward both matrix and intermembrane space, and the electron transfer flavoprotein (ETF), which is involved in the oxidation of fatty acids and transfers electrons to CoQ through ETF:CoQ oxidoreductase (245). Superoxide produced by the latter system is directed to the matrix space, similar to the superoxide produced by the α-ketoglutarate and pyruvate dehydrogenase complexes, other known sites of ROS generation in mitochondria (329, 348). The flavoenzyme monoamino oxidase, located on the outer mitochondrial membrane, on oxidation of catecholamines or other monoamines, produces H2O2, which is directly released to the cytosol (186). An enzyme (p66Shc), acting as a lifespan regulator and specifically dedicated to the formation of ROS, has recently been shown to lie in the mitochondrial intermembrane space (127). This protein takes up electrons from the respiratory chain at the level of cytochrome c and transfers them to oxygen, forming H2O2 (127). p66Shc occurs as a reduced inactive dimer or an active tetramer with disulfide bridges, and the equilibrium between the two forms is controlled by the redox state of thioredoxin and glutathione (125). The oxidation from the inactive dimer to the active tetramer potentially depends on a thiol/disulfide exchange with the protein Mia40, located in the intermembrane space and in turn reoxidized by the sulfhydryl oxidase (SOX) Erv1 (125) (see also section III.A.2.a.).
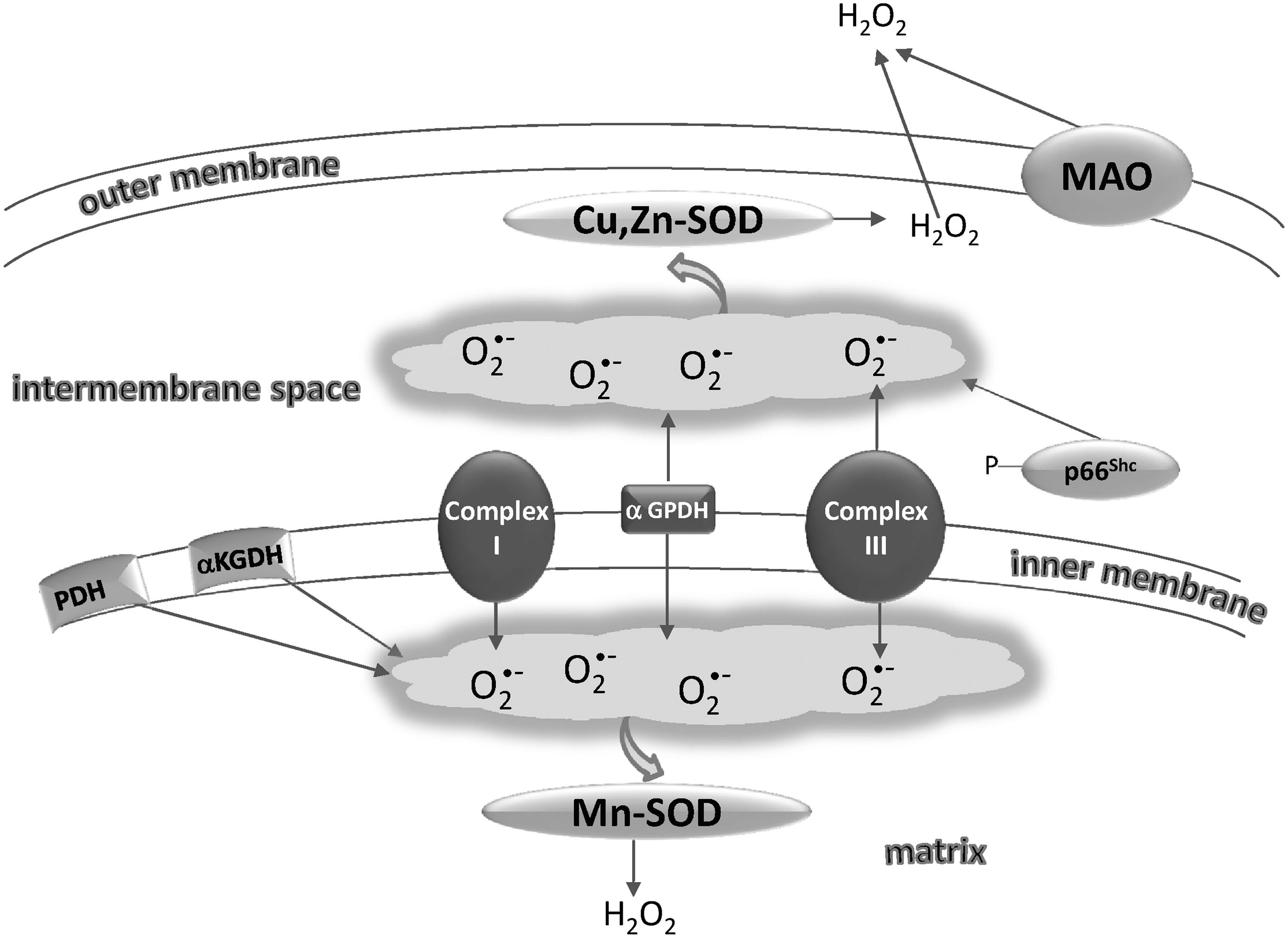
ROS modulator 1 (Romo1), a recently described protein located in mitochondria, can stimulate ROS production (66) and links TNF-α signaling to mitochondrial ROS production and apoptotic cell death (193). Of note, mitochondria are both producers and consumers of H2O2 (294, 380) and, consequently, the resulting net concentration of H2O2 released by mitochondria is closely regulated by the balance deriving from its production and removal (5). As described below, GPx and peroxiredoxin (Prx) occur both in the mitochondrial matrix and in the intermembrane space and effectively reduce H2O2 by using the reducing equivalents supplied by NADPH. This circumstance is important in the signaling exerted by H2O2 of mitochondrial origin. It should be also noted that Prxs may be inhibited by high concentrations of H2O2 (288, 366), leading to an increased efflux of mitochondrial H2O2.
Mitochondria are a significant source of ROS, although the general assumption that they are the main cellular source of ROS has been criticized (42, 50), as there are many other systems with a great capacity to produce ROS and only the net mitochondrial production of ROS should be taken into account.
C. Peroxisomes
Several enzymes able to produce H2O2 such as acyl-CoA, D-aminoacid, L-α-hydroxyacid, polyamine, and xanthine oxidases are located in peroxisomes. The inducible NOS can also be found among the peroxisomal enzymes, therefore accounting for the production of nitric oxide by this organelle (118 and references therein). However, peroxisomes are endowed with catalase and all the enzymes removing oxidant species such as SOD, glutathione S-transferase (GST), and Prx 5 (118), so that, as in mitochondria, a balance between ROS production and decomposition occurs. In spite of their high catalase content, peroxisomes still release a significant proportion of H2O2 (118). Peroxisomal respiration in the liver accounts for about 20% of the total oxygen consumption used by peroxisomal oxidases (174), and these organelles are thus potentially important sources of cellular H2O2. Redox communication with other subcellular components such as mitochondria also takes place (118, 174), as shown in catalase-deficient cells, where an excess of ROS generation by peroxisomes can perturb the mitochondrial redox balance (174).
D. Xanthine oxidase
The last two steps of purine catabolism are catalyzed by xanthine oxidoreductase (XOR), which converts hypoxanthine to xanthine and xanthine to uric acid (Fig. 4). XOR from animal sources are homodimers of about 300 kDa, and each subunit is endowed with a molybdopterin cofactor, two iron sulfur clusters, and a flavin adenine dinucleotide (FAD) molecule, which reoxidizes the enzyme using NAD+ or oxygen as final electron acceptors (256 and references therein). In the freshly isolated enzyme, NAD+ is reduced to NADH, thus characterizing the enzyme as a dehydrogenase (XDH). However, if treated in various ways, for example, storage at −20°C, heating at 37°C, anaerobiosis, proteolysis, or thiol-modifying reagents, the enzyme is converted to an oxidase (XO) (86) that can form both O2 •− and H2O2 (120). The relative amounts of O2 •− or H2O2 produced depends on the specific experimental conditions (120). In normal aerobic conditions, H2O2 is the major ROS produced by XO, and the amount of H2O2 with regard to O2 •− further increases in pathophysiological conditions characterized by lower oxygen tension. This feature emphasizes the critical role played by XO-generated H2O2 in processes such as those occurring in vascular inflammation (187). While proteolytic treatment leads to irreversible transformation of the enzyme to oxidase, treatment with thiol-modifying reagents or oxidation to disulfide of some specific cysteines of the enzyme converts the dehydrogenase into an oxidase, which, however, may revert to its original form on reduction by various thiols (86). In particular, Cys535 and Cys992 have been reported to be critical for the conversion of the rat liver dehydrogenase into an oxidase (256 and references therein).
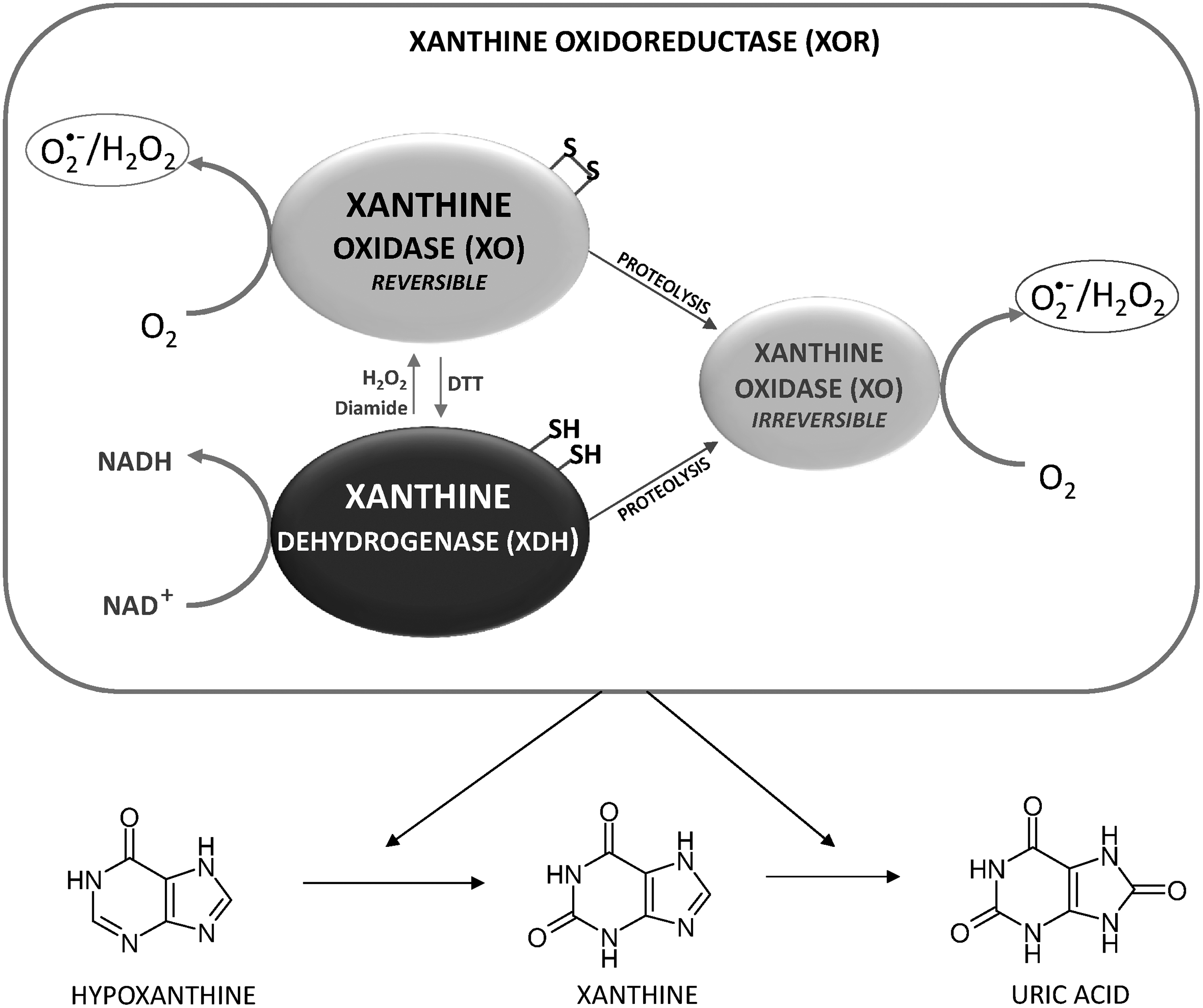
Perfusion of isolated rat heart in the presence of H2O2 or diamide leads to a significant conversion of XDH to the reversible XO, which, however, can be reconverted to the native form on removal of the oxidizing agents from the perfusion medium, indicating the sensitivity of the enzyme to cell redox conditions (31). XOR can, therefore, be a target for redox regulation by oxidants in a process whereby the enzyme reversibly switches from a dehydrogenase form (XDH) to an active, ROS-producing species (XO) (Fig. 4). Oscillatory shear stress increases the endothelial production of both O2 •− and H2O2 in a process prevented by oxypurinol (237), a typical inhibitor of the xanthine-converting enzyme. Of note, endothelial cells deficient in p47phox show very low XO activity and a marked decrease in O2 •− production (237), suggesting that the high ratio of XO to XDH, responsible for ROS production, is maintained by NADPH oxidase activity (237). These observations provide an attractive example of ROS modulation exerted by NADPH oxidases on XOR (237), framed within the general concept of an active ROS-mediated redox communication in cells (90, 378, 382).
E. Sulfhydryl oxidases
SOXs can catalyze the O2-dependent oxidation of thiols to disulfides with the concomitant production of H2O2 according to equation 3:
These FAD-dependent enzymes play an important role in the formation of disulfide bonds in eukaryotes and, as they generate H2O2, oxidative stress may occur, particularly in cells with elevated secretory loads of protein containing disulfide bonds (72). SOXs are subdivided into several families, including Erv/ALR, Ero, quiescin, and fungal-secreted SOXs, each characterized by a different domain organization (95). In particular, members of the quiescin SOX (QSOX) family have been identified in several organs and tissues (95, 197). In the nervous system, QSOX is considered an important source of H2O2 that may play a role in ROS-mediated redox regulation (226). Other SOXs contributing to H2O2 formation in the cell, such as Ero and Erv, are described in section III.A.2.a.
F. Other sources of ROS
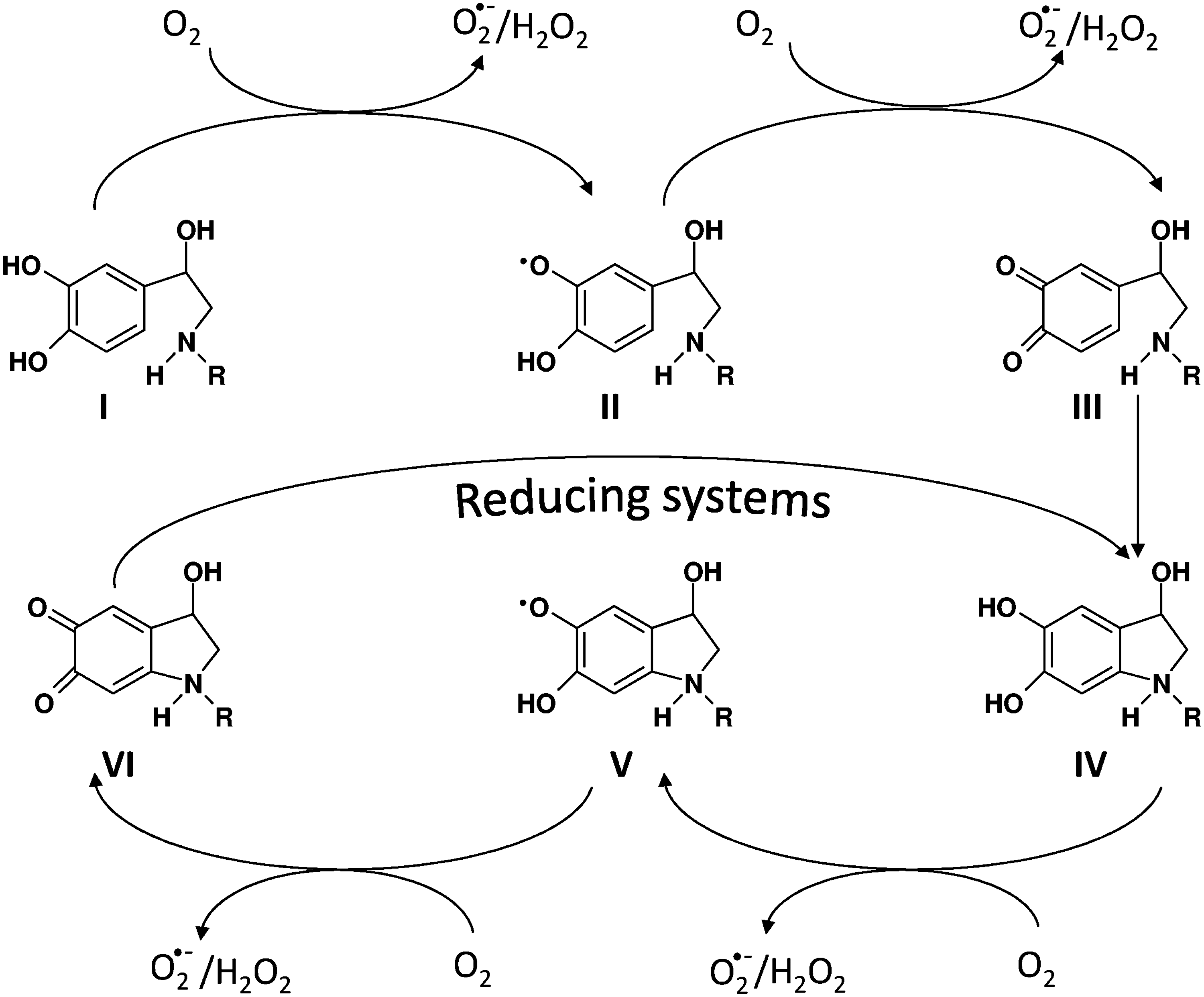
In addition to their normal catabolism, catecholamines can also undergo nonenzymatic oxidation, depending on the presence in their molecule of the catechol group (35, 73). At physiological pH, and in the absence of catalysts, the catecholamine oxidation process is extremely slow and, therefore, of little significance as a source of ROS (180). However, in the presence of trace metals, their oxidation is markedly stimulated and generates various quinone derivatives, semiquinone free radicals, and ROS (35, 73 and references therein) (Fig. 5). Once oxidized to the corresponding ortho-quinones, catecholamines undergo an irreversible 1,4-intramolecular cyclization reaction, leading to the formation of aminochromes (2,3-dihydroindole-5,6-quinones) (35 and references therein), which are very effective in eliciting a redox cycling process producing O2 •− and, secondarily, H2O2 (Fig. 5). The pathophysiological role of catecholamines, their oxidation products in the heart, and their relationship with oxidative stress have been extensively examined in a recent review article (73). The potential involvement of catecholamine oxidation in neuronal signaling mechanisms, in which H2O2 formation may play a vital role, has recently been hypothesized (321).
The cytochrome P450 enzymes of the ER are versatile proteins involved in the oxidation of xenobiotics and in drug metabolism (141). In appropriate conditions, they can also release O2 •− and H2O2 (141). Of note, in cells over-expressing CYP2E1, the ethanol-inducible cytochrome P450 2E1, ROS, and particularly H2O2, produced during the catalytic cycle of this enzyme, can initially activate the transcription factor Nrf2 (see section V.E.) that drives the expression of genes forming several antioxidant enzymes such as glutamate cysteine ligase, GST, and heme oxygenase-1, highlighting the occurrence of an adaptive response toward increased oxidative stress (58). However, extensive production of these oxidants may overwhelm the regulatory mechanisms and instigate toxicity (58).
The endothelial NOS (eNOS) is the enzyme that generates •NO in endothelial cells and, in the absence of the essential cofactor tetrahydrobiopterin (BH4) or on its oxidation, may be converted into an “uncoupled” form, which produces O2 •− instead of •NO by using oxygen as the final electron acceptor (203). In redox signaling, the capability of NOS to form O2 •− is a significant issue. First of all, O2 •− readily reacts with •NO, forming peroxynitrite (251), which may be toxic but that is also active in signaling cascades (45). In addition, since the signaling pathways of O2 •− and H2O2 differ from those of •NO (102, 204), all the conditions stimulating the conversion of eNOS to an oxidase may profoundly alter the intracellular message originally depending on •NO. Intriguingly, activated NADPH oxidase can produce ROS that is able to oxidize tetrahydrobiopterin and uncouple eNOS, suggesting an interaction between the two enzymes, leading to the increased generation of ROS and impaired production of •NO (203).
LOX and COX, in the presence of a suitable reducing substrate, can produce O2 •− as a side-chain reaction (199). These enzymes are particularly important producers of the lipid peroxides involved in signaling processes, where GPx4 also plays a critical role (69, 129) (see also section IV.A.3.).
Other sources of ROS in various organisms, cells, and subcellular compartments are described, showing both the complexity of enzymatic ROS production in aerobic life forms and the potential difficulty of distinguishing the many redox signaling pathways.
III. Protein Targets Sensitive to Oxidant Species
In principle, in biological systems, any proteins that are potentially subject to reversible redox reactions may act as sensors of oxidant signaling. For instance, proteins containing Fe-S clusters are critical in sensing ROS and regulating cellular functions (192). However, there is currently a large consensus in considering specific cysteine residues as the major targets of the oxidants involved in redox signaling.
A. Chemistry and biochemistry of thiol oxidation
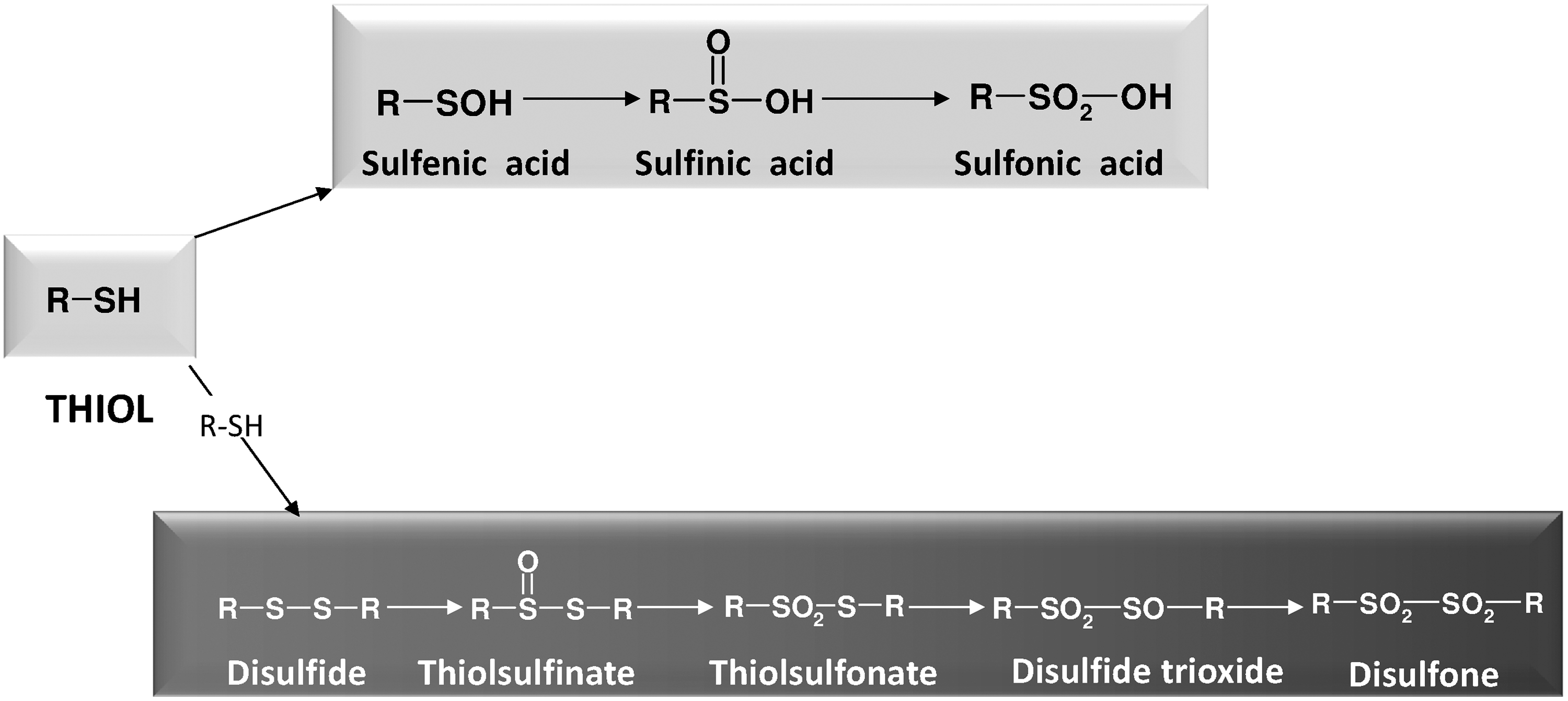
Organic molecules bearing thiol groups and including cysteine residues in proteins easily undergo oxidation, and a vast number of reactions and conditions can stimulate this process (181). The direct reaction of thiols with oxygen is a spin-forbidden reaction. Consequently, this reaction is very slow and, in order for it to occur to a significant extent, requires the intermediacy of transition metals that facilitate the electron transfer from the thiol to oxygen with the formation of H2O2 (181). In biochemistry, the formation of disulfides in polypetide chains contributes to the tertiary structure of proteins. However, this is not the only function of thiol oxidation, as reversible thiol redox changes are also crucial for the modulation of enzyme activity and the occurrence of redox signaling. Oxidation of thiols is a complex process (175) leading to different oxidation states of sulfur, as shown in Figure 6. The two oxidative chains of that figure are not to be viewed as rigorously separated into “dimeric” or “monomeric” products, but the various sulfur intermediates can undergo many interconversions in which the disulfide bond can be formed or broken. The lowest oxidation states (disulfide and sulfenic acid) are the most important for biological processes, including redox signaling events, and will be discussed in detail in the following sections.
1. Sulfenic acid
Sulfenic acid (sulfur oxidation state 0), the first step in the oxidation of a single thiol forming oxygen derivatives (Fig. 7), occurs in proteins playing roles in signaling processes.
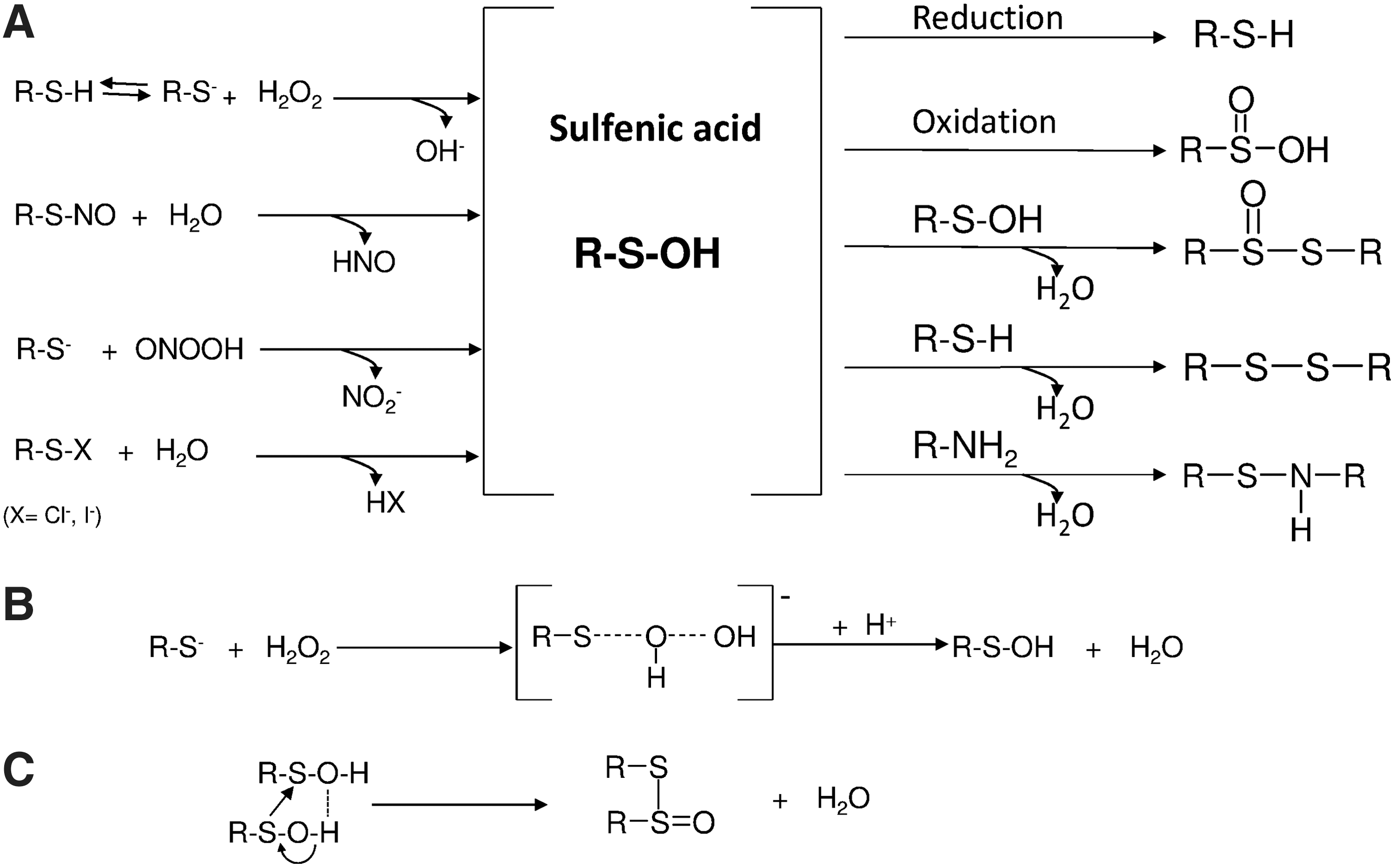
Sulfenic acid is a quite unstable and elusive species (164, 191), commonly considered a transient intermediate formed during the oxidation of thiols to disulfides or sulfinic acid. The functional group of sulfenic acid is, in fact, highly reactive, partly because it shares both nucleophilic and electrophilic properties (2). For instance, due to the electrophilic nature of sulfenyl sulfur, mild oxidation of low-molecular-weight thiol compounds inevitably leads to the formation of disulfides, as the transient sulfenic acid intermediate rapidly reacts with the parent thiol (2). The high reactivity of sulfenic acids is also due to their facility to self-condensate, forming thiolsulfinates (83). Consequently, only a limited number of small organic sulfenic acid molecules, sufficiently stable to be isolated and crystallized, have been described (2, 67, 191). Interestingly, a stable sulfenic acid isolated as a crystalline solid was obtained by direct oxidation of the corresponding thiol. In this case, the sulfenic residue is surrounded and protected by a bowl-shaped substituent, thus preventing its self-condensation (136). This condition is reminiscent of that found in proteins in which the geometry of the active site helps stabilize the sulfenic group.
Sulfenic acid or sulfenyl derivatives in proteins have long been described in various enzymes (2, 215), and sulfenic acids have also been directly shown by X-ray spectroscopy in crystallized NADH peroxidase (369). However, methods such as those applying the proteomic approach are currently exploited to identify sulfenic acids in proteins and to establish their role in enzyme catalysis and oxidative stress (190 and references therein).
Specific criteria have been defined to characterize the stabilization of sulfenic acid residues in proteins (67, 369): They are briefly summarized here: (i) absence of a thiol group in close proximity to the sulfenic acid residue. This requirement is probably the most important, as a rapid reaction leading to a disulfide formation is excluded. The prevention of disulfide formation may also depend on a topological constraint, due to a specific secondary structure of the peptide at the active site (67); (ii) formation of an intramolecular hydrogen bond; (iii) ionization of sulfenic acid to the corresponding conjugate sulfenate (R-SO−); and (iv) existence of an apolar micro-environment that is associated with a limited solvent accessibility.
a. Formation of sulfenic acid by reactions of thiols with peroxides
Unlike the case with oxygen, the reaction of thiols with H2O2 is a spin-allowed reaction and is, therefore, not subject to kinetic barriers. The reaction proceeds by a bimolecular nucleophilic substitution (SN2) reaction (247) according to equation 4:
A transition state occurs and is characterized by one oxygen atom of H2O2, partially bound both to the sulfur atom and to the OH leaving group (Fig. 7). Similar to H2O2, peroxynitrite (see also section III.A.1.c.) or alkyl hydroperoxides originating from the activities of LOXs and COXs or from lipid peroxidation can also oxidize thiols, with the formation of sulfenic acid, following a pattern essentially similar to that of H2O2. Renewed interest has recently been shown in the reaction leading to peroxide-dependent sulfenic acid formation reaction, as in addition to the role of intermediate in the catalytic cycle of peroxidases, it is also a critical step in the signaling processes mediated by H2O2 (45, 78, 362).
Although oxidation of thiols by H2O2 is markedly catalyzed by trace metals, it may also take place in their absence, in a process leading to disulfide formation (262). The reaction proceeds through the initial formation of sulfenic acid, which, in the presence of excess thiol, quickly leads to the final generation of disulfides (364). However, the kinetics of this reaction have revealed that the nonenzymatic reaction of low-molecular-weight thiols with H2O2 is relatively slow, as thiols such as glutathione, cysteine, cysteamine, dithiothreitol (DTT), penicillamine, and N-acetylcysteine exhibit second-order rate constants, ranging from 18 M −1 s−1 for DTT to 26 M −1 s−1 for cysteine (364). In view of this kinetic behavior, it is clear that this reaction is not of great importance in a biological context. It has also been shown that the relative reactivity of thiol compounds is inversely related to their pKa value, suggesting that the thiolate is the reactive species (364). In proteins, sulfenic acid formation after the interaction of H2O2 with cysteine residues has long been known (215). For instance, glyceraldehyde-3-phosphate dehydrogenase can be inactivated by H2O2 in a reaction that does not involve disulfide formation but a sulfenic acid derivative (215). The enzyme is, in fact, reactivated by the rapid addition of low-molecular-weight thiols, while delayed treatment is ineffective, indicating the occurrence of an irreversible process due to the oxidation of cysteine to the corresponding sulfonic acid (215). Of note, this reaction depends on pH, showing that the active species decomposing H2O2 is a thiolate group (215). Several proteins and enzymes appear to interact with H2O2 through cysteine thiolates in their active site, although their second-order rate constants are relatively low, as shown for papain (62 M −1 s−1) and glyceraldehyde-3-phosphate dehydrogenase (100 M −1 s−1) (331 and references therein). Similarly, mammalian enzymes proposed as H2O2 sensors, such as protein phosphatase PTP1 and Cdc25B, exhibit second-order rate constants that are comparable to those shown for the above proteins (87, 322). Examination of the inhibitory effect of H2O2 on three distinct PTPs shows inactivation constants ranging from 9.1 to 17.9 M −1 s−1 (87), whereas Cdc25B phosphatase rate of inactivation proceeds with a rate constant of 164 M −1 s−1 (322). In contrast, the reaction kinetics with H2O2 is dramatically accelerated by GPxs (347), Prxs (367), and the bacterial H2O2 sensor OxyR (10). The rate constant of the latter is reported to be approximately 107 M −1 s−1(10). GPx is an enzyme that is characterized by a selenocysteine at the active site and has rate constants higher than 107 M −1 s−1 (347). Prxs, characterized by a sulfur atom at their active site, show catalytic efficiencies for the reduction of hydroperoxides exceeding 107 M −1 s−1 (261). Both catalytic chacolgens (Se or S) at the active site show similar although not identical patterns in their reactivity toward H2O2 or hydroperoxides The reduction of peroxynitrite by Prxs also shows remarkable rate constants, with values higher than 107 M −1 s−1 (98); a kinetic study of the GPx reaction with peroxynitrite demonstrates that it can efficiently reduce peroxynitrite/peroxynitrous acids with a second-order rate constant estimated to be 8×106 M −1 s−1 (47). In addition to H2O2, peroxidized lipids can also mediate cellular PTP oxidation, depending on the disruption of Gpx4 (71). This is directly apparent by the in vitro observation that 15-HEPETE (15-hydroperoxy-eicosatetraenoic acid) at nanomolar concentrations was more effective than H2O2 in inducing PTP oxidation, as micromolar concentrations of H2O2 are needed to obtain a comparable oxidizing effect (71). Therefore, the control of receptor tyrosine kinase signaling by peroxidized lipids also appears regulated by the concerted action of 12/15 LOX and Gpx4 (71).
A simple comparison of the above second-order rate constants, particularly those of PTP1 and thiol peroxidases, indicates that the low pKa of the cysteine residue (Fig. 8) is not in itself a condition that is sufficient to significantly accelerate the reaction between the thiol and H2O2, to make it efficient for the needs of a physiological process and in particular for a signaling event. According to Flohé (105), in a biological context, a protein thiol can act as a primary sensor for H2O2 only at rate constants no lower than 104 M −1 s−1. Therefore, to accelerate the reaction, the protein thiolate at the active site requires specific aminoacid residues creating a micro-environment that is capable of stabilizing the transition state (331). In both selenium and nonselenium GPxs, the specific environment around the catalytic cysteine or selenocysteine is the critical condition explaining their high reactivity (107). To reach these appreciably high bimolecular rate constants, the following conditions should be fulfilled (107, 347): (1) The cysteine (or selenocysteine) should be in its dissociated form, that is, thiolate (or selenolate), which is the reactive nucleophilic species; (2) the oxygen–oxygen bond of the peroxide molecule should be polarized to favor nucleophilic attack; and (3) a proton should be available to interact with the leaving group and to form a water (or alcohol) molecule. Flohé et al. (107) compared thiol peroxidase mechanisms and showed that the GPx family is characterized by a specific tetrad formed by the active site Cys (or SeCys), Asn, Gln, and Trp, whereas the Prx catalytic site is composed of a triad including Cys, Thr (or Ser), and Arg. In selenium-dependent GPxs, the amino acids Asn, Gln, and Trp create an effective hydrogen bonding network that favors the dissociation and polarization of the nucleophilic selenol, which attacks the electron-poor oxygen belonging to the peroxide moiety (347). Lastly, a labile proton associates with the leaving group, forming H2O or R-OH (347). Similarly, in Prxs, stabilization of the transition state is achieved by alignment of the peroxide substrate with the active site thiolate along a Cys-O-O axis, which is further stabilized by hydrogen-bonding interactions (145). This arrangement favors nucleophilic displacement, finally leading to the formation of sulfenate (145). As a complementary mechanism, Ferrer-Sueta et al. (98) proposed that the approaching peroxide molecule induces a shift of the hydrogen bond from the thiolate to itself, creating favorable conditions such as increased nucleophilicity of the thiolate anion, which stimulates the overall process. In human Prx2 and Prx3, two highly conserved arginine residues are critical for the peroxidase activity of these enzymes, as the point mutation of either arginine causes a decrease in reactivity of five orders of magnitude (247). In conclusion, H2O2, lipid hydroperoxides, and peroxynitrite interact with reactive thiols in their thiolate (or selenolate) form, modify the sensor molecule, potentially enabling it to transmit the signal. Sulfenic (or selenenic) acids are formed as critical intermediates.
b. Formation of sulfenic acids from sulfenyl halides
Activated phagocytic cells play a bactericidal role by producing O2
•− and H2O2, and are also endowed with the enzyme myeloperoxidase, which uses H2O2 and chloride to generate secondary oxidants such as hypochlorous acid (HOCl) and N-chloramines (RNHCl) (146), both of which can efficiently oxidize thiols (267, 277, 363). With thiols, chloramines exhibit reaction rates lower than HOCl, but they are more specific (363 and references therein). After a reaction with thiols, both chloramines and HOCl generate sulfenic acid, after the initial formation of a sulfenyl chloride. Equation 5 shows the reaction of a thiolate, more reactive than the corresponding thiol, with HClO, leading to the formation of a sulfenyl chloride. The latter is subsequently hydrolyzed to sulfenic acid, as shown in equation 6:
Chloramines are important physiological oxidants produced during inflammation, although their reactivity with Prx is low in comparison with H2O2 (266). They also appear to react preferentially with low pKa thiols, so that their participation in different signaling pathways where protein thiolates act as sensors is conceivable (363 and references therein). The potential role of these reactive oxidants of pathophysiological origin in signaling pathways has recently been discussed by Winterbourn and Hampton (363).
c. Formation of sulfenic acid from nitrosothiols
As previously described (section I.C.), the formation of nitrosothiol derivatives (R-SNO) from sensitive cysteines in proteins is a critical step in signaling (156). Once formed, nitrosothiols can lead to the formation of sulfenic acid (327) in a hydrolytic reaction:
Nitric oxide reacts with O2 •− at a near-diffusion limit rate, forming peroxynitrite (ONOO−) (251), an extremely efficient oxidant acting on both low-molecular-weight and protein thiols in a reaction that also gives rise to oxidation products beyond the sulfenic acid species (282), which is therefore an intermediate in this reaction.
Both peroxynitrite anion (ONOO−) and the corresponding conjugated peroxynitrous acid (ONOOH) readily react with low-molecular-weight thiol compounds with second-order rate constants of approximately 103 M
−1 s−1, which are higher by more than three orders of magnitude than those observed for the reaction of the same compounds with H2O2 (3, 282, 350) (see also section III.A.1.a.). The reaction of peroxynitrous acid with thiolate anion (reaction 8) or of peroxynitrite anion with protonated thiol is a two-electron oxidation process that is strongly dependent on pH and leading to the formation of sulfenic acid, which can further interact with another thiolate, forming a disulfide (282, 350).
However, this oxidation process can also proceed by a one-electron pathway that is mediated by the homolytic decomposition of peroxynitrous acid, forming hydroxyl and nitrogen dioxide radicals (279). As previously reported, Prxs very efficiently catalyzes the reduction of peroxynitrous acid in a two-electron process, involving their active site thiolate and leading to the formation of nitrite and a protein sulfenic acid (98, 99). The rate constants of this process are up to 105 times faster than those observed with low-molecular-weight thiols (99).
The formation of sulfenic acid in proteins reacting with peroxynitrite has also been described for the bacterial Prx AhpC, as evidenced by the formation of NBD adducts (see below) with the protein (52) and for bovine 1-Cys Prx according to reactivity with 2-nitro-5-thiobenzoic acid (TNB) (see below) (265).
All the above processes of the formation of sulfenic acid, along with additional mechanisms such as the hydrolysis of disulfides, reactions with thiolsulfinates, and generation from thiol radical species, have been amply covered in a recent review (190 and references therein).
d. Reactions of sulfenic acid
The reactivity of sulfenic acid depends on its dual character as a nucleophilic and electrophilic compound (2). For instance, sulfenic acids have been shown to react with the electrophilic compound 7-chloro-4-nitrobenzo-2-oxa-1,3-diazole (NBD-Cl), or nucleophilic molecules such as dimedone (5,5′-dimethyl-1,3-cyclohexanedienone) and its derivatives, or the reduced form of the thiol reagent 5,5′-dithiobis(2-nitrobenzoic acid). All these compounds have been used to detect cysteine sulfenic acid residues in proteins (2, 87, 92, 190, 271, 272). The most important reaction of sulfenic acids in proteins is probably that with nearby cysteinyl residues, for example, in the catalytic cycle of Prxs (288) or with glutathione, present in the cell at high concentrations and forming a mixed disulfide in a reaction favored both kinetically and thermodynamically (111) and occurring according to equation 9:
The formation of a mixed disulfide with glutathione (glutathionylation) plays a critical role in redox-regulated processes and also prevents further oxidation of the cysteine residue to higher oxidation states (81) (see below). A similar protection from over-oxidation is also offered by the reaction of the sulfenic acid moiety with the protein backbone, forming a sulfenyl amide (306, 356). Sulfenic acids can easily undergo a self-condensation reaction, forming thiolsulfinates (67, 164, 191) or may be further oxidized to sulfinic and sulfonic acids (see below).
e. Sulfinic and sulfonic acids
Sulfinic acids, unlike sulfenic acids, are stable oxidation derivatives that may be isolated and crystallized. Sulfinic acids of biological interest such as hypotaurine and homocysteine sulfinic acid are well known (181). Although oxidation of proteins to sulfinic acid is commonly considered irreversible in the cell (288), its formation has acquired importance in view of the redox control of Prxs (366, 368). On interaction with H2O2, the peroxidatic cysteine of these enzymes is oxidized to sulfenic acid, which, in turn, forms a disulfide by reacting with the resolving cysteine. However, at elevated concentrations of H2O2, the sulfenic acid can further interact with H2O2, forming a sulfinic acid species that cannot be reversed to thiol by the reducing action of thioredoxin (179, 288). However, a highly specific enzyme, sulfiredoxin (38), has been shown to revert the process in a series of reactions requiring ATP, magnesium, and a reducing agent (38, 365). The existence of an enzymatic pathway reversing the process highlights the potential importance of the over-oxidation of Prx in redox signaling events. According to the “floodgate” hypothesis (366), this feature of Prx permits the accumulation of H2O2 that is able to propagate its signal (288, 366). In addition to Prx, oxidation of cysteinyl residues of proteins to sulfinic acid also occurs in several other enzymes (288) (and references therein). Sulfinic acids can easily oxidize to the corresponding sulfonic acids, which represent the highest oxidized forms of both thiols and disulfides (Fig. 6). Sulfonic acids such as taurine, isethionic, and methanesulfonic acids occur naturally (181).
2. Disulfides
Disulfides represent the first oxidation step of thiols that is characterized by a transition from oxidation state −2 of the thiol to oxidation state −1 of the disulfide (Fig. 9). This occurs according to the following general reaction:
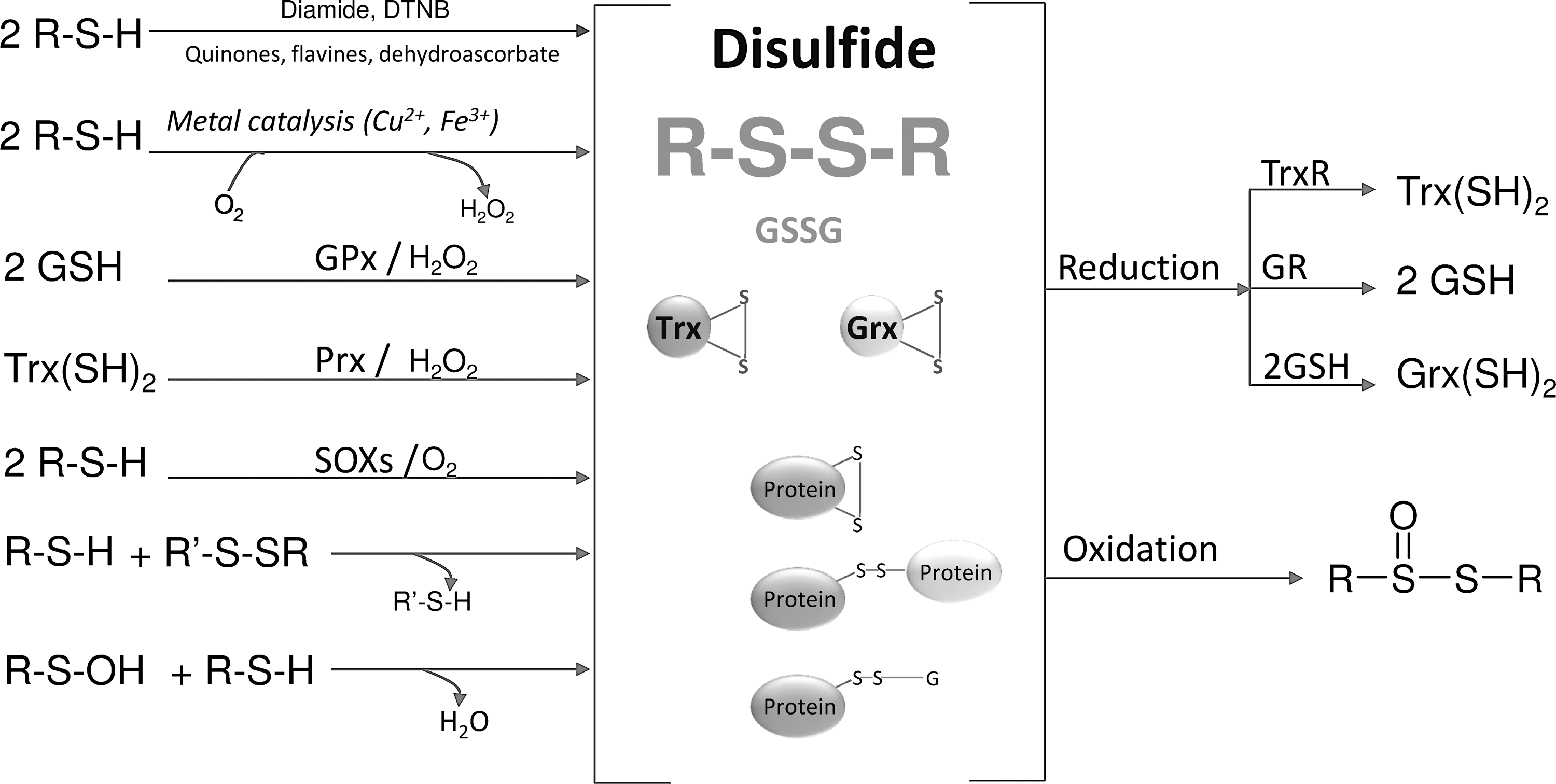
The reversible thiol/disulfide transition is a quite familiar biochemical redox process that takes place in mild conditions, and the resulting disulfide bond is relatively stable. Well-known thiol/disulfide couples in biochemistry are GSH/GSSG, dihydrolipoamide/lipoamide Trx(SH)2/Trx(SS), and Grx(SH)2/Gr(SS) (181, 211).
a. Formation and role of disulfide bonds in proteins
In proteins, disulfide bonds can be generated anew by SOXs (See section II.E.), which use oxygen or can be formed by thiol/disulfide oxidoreductases acting according to a thiol/disulfide exchange and shuttling the disulfide bonds in proteins (289). Specific disulfide bonds increase the stability of the tertiary structure in secreted and cell-surface proteins (289, 361), and the reversible thiol/disulfide transition also occurs in enzyme catalysis and redox-regulated signaling processes (264). In developing sperm cells, characterized by a very low content of GSH, GPx4 also acts as a protein thiol peroxidase, leading to the formation of disulfide bridges (69, 354).
In eukaryotic cells, protein disulfide bond formation takes place in the ER and the inter membrane space of mitochondria (IMS), whereas in bacteria, this process occurs in the periplasmic space (155, 289, 317, 361). The ER comprises a specialized redox environment that is endowed with enzymatic systems supporting the corrected disulfide formation in proteins (361). In the ER lumen, members of the protein disulfide isomerase (PDI) family, in their disulfide form, catalyze the oxidation of thiols of newly formed proteins. PDI can isomerize proteins with incorrect disulfide bonds, helping them attain their native position (361). This occurs by an intramolecular thiol-disulfide exchange or through repeated cycles of misfolded protein reduction and reoxidation, occurring until the correct configuration is obtained (361). PDI belongs to the dithiol/disulfide oxidoreductase family, which contains one or more domains showing a sequence similarity to Trx (361). The two thioredoxin domains of PDI, characterized by a Cys-xx-Cys motif, are active in the redox/isomerization process (361). It is now well established that the dithiol motif of PDI, resulting after oxidation of the imported peptide substrate, is reoxidized back to a disulfide by the flavoprotein Ero1, which shuttles the electrons to oxygen with the final formation of H2O2 (289, 351)
In the ER lumen, the GSH/GSSG redox pair constitutes the primary redox buffer (152) and, in comparison with the cytosol, it contains a higher concentration of oxidized glutathione, as about 25% of the glutathione is in the disulfide form (152, 169). In vitro, the oxidized form of glutathione is an efficient oxidant of PDI and, in the cell, may perhaps act as a source of oxidizing equivalents for PDI as an alternative to Ero1. However, in its reduced form, it may also maintain the reduced state of PDI family proteins and directly perform corrective reduction of non-native disulfide bonds of misfolded proteins of the ER (8, 59, 152). Therefore, considering the whole process, it is apparent that nascent polypeptides and GSH enter the ER from the cytosol compartment by translocation or facilitated diffusion, while mature correctly folded proteins and oxidized glutathione are exported in a process in which glutathione is a net source of reducing equivalents (8, 77).
Recently, a novel pathway parallel to that of Ero1 in mediating oxidative protein folding in ER and involving Prx 4 has been proposed (53, 340, 379), as Ero1-deficient mammalian cells are subject to only a modest delay in their capacity to form disulfide bonds (379). In this model, PDI is oxidized by the catalytic disulfide of Prx4, which, in turn, is reoxidized by H2O2. The contribution to disulfide bond formation in the ER, therefore, appears to be a new function for Prx4, in addition to its classical role as a H2O2 scavenger (97). H2O2 used by Prx4 may derive not only from Ero1-mediated protein oxidation but also from NADPH oxidase and actively respiring mitochondria (379). Therefore, the protein folding process of eukaryotes appears to be closely regulated by the availability of H2O2, which, according to this model, is effectively used in the ER to support oxidative folding (53, 379). The combined action of the Ero1- and Prx4-dependent pathways also represents a very efficient process, as two disulfides are formed with the concomitant reduction to water of a single molecule of oxygen (97, 379).
The intermembrane space of mitochondria houses a complex system, which, as in the ER, leads to the formation of disulfide bonds in newly synthesized proteins (289 and references therein). In this case, oxidation is catalyzed by the oxidoreductase Mia40, which is endowed with a redox-active disulfide bond that is able to oxidize peptide thiols (240). In this process, Mia40 is reoxidized to its active state by another protein, Erv1, which, similar to Ero1, is an FAD-dependent member of the SOX enzyme family (290). Of note, Erv1 has been shown to be able to transfer electrons to oxygen, forming H2O2 in a manner similar to Ero1. However, reducing equivalents can also be delivered to cytochrome c, with the final reduction of oxygen to water through the respiratory chain. In this way, H2O2 generation is prevented, and the disulfide forming oxidative pathway is markedly stimulated (289, 290).
In addition to ER, mitochondria also exhibit PDI activity, which appears to be mainly located on the outer membrane, where it may play a potential role in the regulation of redox processes such as mitochondrial-dependent apoptosis (291, 292). In addition, mitochondrial PDI can activate the Cu, Zn-SOD located in the mitochondrial intermembrane space (see also section V.H.) in a process that, however, requires disruption of the mitochondrial outer membrane (171).
Recently, PDI activity has been shown to be located in the mitochondrial-associated endoplasmic reticulum membrane (MAM) compartment (163), formed by the direct apposition of the ER membrane to the mitochondrion (123). PDI increase in response to increased levels of misfolded proteins can stimulate cell death pathways. Consequently, the increased activity of PDI at MAM appears to be responsible for permeabilization of the mitochondrial outer membrane, triggering apoptotic cell death, which may depend on Bak oligomerization caused by oxidation of cysteine residues (163). Therefore, in addition to its classical role of forming disulfides in newly synthesized proteins, PDI also appears to play a role in the induction and redox regulation of apoptotic cell death.
Several PDI homologs have been found in the bacterial periplasm. However, at variance with the protein disulfide isomerase of ER, the oxidative and reductive functions are located in separate proteins (361).
All these disulfide forming systems, although operating in differing subcellular compartments, appear to use oxygen as the final electron acceptor and are connected to the electron transport chain in the mitochondria and periplasmic space of bacteria (155). A major difference is with regard to the different redox condition of the ER in comparison with other cell compartments, as recently shown by Malinuski et al. (228) in HEK-293 cells by means of a genetically encoded H2O2 bioindicator, which was found to be oxidized in the ER but reduced in both the cytosol and the mitochondrial intermembrane space.
In conclusion, reversible disulfide bond formation regulates a vast number of biological processes, including metabolism, redox signaling, transcriptional regulation, and chaperone activity, as recently reviewed by Paulsen and Carroll (264).
b. Oxidation of disulfides
Thiolsulfinates are the first oxidation products of disulfide oxidation (Fig. 9). The best-known biological member of this class of compounds is probably the component of garlic allicin (diallyl disulfide monoxide) (181). In proteins, a thiolsulfinate group linking Prx to sulfiredoxin (Prx-S(O)-S-Srx) acts as an intermediate in the mechanism of reduction by sulfiredoxin of over-oxidized Prx (38, 288). As reported above, thiolsulfinates may derive from the self-condensation of two molecules of sulfenic acids (see above) (67, 164, 191). Thiols can easily reduce thiolsulfinates to the corresponding disulfides, with the intermediate formation of a sulfenic acid, suggesting potential biological importance for this reaction (181, 191). Further oxidation leads to thiolsulfonates, which, similar to thiolsulfinates, readily react with thiols (191).
IV. Control of Cellular Levels of Peroxides and the Thiol Redox Balance
A. Thiol-dependent systems involved in cellular redox balance
The cellular thiol redox balance and the related regulation of H2O2 levels are controlled by two major redox systems, that is, the glutathione and thioredoxin systems (Fig. 10). In these systems, glutathione and thioredoxin play different key roles as, in addition to their different chemical features, their cellular concentrations differ by orders of magnitude. The cell concentration of glutathione is in the millimolar range (89, 181), far greater than that of thioredoxin, which is estimated to be present in micromolar concentrations (140, 168). In addition, the two redox couples GSH/GSSG and Trx(SH)2/Trx(SS) are not in equilibrium with each other (133, 150) and, therefore, operate independently in the pathways that regulate growth or apoptosis, fulfilling different roles in cell redox metabolism (133, 150, 263, 349). However, genetic studies on mouse embryonic fibroblasts have indicated the existence of a partial overlap involving the thioredoxin and glutathione systems (69). TrxR2-deficient embryonic fibroblasts are especially sensitive to oxidative stress when GSH synthesis is inhibited by L-buthionine sulfoximine (BSO) in a process whereby TrxR2-/- fibroblasts are rescued by N-acetylcysteine (NAC). These results highlight the importance of TrxR2 for cell survival in conditions of limited availability of GSH (70).
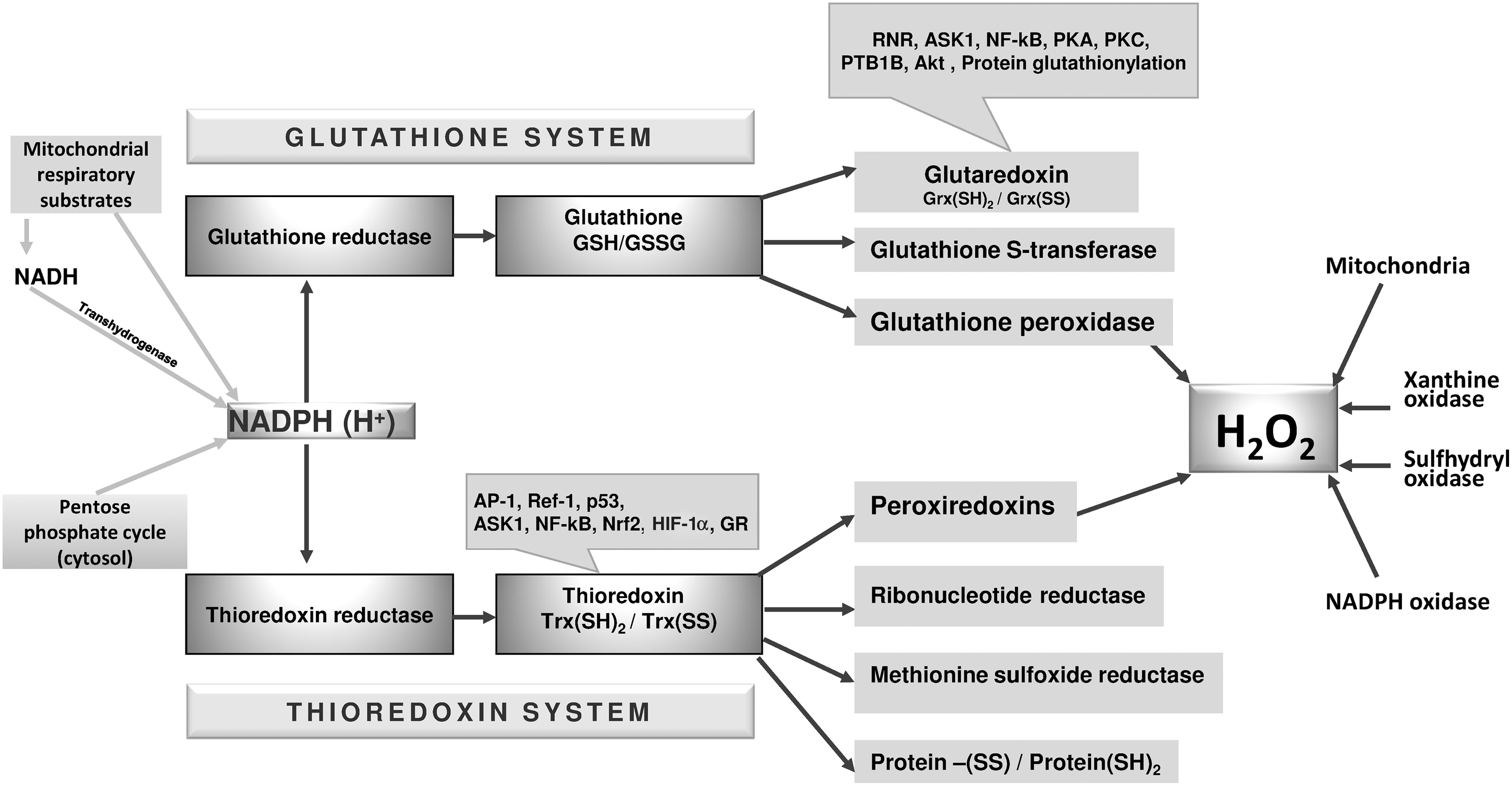
1. The thioredoxin system: characteristics and functions
The thioredoxin system comprises a functional sequence consisting of NADPH, thioredoxin reductase (TrxR), and thioredoxin (Trx), in which electrons are transferred from the reduced pyridine nucleotides to TrxR and hence to Trx (9, 167) (Fig. 10). TrxR is a flavoprotein homodimer that is organized in a head-to-tail arrangement and characterized by a dithiol redox couple in a CVNVGC motif at the N-terminus of one subunit, which interacts with a selenocysteine/cysteine motif at the C-terminus of the second subunit (63). TrxR is found in the cytosol (TrxR1) and subcellular compartments such as mitochondria (TrxR2) (211 and references therein). Similarly, Trx, the natural protein substrate of TrxR, is found in both the cytosol (Trx1) and mitochondria (Trx2) (211 and references therein). TrxR, in addition to its natural substrate, Trx, can also reduce a large number of other molecules, indicating a broad substrate specificity (9). Both cytosolic and mitochondrial TrxR genes are essential for embryonic development, as their inactivation results in embryo death (70, 176). However, distinct phenotypes have been observed. Mice with heart-specific inactivation of TrxR1 are healthy and develop normally (176), whereas TrxR2−/− embryos show altered heart development and impairment of hematopoiesis (70). Differences in the phenotypes of cytosolic and mitochondrial thioredoxin reductase knockout mice clearly suggest the cell type- and tissue-specific functions of these enzymes (9). In addition, in a mouse model, Trx1 and Trx2 null embryos were shown to be unable to develop (233, 257).
Well-known enzymes using Trx as a reducing substrate are ribonucleotide reductase, methionine sulfoxide reductases, and Prxs (211 and references therein). In general, Trx is considered a broad-spectrum protein-disulfide reductase (166, 167, 273). Thioredoxin is involved in cellular redox signaling, as its redox state modulates the functions of many transcription factors that depend on redox-sensitive cysteines (Fig. 10). Trx acts by maintaining critical cysteine residues in a reduced state, which allows binding to DNA (242). Transcription factors that are sensitive to Trx are nuclear factor κB (NF-κB) (see section V.D.), tumor suppressor p53 (352), glucocorticoid receptor (227), hypoxia-inducible factor 1α (see section V.F.) (360), and the AP1 protein complex (161).
The reduction exerted by Trx on Prxs is of special importance, as Prxs play a primary role in redox signaling, acting as sensors of peroxides for other proteins, in addition to their well-known peroxidase activity (45, 288). Prxs are a family of peroxidases using a conserved cysteine residue (peroxidatic cysteine) to reduce H2O2 and other peroxides (288). The mechanism of peroxide reduction proceeds with the initial formation of a sulfenic acid at the peroxidatic cysteine, due to the reaction of Prx with H2O2. Next, the sulfenic acid group reacts with a second cysteine (resolving cysteine), forming a disulfide that can be reduced back by the thioredoxin system in a cyclic process (288). Six isoforms of Prx (Prx1 to Prx6) are expressed in mammalian cells (288 and references therein), distributed into three subgroups, identified as 2 Cys Prxs (Prx1 to Prx4), atypical 2 Cys (Prx5), and 1 Cys peroxiredoxin (Prx6). The peroxidase cycle of the first two groups involves the formation of an intersubunit (Prx1 to Prx4) or an intrasubunit disulfide (Prx5) (288 and references therein). In both cases, the disulfide is reduced by Trx (288), which thus completes the peroxidase cycle. Therefore, most of the Prxs are in closed redox communication with the thioredoxin system, and are consequently closely dependent on its efficiency for accomplishing their reducing action on peroxides. Of note, mitochondrial Prx (Prx3) is not only reduced by Trx2 but also reduced by Grx2, which thus combines the properties of thioredoxins with those of glutaredoxins and suggests the occurrence of interplay between the thioredoxin and glutathione systems in mitochondria (148).
2. The thioredoxin system and redox-regulated processes
It should be recalled that the Prx -dependent oxidation of Trx is in itself a signaling event, as occurs, for instance, in the activation of apoptosis signaling kinase-1(ASK1)(216). Reduced Trx is well known to exert a negative regulation on the kinase activity of ASK1, and c-Jun NH2-terminal kinase (JNK) activation by H2O2 is mediated by ASK1 phosphorylation and Trx oxidation (304, 323). In alveolar macrophages, stimulated NADPH oxidase can activate the ASK1/MKK4/JNK signaling pathway, in a process initiated by H2O2 produced during the respiratory burst and acting as a second messenger. Interestingly, on ADP activation of the macrophages, phosphorylation of ASK1 is transient, peaking after 5 min and then decreasing. In addition, after ADP stimulation, Trx remains in its reduced state, and only bolus addition of H2O2 can give rise to rapid and transient oxidation of Trx. Treatment with aurothioglucose, an inhibitor of TrxR, stimulates the phosphorylation of ASK1 and enhances the dissociation of Trx from ASK1(216). The transient but substantial calcium-dependent oxidation of Trx has been shown in HaCaT keratinocytes after the addition of bradykinin or EGF (131), and this oxidative step appears to be critically associated with the growth initiation of these cells (131). In addition, on treatment with EGF, cytosolic thioredoxin (Trx1) is selectively oxidized in the same type of cells (HaCaT), whereas the mitochondrial isoform (Trx2) is relatively unaffected, indicating that the cellular effects of EGF depend on the compartmental oxidation of Trx1 (147). TNF-α is well known to stimulate ROS production by mitochondria triggering apoptosis (135, 193). Interestingly, treatment of HeLa cells with TNF-α leads to the predominant oxidation of Trx2, with Trx1 remaining unaffected (151). Treatment of HeLa cells with cadmium and mercury chloride or sodium arsenite also shows substantial oxidation of both Trx1 and Trx2, but to an extent that is greater for the mitochondrial than the cytosolic isoform (150). The different redox response to growth factors, cytokines, or metals of thioredoxins located in the cytosolic (Trx1) or mitochondrial (Trx2) compartments clearly indicates that the source and duration of oxidant stimulation drives the cell to different and even opposite signaling responses such as proliferation or apoptosis. These observations raise the question of the different action in cell signaling exerted by oxidants generated from different sources (34), although dynamic cross-talk involves the various cellular ROS sources (see section V.I.). ROS produced in various compartments may result in different signaling responses. A number of conditions that increase mitochondrial ROS production prevent cell growth or stimulate cell death (209, 259, 280) so that the sustained and long-lasting mitochondrial generation of ROS appears to stimulate more growth arrest or apoptosis than cell proliferation—unlike NADPH oxidases—characterized by short-lived ROS production which drives the cell toward growth.
Metals that are capable of inducing the oxidation of Trx in cells are potent inhibitors of thioredoxin reductase (36, 41, 150) and generally belong to the “soft” class of chemicals. According to the principle that soft acids “prefer” to associate with soft bases, they preferentially react with the thiol/selenol motif of TrxR (36, 353). Inhibition of thioredoxin reductase, which hinders electron flux through the thioredoxin system, changes the redox conditions of that system, so that components upstream of TrxR such as pyridine nucleotides are completely reduced, whereas elements downstream of TrxR (Trx, Prx) are essentially oxidized (36, 62, 76). Over the years, a very large number of inhibitors and substrates of TrxR of natural or synthetic origin have been examined (9, 55, 96, 353). For instance, both cytosolic and mitochondrial isoforms of thioredoxin reductase are potently inhibited by gold complexes (139, 160, 295)—an effect that is essentially dependent on the binding of gold atoms to the cysteine and selenocysteine residues of the enzyme (36, 353). In isolated mitochondria, the inhibition of TrxR by the gold compound auranofin leads to abundant production of H2O2 (293) that is associated with a calcium-dependent mitochondrial membrane permeability transition (MPT) observed as swelling, specifically inhibited by cyclosporin A (CysA) and concomitant with a decrease in membrane potential (295). The MPT induced by the inhibition of TrxR is responsible for the release of the proapoptotic factor cytochrome c (296). Interestingly, after treatment of mitochondria with auranofin, no oxidation of pyridine nucleotides is observed, and only a slight decrease in total thiol groups takes place (296). The inhibitory effect of auranofin on mitochondrial thioredoxin reductase occurs at concentrations that do not affect GR, GPx, or the respiratory chain function (296), indicating that the observed effects are mainly attributable to an alteration in the mitochondrial thioredoxin system.
As previously reported, mitochondria are an important source of O2 •−, which dismutes and forms H2O2 (see section II.B.). Consequently, in mitochondria, the inhibition of TrxR2 by gold complexes leads to the build up of large amounts of H2O2 that cannot be detoxified by Prx, which, in these conditions, is unable to receive reducing equivalents from the thioredoxin system (36). However, as previously noted (section IV.A.1.), Prx3 can also receive electrons from Grx2 (148), therefore mitigating the effects due to inhibition of TrxR. The inhibition of the latter also leads to a shift in the redox state of thioredoxin, which is mostly found in its oxidized state (36, 51, 133) and may be responsible for the observed alteration of the permeability conditions of the mitochondrial inner membrane. ANT, a major component of the mitochondrial permeability transition pore, is known to be endowed with oxidation-sensitive cysteine residues located on the matrix-facing loops (see section V.I.). Although thioredoxin essentially acts as an effective reducing protein (166), in its oxidized form, it may reverse this role and act as an oxidant, which is able to modify the redox conditions of proteins (330). It has been shown in Escherichia coli that mutant strains lacking TrxR accumulate oxidized Trx, which can actively catalyze disulfide bond formation (330). A process whereby oxidized Trx can transfer its disulfide bond to a protein like reduced ribonuclease has previously been shown in vitro (221, 269). Therefore, accumulation of oxidized Trx may endanger the structures of the cell or alter its functions in compartments such as the cytosol or mitochondria. However, in mammalian Trx1, in addition to the cysteine couple at the active site, other cysteines are also present, which, on oxidation to disulfide bonds, inactivate Trx (284). This gives rise to the suggestion that the generation of non-active site disulfide bonds in Trx represents an autoregulatory function, preventing the formation of disulfide bonds in proteins (330). It should be noted that mitochondrial thioredoxin, which only possess the dithiol couple at the active site, is not subject to this type of regulation.
When cells are treated with TrxR inhibitors, the large concentration of H2O2 forming inside the mitochondrion can easily cross the mitochondrial membrane and diffuse to the cytosol compartment, from which it cannot be removed, as cytosolic thioredoxin reductase is also inhibited similar to the mitochondrial isoform. The first consequence of this is an increase in the oxidized condition of cytosolic thioredoxin (36 and references therein). This alteration can activate various signaling pathways. For instance, by oxidizing the Bax protein after interaction with the conserved Cys62 residue, H2O2 induces a conformational change with the subsequent translocation to mitochondria and oligomerization of Bax (253), which is enabled to release the propapoptotic factors located in the intermembrane space. The MAP kinase pathway is especially sensitive to these oxidizing conditions (260, 303) and, in particular, the oxidation of Trx removes its inhibitory effect on the apoptosis signal-regulating kinase (ASK1), thus favoring the apoptotic process (304). Of note, the complex gold(III)-ethylsarcosine dithiocarbamate, another inhibitor of TrxR, can induce apoptosis in HeLa cells by stimulating rapid phosphorylation of ERK1/2, in a process depending on thiol oxidation, as shown by the complete inhibition exerted by N-acetylcysteine (303). Although activation of the ERK1/2 route is commonly considered a cell survival pathway, there is also evidence that ERK1/2 can promote cell death (232), particularly after prolonged activation (305), for example, in the conditions occurring after complete inhibition of TrxR.
In TrxR, a truncated form lacking the two last C-terminal amino acids or the targeting of selenocysteine with electrophiles leads to the formation of SecTRAPs (selenium compromised thioredoxin reductase-derived apoptotic proteins), a modified form of TrxR that is able to trigger the formation of ROS and induce cell death (6). In these conditions, the activity of TrxR is completely lost, and it turns into an oxidase that can generate a large extent of oxidants (6).
On inhibition of TrxR, not only Trx but also Prx may shift toward more oxidized conditions. The redox state of Prxs after exposure to the TrxR inhibitor auranofin was estimated by Cox et al. in Jurkat T cells (76). In that study, the mitochondrial isoform of Prx (Prx3) appears to be considerably more sensitive to oxidation than its cytosol counterparts (Prx1 and Prx2), probably because of the proximity of Prx3 to the respiratory chain complexes, which are the sites of H2O2 production. In the apoptotic process induced by auranofin inhibition of TrxR, it has also been shown that Prx3 oxidation is an early event and apoptosis appears to be mediated by the Bax/Bak-dependent pathway, as it is inhibited by over-expression of Bcl-2. However, it should be noted that Prx3 can also be reduced by Grx2 (148), here too indicating dependence on the glutathione system.
Prxs are major sensors of H2O2 signaling (45, 116, 243, 288), and hence, in view of their marked oxidation observed in cells exposed to TrxR inhibitors (76), these enzymes might directly transmit their oxidized state to proteins involved in apoptosis (Fig. 11). All these observations emphasize the important role played by Trx, mediated by the action of TrxR, in maintaining a reduced Prx that is able to modulate the signaling action of peroxides. Therefore, when TrxR is inhibited, oxidized Trx and Prx accumulate, and may activate the apoptotic signaling pathways promoting disulfide bond formation associated with increased concentrations of H2O2 (Fig. 11).
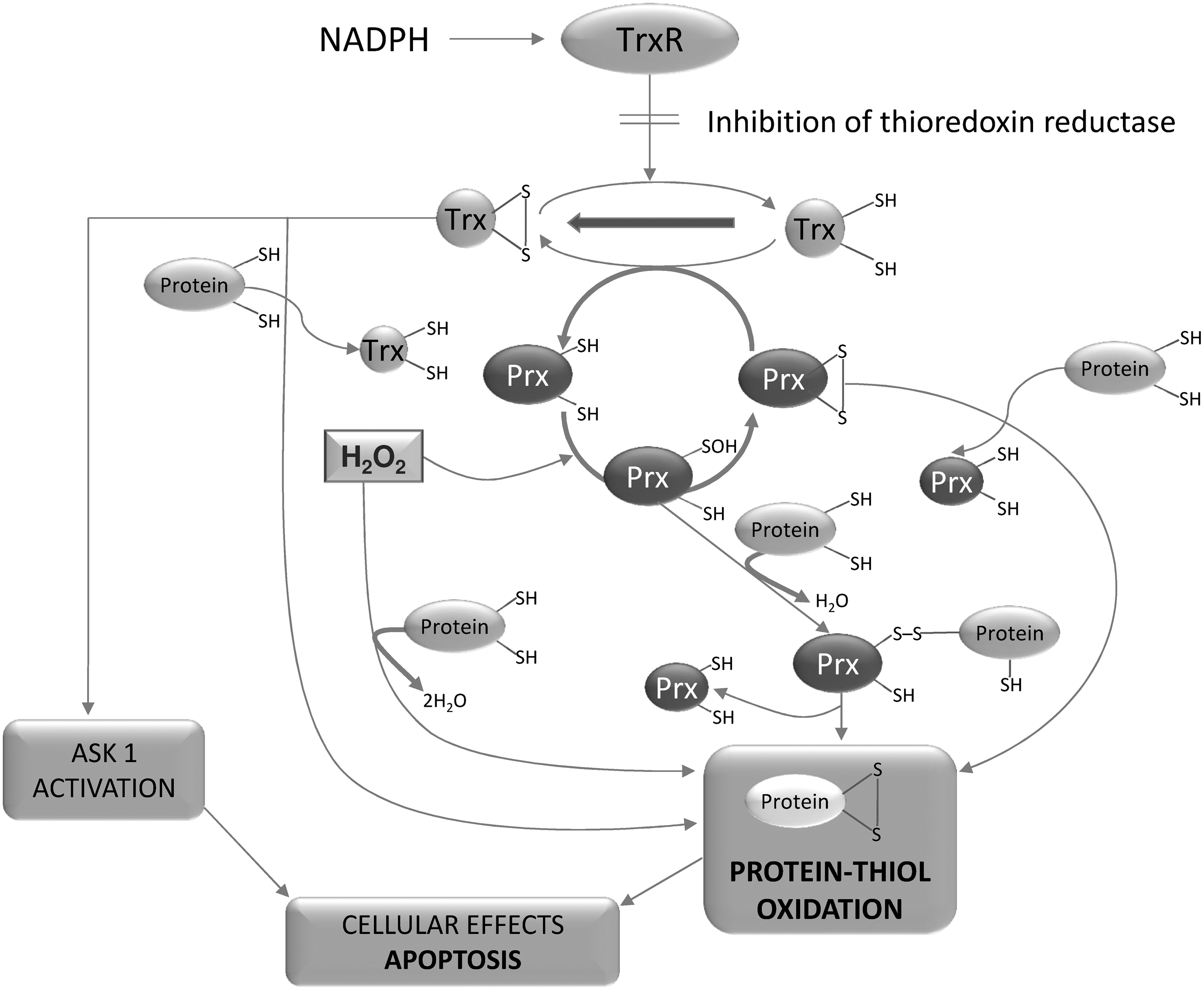
Cytokines and growth factors through NADPH oxidase induce the rapid and transient formation of H2O2, which oxidizes Trx(SH)2 to Trx(SS). However, the latter rapidly returns to its reduced condition and triggers a proliferative process (131, 147). Conversely, in mitochondria, inhibition of TrxR may give rise to a sustained and long-lasting production of ROS, therefore signaling the apoptotic fate of the cell. However, this distinction between mitochondria and NADPH oxidase actions is not absolute, as the active cross-talk between them and with many other sources of ROS should be taken into consideration (section V).
3. The glutathione system
The glutathione system is formed of the sequence NADPH, GR and glutathione (Fig. 10). Glutathione is the most abundant nonprotein thiol, and its biosynthesis takes place in two steps, both requiring ATP. The first reaction is catalyzed by γ-glutamylcysteine ligase, and the second requires glutathione synthase (181). Glutathione plays a critical role in controlling the apoptotic process, as increased GSH levels prevent apoptotic cell death; whereas GSH down-regulation by the specific inhibitor of glutathione synthesis BSO induces apoptosis in leukemia cells (122).
Cellular glutathione is mainly found in its reduced form, as the GSH/GSSG ratio ranges from 30:1 to 100:1 (169). In contrast to other cell compartments, the ER is a more oxidizing environment, as the GSH/GSSG ratio ranges from 1:1 to 3:1 (169). The highly reduced condition of glutathione is maintained by GR, which, in turn, uses reducing equivalents that are derived from NADPH. Although GR effectively reduces GSSG in basal conditions, any marked alteration in GSH/GSSG ratio during oxidative stress may lead to a disruption in redox homeostasis (12). Variations in the ratio may influence the signaling pathways involved in cell proliferation, apoptosis, autophagy, and gene expression (37).
GSH can deliver its reducing equivalents to a range of enzymes, including GPx, GST, and glutaredoxin (Grx) (89, 181, 210). GSH acts as a substrate of the GPx selenoproteins, which exist as distinct isoenzymes exerting cell- and tissue-specific functions in biochemical pathways and that are characterized by a selenol group which is very reactive toward peroxides (43, 45). GPxs are a family of up to eight distinct enzymes in which GPx1 to GPx4 are selenoenzymes together with GPx6 (depending on species); a cysteine replaces the selenocysteine at the active site in the remaining isoforms (347).
Although there are no reports stating that selenols in selenoproteins play a role in sensing hydroperoxides (45), in GPx4, selenenic acid, in addition to the ordinary two-step reduction by GSH, can also react with the cysteine residues of various proteins, thus transducing the oxidant signal (45). This event represents the molecular mechanism that takes place during sperm maturation in which GPx4 is specifically transformed into a structural protein (354). During sperm cell maturation, the absence of GSH drives GPx4 to accept electrons from the cysteine residues of proteins, forming disulfide bridges and cross-links with other proteins (69, 354). The sperm-associated cysteine-rich protein (SMCP) is involved in this process, as adjacent cysteine residues in SMCP peptides have been shown to undergo oxidation to disulfides by the action of GPx4 (225). This adjacent cysteine motif gives rise to an unusual eight-membered ring with a disulfide that is highly prone to reshuffling, in a process involving different cysteine-containing proteins (225).
Similar to GPx, glutathione also delivers reducing equivalents to the protein Grx, which, in mammalian cells, is located in both the cytosol (Grx1) and mitochondria (Grx2 and Grx5) (210). The mitochondrial isoform of glutaredoxin (Grx2) can react with GSH or TrxR, showing biochemical features of both Grx and Trx (182) and suggesting a link between glutathione and thioredoxin pathways in mitochondria.
One major function of Grxs resides in their ability to catalyze the reversible formation of mixed disulfides involving cross-links between glutathione and proteins (21, 25) and thus acting as sensors of the glutathione redox state. Glutathionylation in proteins may occur between a cysteine sulfenic acid residue and GSH, resulting in either a regulatory process or a defensive mechanism, preventing any further oxidation of the protein thiol involved (80). In proteins, the reversible formation of mixed disulfides with glutathione is a major event in redox regulation and signaling that may be compared with phosphorylation/dephosphorylation (210). Although the reduction of protein mixed disulfides with glutathione catalyzed by Grx is favored, the reaction may also take place in the opposite direction with the formation of mixed disulfides, particularly in conditions where GSSG increases (210, 302).
In cells subjected to oxidative stress and analyzed by a proteomic approach, several proteins undergoing glutathionylation have been identified, including cyclophilin A and Prx5 (119). Of note, human Trx can undergo glutathionylation at a site identified as Cys72 and its activity as insulin reductase in the presence of NADPH and TrxR is consequently abolished. However, the process is reversible, indicating that the GSH/GSSG ratio can regulate Trx functions, therefore establishing cross-talk between the glutathione and thioredoxin systems (56). A rapid, efficient mechanism for both the formation and reversal of glutathionylation takes place, suggesting the potential regulation of several different proteins, whereas alterations in protein-SSG conditions occur in disease conditions such as diabetes and cardiovascular, lung and neurodegenerative diseases (241). Similar to Trx, Grx is a substrate of ribonucleotide reductase and it also interacts with a number of factors involved in cell signaling and transcriptional activation such as PTP1B, PKA, PKC, Akt, and ASK1 (220 and references therein).
An important family of proteins using GSH is represented by GSTs, a superfamily of phase II enzymes involved in detoxification processes. They catalyze the conjugation of GSH with a large number of xenobiotics and are therefore involved in the elimination of toxic substances and potential carcinogens. GSTs are found in the cytosol, mitochondria, and microsomal membranes (235). They can also mediate the conjugation of GSH to several anticancer drugs; so, their action can be linked to the development of drug resistance in many tumor cells (235). Of interest, GSH-saturated πGST heterodimerizes with Prx6 and catalyzes glutathionylation of the oxidized catalytic cysteine of Prx in a process followed by reduction of the mixed disulfide (230). This suggests the synergy of the two proteins in counteracting oxidative stress (230). The Alpha class of GSTs show GPx activity toward lipid hydroperoxides, although GSTs do not act on H2O2 as a substrate. In addition to the reduction of hydroperoxides, GSTs can conjugate 4-hydroxynonenal (4-HNE), forming derivatives that can be transported out of the cell (11 and references therein). Since 4-HNE is a crucial molecule transducing apoptotic signaling, control of its level by GSTs represents a regulatory step in stress-mediated signaling (11 and references therein).
4. Importance of the inhibition of thioredoxin and glutathione systems for cancer chemotherapy
TrxR and Trx are over-expressed to high levels in many types of tumor cells, a condition that prevents apoptosis and favors cell growth. Their inhibition is consequently considered an important target in cancer chemotherapy (9, 36, 140, 220, 274, 345). This is further supported by the many observations that inhibition of TrxR leads to cell death. In the long list of inhibitors of TrxR (55), there are many recognized anticancer agents. However, a major problem may result from the lack of selectivity of many molecules between normal and tumor cells. However, some new gold(I) complexes specifically against cancer cells are now emerging (157, 281). Their effects depend on the inhibition of both Trx and TrxR, and this inhibition is more pronounced in cancer cells than in normal ones (281). Careful selection of ligands to the gold ion can provide these complexes with physico-chemical properties that specifically choose solely cancer cells for apoptosis (26). In particular, the preferential accumulation of gold compounds inside mitochondria that is achieved by exploiting the elevated transmembrane potential appears to be a valid means of specifically stimulating the death of cancer cells (26 and references therein).
Both thioredoxin and glutathione systems transfer reducing equivalents for reduction of peroxides, maintenance of reduced thiols in proteins, and functioning of ribonucleotide reductase (Fig. 10). Their actions are therefore intertwined, and, according to condition, a compensatory effect may take place if one of the two pathways is compromised. According to Mandal et al. (229), the genetic ablation of thioredoxin reductase does not change the behavior of tumor cells, such as proliferation, cell cycle distribution, and clonogenic potential, due to compensatory up-regulation of the enzymes involved in GSH synthesis, thus reflecting a redundancy between the thioredoxin and glutathione systems. Therefore, tumors defective in TrxR are highly sensitive to drugs leading to glutathione depletion such as BSO (229). It was recently shown that hepatocyte proliferation in mice requires a functional allele of the thioredoxin reductase 1 gene or the presence of GSH, indicating that both TrxR1- and GSH-dependent pathways can support the proliferation of hepatocytes (275). It should be noted that TrxR2 cannot sustain the proliferation of hepatocytes genetically lacking in TrxR1 (275). In some cancer cell lines, simultaneous targeting of glutathione metabolism by BSO and TrxR by auranofin significantly increases the toxicity generated by ROS production that is associated with inhibition of glucose metabolism (309). This indicates that both glutathione and thioredoxin systems should be compromised in order to sensitize cells to chemotherapeutic agents, whereas the sole inhibition of either glutathione or thioredoxin metabolism is not very toxic in the cell lines examined, indicating that the inhibited pathway may be compensated by the nontargeted one (309).
V. Redox-Regulated Systems and Their Role in Signaling Pathways
A. Transcription factor OxyR: a classic example of redox signaling
OxyR represents the classic and probably the model example of H2O2 sensing by a specific protein. In this transcription system, which is found in bacteria, H2O2 stimulates the formation of an intramolecular disulfide bond that induces a conformational change and the protein, in this oxidized form, can bind DNA and switch on the transcription of genes coding for several antioxidant enzymes (333, 376). A specific cysteine (Cys199) has been shown to form a sulfenic acid after interaction with H2O2. This reaction occurs with an apparent second-order rate constant of about 107 M −1 s−1 (10), that is, a relatively high rate, which is compatible with a signaling event. A further reaction with a second cysteine (Cys208) forms a disulfide link (376). The process is reversible, as the disulfide bond is reduced by Grx1, which thus deactivates OxyR (376). This is a well-defined and instructive mechanism of redox regulation of gene expression, offering the basis for understanding the more complex mechanisms occurring in mammalian cells.
B. The Gpx3/Orp1-Yap1 system in yeast. Peroxidases may act as sensors of peroxides
In Saccharomyces cerevisiae, transcription factor Yap1 is oxidatively activated by an increase in peroxide levels (85 and references therein). However, it does not act as a direct sensor of peroxides, but a second component, a GPx-like enzyme, Gpx3/Orp1, reacts with H2O2 to form a sulfenic acid that generates an intermolecular disulfide bond with the transcription factor Yap1. The Orp1-Yap1 disulfide cross-link is then converted into an intramolecular disulfide active form of Yap1, which can stimulate the expression of genes encoding antioxidant enzymes (85). Reduced Trx switches off the signal stimulated by Yap1 activation by reducing back the intramolecular disulfide (85). The dual role of thiol peroxidases in acting both in H2O2 scavenging and in redox signaling is discussed in a review article by Fourquet at al. (116).
C. Protein kinases and phosphatases: relationship between phosphorylation processes and redox signaling
In the cell, most biological processes are regulated by phosphorylation/dephosphorylation, which, by activating or inhibiting specific biochemical pathways, act as critical events in cell signaling. Both protein kinases and phosphatases are endowed with cysteines that are able to sense H2O2 and hence are under redox control (45, 65, 196, 337). There is a substantial agreement on the concept that oxidants act in opposite ways, stimulating kinases while inhibiting phosphatases (65, 342, 346) (Fig. 12).

An appropriate level of oxidants has been shown to increase phosphorylation, dependent on specific cysteine oxidation, and is observed in both serine/threonine and tyrosine residues (196, 250, 357). For instance, the activity of c-Src, a member of the Src family of kinases, is stimulated by oxidation involving Cys245 and Cys487, as shown by mutant insensitivity to oxidative effects (126). Of note, the activation of protein kinases by oxidants may also be an indirect effect due to the inhibition of protein phosphatases (65 and references therein). The molecular mechanisms of redox regulation of protein kinases are discussed in a recent review (196).
With reference to redox signaling, particular attention has been devoted to protein phosphatases, which, in contrast to kinases, are easily inactivated by ROS-induced thiol alterations (65, 206). The active site cysteine of PTP, critical for its enzymatic function, is characterized by a low pKa (218, 337, 375). For instance, the pKa of the active site cysteine of Yersinia PTP is 4.67 (375), whereas that of PTP1 involving Cys215 shows a value of 5.57 (218). A sulfenic acid is formed after an interaction of the catalytic active site thiolate with H2O2, as shown when the electrophilic reagent NBD-Cl is used (87). Crystallographic studies of PTP1B have also revealed the occurrence of a sulphenyl amide bond formed by nucleophilic attack of the backbone nitrogen atom of Ser216 on the sulfur atom of Cys215 previously oxidized to a sulfenic acid species (306, 356) (Fig. 12). However, the in vitro reactivity of the thiolate group at the active site of PTP1B with H2O2 exhibits a relatively low rate, ranging from 9.1 to 42.8 M −1 s−1 (17, 87) and is thus comparable to that observed for low-molecular-weight thiols (364) (see also section III.A.1.a.). Therefore, in a cellular environment, the unfavorable competition of PTP1B with glutathione, present at a concentration which, even in its dissociated form, exceeds that of PTP1B, can be predicted (113). Nevertheless, in a rat alveolar macrophage cell line producing H2O2 on stimulation of the respiratory burst, the formation of a disulfide bond between PTP1B and GSH has been observed, demonstrating physiological glutathionylation of PTP1B (299). All these observations indicate that the mechanism of peroxide-mediated inactivation of PTPs is more complex. It has been proposed that H2O2 is converted to more effective peroxides (337 and references therein) such as, for instance, peroxymonocarbonate (377) or that it has an indirect effect after a reaction with a primary protein sensor (362). Brigelius-Flohé and Flohé (45) discuss a mechanism whereby a peroxide is first sensed by any of the various thiol peroxidases with the consequent formation of a disulfide bond with the active site thiol of the phosphatase. In the following step, GSH cleaves the disulfide bond, generating a glutathionyl residue at the level of PTP (45). The process is made reversible by the reduction of the mixed disulfide by the various reducing systems and in particular by Grx, which acts as a dethiolating reagent (18). It has also been proposed that the in vivo process may differ from that in vitro due to a protein/protein interaction which can increase the rate constant of the enzyme toward H2O2 (45). The chemical and structural aspects of redox-regulated PTPs have been recently illustrated in a review article by Tanner et al.(337).
D. NF-κB: the first mammalian transcription factor shown to undergo redox regulation
NF-κB, a transcription factor stimulated by a variety of mediators and playing a crucial role in the inflammatory response (45, 132 and references therein), was the first transcription factor shown to undergo redox regulation (326), as it is potently and rapidly activated when cells are treated with H2O2 (311). The role of ROS and in particular of H2O2 in activating NF-κB is exerted essentially through the classic IKK-dependent pathway (132), in which phosphorylation of the inhibitory protein IkB stimulates its degradation by the proteasome, favoring the migration of NF-κB from the cytosol to the nucleus (Fig. 13, left). Reduced Trx antagonizes NF-κB activation in the cytosol (106) but acts in the opposite way in the nucleus, activating NF-κB by promoting its binding to DNA (185, 234). A critical cysteine (Cys62) of the p50 subunit, which is essential for binding to DNA, is particularly sensitive to oxidation, probably because it is surrounded by three basic arginine residues (255) (see also Fig. 8).

Similar to Trx1, the thioredoxin-related protein 14 (TRP14), a new member of the thioredoxin family, can also inhibit the signaling pathways depending on NF-κB (178). The direct target of TRP14 has been identified as the dynein light chain 8 (LC8), which is maintained in its dithiol reduced form by TRP14. In this form, LC8 binds to IκB, preventing its phosphorylation and consequent degradation. However, the formation of an intermolecular disulfide bridge in LC8, which can thus act as a sensor of the oxidant signaling condition, leads to its dissociation from IκB, which can then undergo phosphorylation by the IKK complex (178).
E. The Keap1-Nrf2-ARE pathway: redox signaling by nutritional phytochemicals
The Keap1-Nrf2-ARE pathway is a clearly characterized, discrete redox signaling pathway, with the prominent feature of responding to signaling molecules acting on specific thiol groups that undergo oxidation or alkylation (188, 200) (Fig. 13, right). A very large number of different and chemically unrelated compounds of natural or synthetic origin activate the expression of the genes regulated by Nrf2 (343), including diphenols and quinones, Michael acceptors, isothiocyanates and many sulfur-containing compounds, selenium compounds, heavy metals, and trivalent arsenicals (343). Despite the chemical heterogeneity of these compounds, one common feature is their electrophilic character, suggesting their potential capacity to modify specific reactive thiol groups of proteins (Fig. 13, right). A classification based on the reaction mechanism of these compounds with the cysteine residues in proteins has been proposed (165). Not only xenobiotics but also compounds of endogenous origin linked to oxidative stress processes, such as H2O2, hydroperoxides, and peroxynitrite, stimulate the adaptive response, depending on the Keap1-Nrf2 system (45). Of note, structurally different electrophilic lipid oxidation products, deriving from both enzymatic and nonenzymatic pathways, have been shown to bind thiols of Keap1 and activate the antioxidant response element (ARE) response (208). They include 15-deoxy-Δ12,14-prostaglandin J2, (15d-PGJ2), 15-A2t-isoprostane (15-A2t-IsoP), and 4-HNE. 15-A2t-IsoP and 4-HNE, bearing only one electrophilic β-carbon, are less effective than 15d-PGJ2, which has two electrophilic sites (208). The Keap1-Nrf2-ARE system is involved in the regulation of hundreds of genes encoding proteins engaged in the maintenance of cellular redox homeostasis and defense against toxic substances (343 and references therein). This signaling complex exists as the association of two major proteins, Keap1 and Nrf2. Keap1 is associated with actin and contains sensitive thiols that can be modified by oxidation or alkylation (189) (Fig. 13, right). Modification of Keap1 causes the dissociation of Nrf2, which translocates to the nucleus and activates transcription, after binding to the ARE in the promoter region of genes encoding for battery of enzymes protecting the organism against endogenous and exogenous stressors (45, 189, 343). Keap1 contains 25 (mouse) or 27 (man) cysteine residues (15), ten of which are especially reactive because of the presence of positively charged amino acids nearby (91) (see also Fig. 8). This makes Keap1 an ideal sensor that is particularly prone to collect messages conveyed by the various inducers described above. Studies based on mass spectrometry of tryptic peptides of Keap1 have shown that the most reactive cysteine residues in Keap1 are Cys257, Cys273, Cys288, and Cys297, all of which are present in the intervening region (IVR) cysteine-rich domain (91). A mutation analysis of the cysteines in the IVR domain shows that modification of Cys273 or Cys288 by substitution of Cys with alanine or serine makes Keap1 incapable of suppressing the activation of Nrf2, which emerges as constitutively active, while modification of the other two residues (Cys226 and Cys257) was unable to disturb the repressor activity of Keap1 (15, 208, 359, 371). These observations demonstrate the critical role played by Cys273 and Cys278 in signaling by oxidants and electrophiles. Another cysteine, Cys151, is necessary to prevent degradation of Nrf2 by the natural product sulforaphane or oxidative stress (371). In a recent study in which oxidant molecules (H2O2, HOCl or nitric oxygen derivatives) were used instead of alkylating agents to stimulate the oxidation of thiol groups of Keap1, the formation of a disulfide bridge linking two molecules of Keap1 via Cys151 was observed in association with Nrf2 stabilization (115). This oxidation turned out to be transient, indicating that the cell reducing systems depending on thioredoxin or glutathione pathways can recycle Keap1 back to its reduced form (115).
Several models describing the regulation of Keap1-Nrf2 activity have been proposed starting from the original sequester and release pathway. The original model suggested that alterations induced by oxidation or alkylation of specific cysteine residues of Keap1 lead to the rapid release of Nrf2 in the cytosol, followed by its relocation to the nucleus. All the proposed mechanisms were comparatively discussed in a recent review article (15). In particular, according to the two-site substrate recognition model (“hinge and latch” or “fixed-ends” model) (15, 236, 344), two different binding sites on Nrf2 were shown to interact with each of the two molecules forming the Keap1 homodimer. The two binding motifs have different affinities, that is, the high-affinity ETGE (“hinge”), and low-affinity DLG (“latch”). On thiol modification, Keap1 changes its conformation and causes the release of the weaker interaction, while the other bond is retained. In this new condition, the ubiquitination and degradation of Nrf2 no longer occurs, and newly synthesized Nrf2 can accumulate. Nrf2 is also endowed with reactive cysteines that are directly involved in the regulation of its transport through the nuclear membrane and binding to DNA (343). In particular, it has been shown that the Cys506 of Nrf2 should be maintained in its reduced form in order to bind efficiently to DNA, as site-directed mutagenesis of Cys506 to serine in the DNA binding domain of Nrf2 lowers its affinity for the ARE (40). In the nucleus, Trx plays a critical role in keeping the Cys506 of Nrf2 reduced and thus maintaining efficient binding to DNA (149). Therefore, signaling by this transcription factor involves an oxidative step in the cytosol, followed by a reductive process in the nucleus (134, 149) (Fig. 13, right). This behavior is similar to that of other transcription factors such as NF-κB, AP-1, and p53, all of which require a reduced cysteine residue to favor their binding to DNA (134).
Nrf2 plays a protective role in contrasting tumorigenesis, as nrf2-deficient mice show an enhanced sensitivity to chemically induced carcinogenesis, while the action of the chemoprotective agent oltipraz is almost totally abrogated (283). Moderate stress make cells more resistant to further and more severe stress by up-regulation of cellular defensive systems (46). In contrast, tumor cells can take advantage of Nrf2-dependent up-regulation of defense systems, making them resistant to apoptosis, autophagy, and anticancer drugs (46), as shown by prevention of tumor growth and the increased efficacy of anticancer drugs by genetic knockdown of Nrf2 (222).
Currently, there is a widespread consensus regarding the correlation between an optimal intake of nutritional phytochemicals deriving from a diet rich in fruit and vegetables, and the prevention of various forms of cancer, cardiovascular and aging-related diseases. Several activators of Nrf2 (Fig. 14) come from diet and essentially belong to the categories of isothiocyanates, polyphenols, and carotenoids (61, 335). These phytochemicals can activate the adaptive response, as they either directly modify the sensitive cysteine of Keap1 by an alkylating reaction or indirectly change the cell redox conditions to a more oxidized state by undergoing autoxidation with the formation of oxidants acting on the thiol groups of Keap1 (44, 246). Cruciferous vegetables are endowed with abundant precursors of isothiocyanates (glucosinolates) and sulforaphane, a product with chemopreventive properties, is the best-known member of this class (44 and references therein). Due to the high electrophilicity of the carbon atom in the isothiocyanate group (–N=C=S), sulforaphane readily reacts with thiol groups to yield a dithiocarbamate (15) (Fig. 14A). Polyphenols are a very large, heterogeneous class of compounds with antioxidant and health-promoting action that exert most of their effects by modulating various cell signaling pathways (117). Some major constituents of tea, such as the flavanol epigallocatechingallate (EGCG), have been shown to activate Nrf2 (246) in different ways. EGCG can, in fact, undergo an autoxidation process (172), forming ROS that can oxidize the cysteine residues of Keap1 (Fig. 14B). In addition, the oxidized form of EGCG can directly bind to the cysteines of Keap1 or conjugate with glutathione (246, 307), creating a transient alteration in the redox state and leading to the activation of kinase cascades, prompting Nrf2 phosphorylation. Consequently, the accumulation of Nrf2 in the nucleus, followed by ARE binding, is enhanced (246). Clearly, these mechanisms can be extended to other polyphenols that are shown to activate the Keap1-Nrf2 system. Molecules such as curcumin, a secondary metabolite derived from turmeric and with two Michael acceptor centers, is another phenol compound that is effective in this system (15 and references therein). Carotenoids are also able to activate the Keap1-Nrf2-ARE transcription system, although their effects should be ascribed not to the intact molecule but to the carotenoid oxidation products that are the active species (214).

F. Hypoxia-inducible factor-1: a critical modulator of oxygen homeostasis
The hypoxia-inducible factor-1 (HIF-1) is a heterodimeric transcription factor formed of subunits HIF-1α and HIF-1β and acting as a regulator of oxygen homeostasis (313). In normoxia, hydroxylation of a specific proline residue in the HIF-1α subunit occurs, and the latter undergoes permanent ubiquitination and degradation (313). In the HIF signaling system, the redox sensor is represented by the enzyme prolyl hydroxylase, which, in the presence of oxygen, α-ketoglutarate, and Fe(II) at the catalytic site of the enzyme, hydroxylates HIF-1α, targeting it for ubiquitination and proteasomal degradation. This leads to a low steady-state level of HIF-1α that is associated with a lack of formation of the transcription complex (27, 45, 57, 93, 313).
In hypoxic conditions, prolyl hydroxylase is inactivated, ubiquitination is prevented, and HIF-1α is consequently able to translocate to the nucleus and elicit transcriptional activation. Target genes are those involved in the cell adaptation to hypoxia (45, 57, 254, 313). ROS may participate in HIF-1α activation and, specifically, the activity of the sensor enzyme prolyl hydroxylase may be modulated by ROS (57, 254 and references therein). Hypoxic conditions stimulate the mitochondrial generation of ROS, which, diffusing to the cytosol, causes post-translational modifications of prolyl hydroxylase, such as a shift in the redox state of iron from Fe(II) to Fe(III). Since iron is a critical cofactor of prolyl hydroxylase, this oxidation limits its activity (22, 57). It was recently shown that, in a human osteosarcoma cell line treated with H2O2, dual regulation of HIF-1α takes place, as at short times stabilization of HIF-1α occurs, while at longer times destabilization is observed. This behavior was shown to be closely related to modulation of prolyl hydroxylase 2 activity (254). Of note, increased Trx1 expression in human breast carcinoma MCF-7 cells significantly augments HIF-1α protein levels in both normoxic and hypoxic conditions, with a concomitant increase in the production of VEGF and tumor angiogenesis (360).
G. Ero1: Disulfide bond formation and maintenance of redox homeostasis in ER
Oxidative protein folding (see section III.A.2.a.) depends on signaling pathways in which regulation of Ero1 activity plays a central role (318). The de novo generation of disulfide bonds leads to the formation of one molecule of H2O2 per molecule of disulfide formed. Clearly, the extent of H2O2 formation needs to be closely controlled in order to prevent oxidative stress (289, 339). This control is exerted on Ero1, which, in addition to catalytic cysteines, also contains regulatory cysteines that are able to form intramolecular disulfide bonds (289, 318, 338). As a result, in oxidizing conditions giving rise to regulatory disulfides, the mobility of shuttle cysteines may be restricted and the activity of Ero1 is consequently depressed (318). Redox control of Ero1 appears to be particularly important in specialized secretory cells in which large amounts of disulfide bond-containing proteins are formed (318 and references therein). On the basis of the ability of Prx4 to oxidize PDI by H2O2 generated from different sources, including Ero1 (see section III.A.2.a.), it has also been speculated that H2O2 generated by NADPH oxidases located in the ER connect the extracellular signaling to the oxidation of proteins (379).
H. SOD: redox control
A Cu, Zn-SOD occurs in the mitochondrial intermembrane space, and can reversibly change from an inactive (dithiol) to an active (disulfide) form according to the level of O2 .−/H2O2 in the mitochondrial matrix or cytosol (170). Inhibition of TrxR1 in the intermembrane space has also been observed to prolong the duration of the active state of SOD1, indicating that the thioredoxin system can reversibly inactivate SOD1 by keeping its critical thiol groups reduced (170).
I. Control of mitochondrial functions and redox regulation of permeability transition
Mitochondria fulfil a number of functions in the cell (Fig. 15). It has long been known that almost all the mitochondrial processes, such as the electron transport chain, oxidative phosphorylation, and active or passive transport, are controlled by the redox state of protein and nonprotein thiol groups (181). In particular, the permeability conditions of the mitochondrial membranes are closely dependent on the integrity of thiol groups, as treatment of mitochondria with thiol oxidants or alkylating agents generally leads to extensive swelling (320). Respiratory substrates appear to control the permeability conditions of the mitochondrial membranes, essentially supplying reducing equivalents to the thioredoxin or glutathione systems existing in the mitochondrial matrix (30, 297).
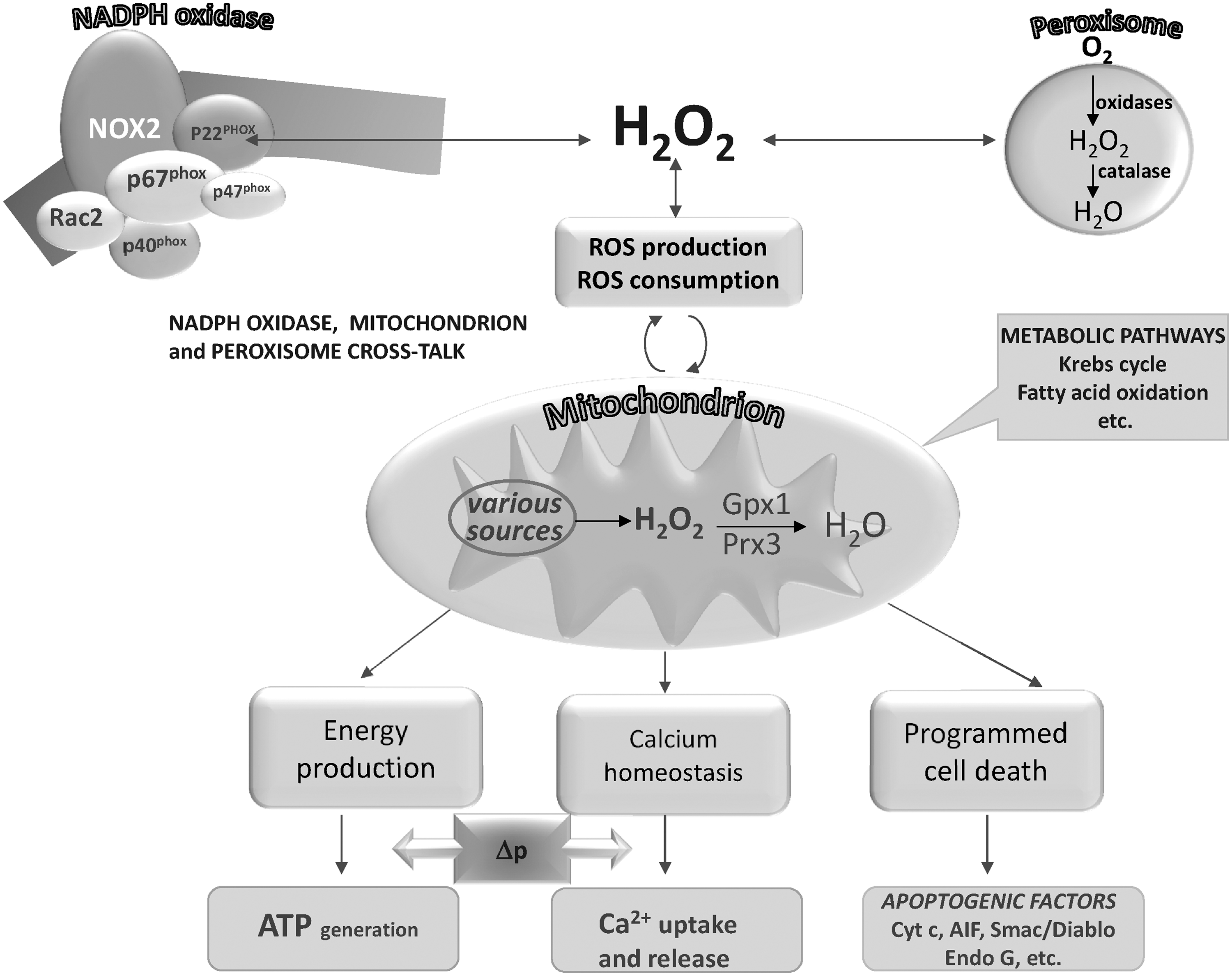
The process of MPT involves a rapid increase in the permeability of the inner mitochondrial membrane, which allows free passage to any solutes with a molecular mass lower than 1.5 kDa (24, 143, 198). This change in permeability properties depends on the opening of a channel in the inner membrane, called the mitochondrial permeability transition pore (143). Although the molecular composition of the pore is controversial (383), several proteins with either structural or regulatory roles are believed to participate in its formation, including the ANT and cyclophilin D (CypD), a critical regulatory protein (198). However, recent genetic evidence shows that previously considered core elements of the pore appear to play a regulatory role instead (383 and references therein). In addition to calcium ions, the opening of the pore is very sensitive to a large number of pro-oxidants, including H2O2, indicating that potential redox regulation by oxidant signaling molecules takes place (198, 381). For instance, oxidation of the cysteine residues of ANT causes pore opening (74). It was also shown that the permeability transition pore may be affected at two different sites (64, 75). The first is in equilibrium with the pyridine nucleotide pool, as oxidation of the latter leads to pore open probability in conditions in which glutathione is kept completely reduced. The second site is a redox-sensitive dithiol that is cross-linked by arsenite or phenylarsine oxide and may be in equilibrium with the glutathione pool (64, 75). According to McStay et al. (238), the site sensitive to oxidation of glutathione mediated by oxidants such as diamide or tert-butylhydroperoxide is represented by a cross-link between the Cys160 and Cys257 of ANT (238). Other observations suggest that pyridine nucleotide oxidation leads to permeability transition in a process that is independent of glutathione oxidation, at least when the process is induced by Ca2+/phosphate or acetoacetate, and in which dithiols such as thioredoxin or lipoamide are probably involved (30, 297). Altogether, these data indicate that both glutathione and thioredoxin systems play a role in the control of MPT conditions in a process triggered by oxidants and sensed by thiols. All the components of the redox networks of thioredoxin (Trx2, TrxR2, Prx3, and Prx5) and glutathione (GSH, Gpx1, Gpx4, and Grx2) are present in mitochondria and contribute, in a parallel fashion, to the control of mitochondrial redox signaling and functioning (373). The glutathione and thioredoxin systems can also communicate: For instance, as reported above, Grx2 can be reduced by both GSH and TrxR2 (182). More specifically, in the liver, GPx1 is the prevailing isoform in the cytosol, although it has also been found in the mitochondrial fraction (94). GPx4 is expressed predominantly in rat testis, where it was found in both cytoplasm and mitochondria, but it also occurs in somatic tissues, although to a lower extent (278, 300).
Thioredoxin 2 acts as a regulator of pore opening by preventing the mitochondrial permeability transition induced by exogenously added peroxides and calcium ions as shown by comparing the extent of swelling of liver mitochondria isolated from wild-type or Trx2 transgenic mice (154). It has been observed that the mitochondria of HEK-293 cells over-expressing Trx2 show an increased membrane potential and are more resistant toward etoposide-induced cytotoxicity (82), suggesting the anti-apoptotic function of mitochondrial Trx. Trx2 can also prevent apoptosis by inhibiting the mitochondrial-located ASK1 protein (374). Deficiency of Trx2 in embryos is lethal, coinciding with the maturation of mitochondria, and leads to massive apoptosis (257). In addition, Trx2-deficient cells accumulate ROS and are subject to apoptosis, in a process accompanied by a decrease in intracellular and intramitochondrial GSH, which is probably used to compensate for the deficiency of the thioredoxin-dependent ROS removal system (336).
The immunosuppressive cyclic peptide CysA is a specific inhibitor of MPT and exerts its effect by interacting with CypD (144). The latter is the mitochondrial isoform of a family of peptidylprolyl isomerases that catalyze the cis-trans isomerization of peptidylprolyl bonds (310). CypD is a critical regulator of the permeability transition pore (68), as also shown by the observation that mitochondria from mice lacking CypD are resistant to swelling (14, 248). According to Linard et al. (213), Cyp-D may act as a redox sensor in mammalian mitochondria. The Cys203 of CypD, acting as an oxidation-sensitive site, plays a major role, and it is also suggested that a disulfide bond involving Cys203 and Cys157 is responsible for the redox regulatory process. Therefore, Cyp-D acts as a redox sensor in mitochondria inducing MPT, which, in turn, is responsible for cell death (213). The critical role of the Cys203 of CypD in the activation of MPT was further highlighted by Nguyen et al. (252), showing that liver mitochondria prepared from mice expressing Cys203Ser CypD were resistant to swelling induced by calcium ions. H2O2 is known to induce pore opening (381), acting in mitochondria as a signaling molecule sensed by specific thiol groups. Other studies (177, 205, 244) clearly show the link involving cyclophilin, Prx, and Trx and highlight the role of this protein in mitochondrial signaling events. In particular, the finding that chloroplast cyclophilin can be targeted and reduced by Trx (244) reinforces the idea of the role of cyclophilin in redox cellular signaling, which adds to its recognized isomerase function.
The presence in mitochondria of all the major components involved in redox signaling responses acquires special significance when we consider the increasingly characterized network of mitochondrial kinase/phosphatase proteins, widely distributed in all mitochondrial subcompartments and potentially subject to redox control, revealing active interplay between phosphorylation processes and redox signaling events (114).
In the integrated cell system, the mitochondria are involved in intensive cross-talk with other cell components (79). ROS produced by NADPH oxidase stimulates ROS generation from mitochondria and, conversely, mitochondria stimulate NADPH oxidase activity through oxidant molecules (378). In conclusion, mitochondria are important generators of redox signaling molecules acting in metabolic adaptation and, according to the duration and intensity of ROS production, are able to signal the induction of apoptosis or autophagy (104).
J. Endoplasmic and sarcoplasmic reticulum calcium movements: a link between redox signaling and calcium signaling
In the sarcoplasmic and endoplasmic reticulum, calcium ions are actively taken up by an energy-dependent calcium pump (SERCA), and their release to the cytosol is mediated by specific calcium-release channels such as ryanodine receptors (RyRs) and inositol 1,4,5-trisphosphate receptors (IP3Rs) (100, 184, 308). The uptake and release of calcium are responsive to changes in redox conditions, as both RyRs and IP3Rs are endowed with a large number of cysteine residues that are prone to oxidative modification (184, 341). Therefore, the calcium channel response to oxidants essentially depends on thiol oxidation, clearly showing that the overall process depends on redox signaling. The ryanodine receptor (RyR) open probability, a condition facilitating calcium release, is increased by oxidation of cysteine residues, which are S-nitrosylated or converted to intraprotein disulfides or mixed disulfides with glutathione (308, 341). Similarly, the IP3R channel is also sensitive to the thiol redox state (184). Treatment of isolated sarcoplasmic reticulum (SR) vesicles with thiol reagents causes rapid release of calcium ions. Of note, while monothiol reagents such as p-chlomercurybenzoate and mersalyl give rise to a marked calcium release, the dithiol reagent diamide is ineffective (33). Not only in isolated SR but also in cell systems, the addition of thiol-oxidizing agents modifies the redox conditions of specific cysteines located in the cytoplasmic domain of RyR channels (231, 341). The results obtained in isolated SR vesicles and in cells indicate that modifications of thiol groups alter the conformation of proteins, therefore facilitating Ca2+ release. Recently, stretch-dependent signal transduction has been examined in cardiomyocytes in which low physiologic stress had been applied and caused Ca2+ release, triggered by local production of ROS (276). This is an attractive example of mechanotransduction of Ca2+ movements, mediated by ROS produced by activated NADPH oxidase. These oxidant species can sensitize and activate the ryanodine receptor by thiol oxidation, and consequently promote the release of calcium from SR (159, 276). The SERCA is also subject to redox regulation, depending on cysteine oxidation as, according to the level of ROS, its activity can be reversibly stimulated or the enzyme can undergo irreversible oxidative modification including either sulfonylation or nitration of specific cysteines or tyrosines, respectively (308). Oxidation to sulfonic acid of the Cys674 of SERCA prevents redox regulation in an irreversible manner (370). Taken together, all these observations indicate the existence of active cross-talk between ROS signaling and the various calcium signaling processes and functions, which may be important in both normal and pathological conditions (158).
VI. Conclusions
Scientific interest in redox signaling processes is increasing considerably, as a review of the literature on the subject clearly shows. Some points have attained broad consensus among scientists in this field. First, there is general agreement that ROS act not solely as noxious agents but also as signaling molecules. Among ROS, H2O2 is a significant oxidizing molecule that is involved in signaling, although superoxides, electrophiles, and other peroxides also play direct roles. Lipid hydroperoxides enzymatically generated by COXs and LOXs are also important signaling molecules. “Reactive” thiols are the main sensors of oxidant signaling, particularly those of Prxs (45). Redox signals, initiated by cytokines, hormones, growth factors, or other events, are transmitted to enzymes and transcription factors and also involve specific proteins of subcellular organelles such as mitochondria and sarcoendoplasmic reticulum, conferring functional significance on signaling.
The production of redox signaling molecules should not be considered as isolated and pertinent to a single source or pathway, as very active interplay between the various ROS sources occurs in the cell (378). Particular attention has been devoted to the redox communications occurring between mitochondria and NADPH oxidase in which ROS production by either source stimulates reciprocal production of oxidants (90, 378). NADPH oxidase has been defined as a “master oxidase” capable of modulating the release of ROS from various cellular sources (237) and highlighting its critical role in signaling.
The presence of a network of redox signaling processes suggests that various pathophysiological conditions such as cancer, neurodegeneration, and aging could be targeted by specific drugs designed to combat them. Translational potentials thus emerge from elucidation of the pathways involved in the many redox signaling processes. For instance, some drugs used or proposed for antitumor therapy can markedly increase ROS in tumor cells and hence change the redox state of critical thiols in proteins and signal for cell death. Another important translational aspect stemming from basic knowledge of signaling cascades is the possibility of devising nutritional guidelines. Many phytochemicals such as polyphenols and isothiocyanates interfere with signaling pathways, particularly those of the Nrf2-Keap1 system. It is hoped that future work will identify all the pathways along which redox signaling occurs and clarify the processes and their importance in cellular physiology and pathology.
Comment on References
The purpose of this review is to give readers a broad-spectrum picture of redox signaling events. However, the literature on this subject is enormous and rapidly increases daily. In addition to primary sources, very many excellent reviews and monographs can be found. In order to describe the subject in a generalized form, the authors have frequently quoted secondary sources, although they realize that, as a consequence, many excellent papers may have been completely missed. They regret this and offer their apologies.
Footnotes
Acknowledgments
This work was supported by the University of Padova and the National Research Council of Italy (CNR).