
Abstract
Introduction
Under normal circumstances, the adverse effects of ROS are counteracted by antioxidants such as the enzymes superoxide dismutase, catalase, and glutathione peroxidases. Recently, a novel endogenous antioxidant, the lipocalin and radical-binding protein α1-microglobulin (A1M), has been described to protect cells and matrix fibrils from Hb-, heme- and ROS-induced oxidative damage (28, 33, 36, 37). A1M is a reductase (2), a binder of heme groups (1d, 22), and a multispecific scavenger of small organic radicals (1c). It is a 26-kDa plasma and tissue protein (1b) mainly synthesized in the liver (45) and in less amounts in peripheral organs (4, 20). A1M is found in the blood at 1–2 μM (5, 8) and is rapidly distributed to the extravascular compartments (23). Adding to the antioxidation properties, an increased synthesis in the liver, blood cells, placenta, and skin keratinocytes is induced by cell-free Hb and ROS (28, 32, 34, 35).
Innovation
Import of the radical- and heme-binding protein α1-microglobulin from the extracellular compartment confers protection of mitochondrial structure and function against oxidative stress during cellular insult.
Cell death, whether accidental, stress-induced, or apoptotic, frequently involves activation of cell degradation and recycling programs (7). Therefore, cell death is an energy-depending process in need of preserved and intact mitochondrial functions. Furthermore, cell death in vivo is associated with generation of redox-active compounds, and the dying cells need to protect themselves and surrounding tissue from oxidative damage. Mitochondria are generally regarded as a major source of ROS, and autophagocytic elimination of mitochondria (mitophagy) is suggested to play a central role during programmed cell death (24). Thus, there is a need of protection from oxidative stress generated by mitochondria and other sources, on one hand, and a need of functional mitochondria, on the other, during programmed cell death, and it is not yet understood how this is accomplished. We hypothesized that enhanced antioxidation activity may play a pivotal role and therefore investigated a possible association of A1M to intracellular structures during cell death. Here, we have found evidence of a specific binding of A1M to a subunit of Complex I, localizing A1M to mitochondria. The results also indicate that this localization confers protection of the mitochondrial structure and function during fatal oxidative insult toward the cell.
Results
Specific binding of A1M to damaged cells
Binding of A1M to apoptotic and healthy cells was analyzed by flow cytometry and compared to untreated cells. First, apoptosis was induced in murine T-cell hybridomas (HCQ.4) by cross-linking of the CD3 molecule with immobilized anti-mouse CD3 antibodies (Fig. 1A, E), or by incubation with 5% ethanol or 10% dimethyl sulfoxide (DMSO) (Fig. 1B). These treatments resulted in DNA fragmentation and uptake of trypan blue after 15–18 h (not shown). A weak binding of A1M could be detected to untreated cells (Fig. 1A, left panel). An additional stronger binding was detected to cells cross-linked with anti-CD3 (Fig. 1A, right panel) or treated with ethanol or DMSO (Fig. 1B). The binding could be correlated to propidium iodide (PI) uptake; that is, only cells that could incorporate PI displayed the stronger binding of A1M (Fig. 1B). Flow cytometry of a murine pre-B-cell line (70Z/3), induced to apoptosis using the benzamide drug declopramide (3-CPA) and incubated with A1M followed by anti-A1M, showed similar results (Fig. 1C), indicating that the binding to apoptotic cells is not restricted to T cells.

To further characterize the A1M binding to damaged cells, the binding was also studied using fluorescence and confocal microscopy of the human erythroid cell line (K562) (Fig. 1D) and the promyelocytic cell line HL 60 (not shown) exposed to heme, and incubated with A1M followed by anti-A1M. Two different types of staining could be seen: a pronounced, intracellular, and uniform staining to a subset (∼6%) of the cells (confocal microscopy, Fig. 1D), and a weak granular staining to the cell surface of most cells (fluorescence microscopy, not shown). Similar results were obtained with the HL 60 cells. These results indicate that the strong binding of A1M to damaged cells is mainly intracellular.
To investigate the specificity of the binding, a competitive cell-binding assay was performed on HCQ.4 cells, induced to apoptosis by CD3 cross-linking, and compared to normal untreated cells. 125I-labeled A1M and an excess of unlabeled A1M, ovalbumin, bovine serum albumin (BSA), or α1-acid glycoprotein (AGP) were added to the cells (Fig. 1E). More A1M was bound to apoptotic cells (Fig. 1E, right) compared to untreated cells (left). Excess of unlabeled A1M blocked the 125I-A1M binding to the same basal level for apoptotic cells as for untreated cells. The reduction was found to be significant (p<0.001). None of the unlabeled control proteins could significantly reduce the binding of 125I-A1M to untreated cells, thus indicating a specific binding of A1M. To the apoptotic cells, there was a small, significant reduction by the control proteins (p<0.05). This small reduction may be due to an increased unspecific background binding to exposed intracellular structures. Accordingly, the results indicate a specific strong binding of A1M to apoptotic cells. From a Scatchard plot, an affinity constant for the A1M binding to apoptotic HCQ.4 cells could be determined to 1×106 M −1. The viability of these cells was 25% according to trypan blue exclusion.
As mentioned above, the A1M-binding cells internalized PI (Fig. 1B). This indicates that the A1M-binding occurred late in the apoptotic process after the cell membranes start to leak. To confirm this result, time studies on the binding of A1M to HCQ.4 cells, induced to apoptosis by anti-CD3 cross-linking, were performed. Flow cytometry of samples taken at various timepoints after induction shows that the PI uptake precedes the binding of A1M (Fig. 2A). This correlation was not seen to cells negative for PI uptake. The same result was obtained when the 70Z/3 were triple-stained with A1M, annexin V (marker for apoptosis), and 7-amino actinomycin D (7AAD, marker dye for cell membrane permeability) (Fig. 2B). Only 7AAD-positive cells showed a strong A1M binding, whereas cells positive for annexin V, but not for 7AAD, did not bind A1M.

Identification of intracellular A1M-binding proteins
To search for cellular proteins interacting with A1M, the yeast 2-hybrid system was used. cDNA coding for A1M was used as a bait to search for A1M-interacting proteins in a human leukocyte library. Approximately 2×106 transformants were analyzed for reporter gene activation. A total of 168 colonies survived on plates lacking histidine, and 13 of them were also positive for β-galactosidase. The His+LacZ+ recombinant library plasmids were isolated and tested in direct two-hybrid assays with bait plasmids encoding only the bait protein as well as the protein fused to unrelated proteins. Eleven recombinant plasmids were shown to encode proteins that interacted with A1M, but not with the bait protein or other unrelated proteins fused to it. DNA sequencing of the inserts revealed that seven of them were a truncated form of the SDAP subunit (NDUFAB1) in mitochondrial Complex I; one was the complete sequence of the same subunit; one was a small nuclear RNA (snRNA)-binding protein; one was N-acetylglucosamine kinase; and one was a colon cancer antigen. All inserts were in frame in the prey vector (Table 1).
α1-
According to the base numbering of the Genebank Accession No. assigned in this table.
Binding of A1M to mitochondrial Complex I
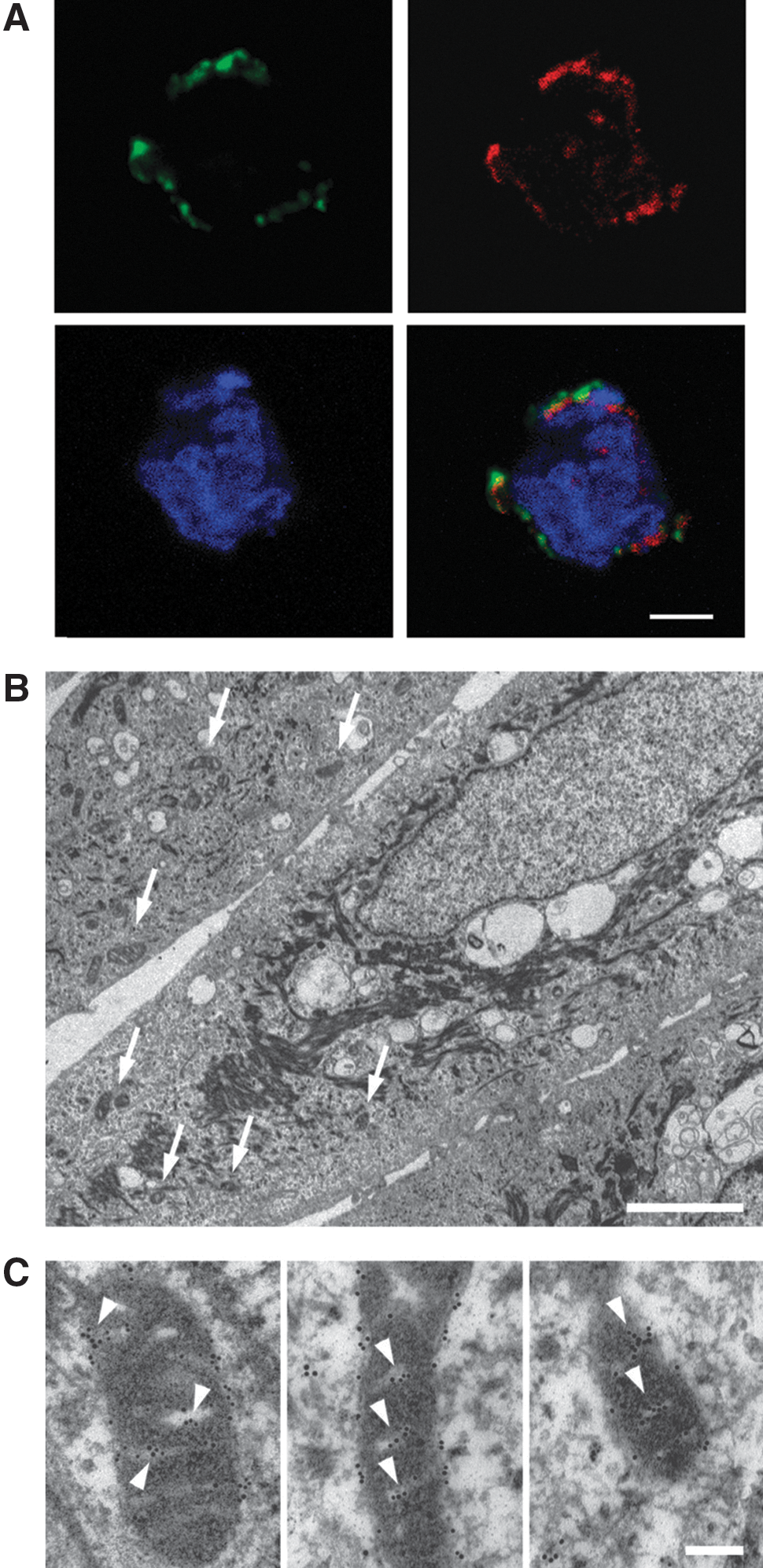
The results from the yeast two-hybrid experiments thus suggest that a subunit of mitochondrial Complex I is a major A1M-binding intracellular protein. Binding to mitochondria and to Complex I, in particular, was therefore investigated in detail using several independent methods: confocal microscopy, electron microscopy, subcellular fractionation, and polyacrylamide gel electrophoresis (PAGE). Using a mitochondrial fluorescent probe (MitoTracker) and confocal microscopy, we evaluated the subcellular localization of the intracellular A1M in K562 cells with or without addition of exogenous A1M (Fig. 3A). Analyzing cells without exogenously added A1M, a very weak unspecific intracellular staining was observed (not shown). However, with the addition of exogenous A1M, an intense, mitochondrial-specific staining was observed. The subcellular localization of the bound A1M was also studied by transmission electron microscopy (TEM) using primary human keratinocyte cultures (Fig. 3B, C). TEM of keratinocytes, containing exogenously added A1M and incubated with gold-labeled anti-A1M, showed a highly specific localization of A1M to the mitochondria (Fig. 3C).
Mitochondrial binding was confirmed and the specificity was verified using purified mitochondria from the mouse liver (Fig. 4A). 125I-labeled A1M was incubated with the mitochondria, with or without an excess of unlabeled A1M or the control protein AGP. Excess of unlabeled A1M blocked the 125I-A1M binding significantly at the two higher concentrations, whereas AGP at the highest concentration had no effect on the binding. Scatchard analysis of the binding data yielded an affinity constant of the binding at 1.2×106 M −1.
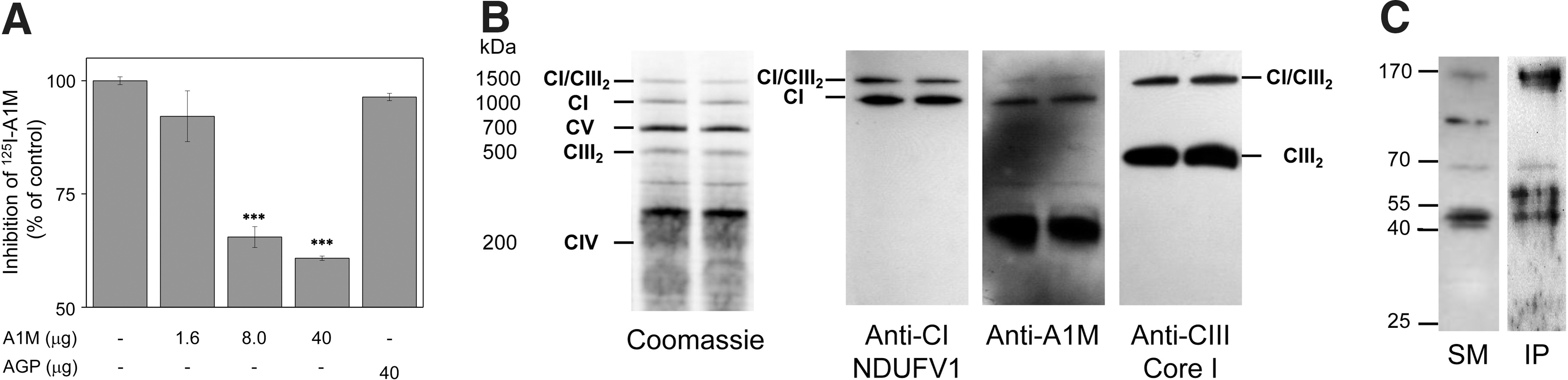
To investigate if endogenous A1M is found in mitochondria and associated with Complex I, mouse mitochondria were purified without freezing, solubilized, separated under nondenaturing conditions, and analyzed by Western blotting using antibodies against subunits of Complex I and III (denoted NDUFV1and Core I, respectively) and against mouse A1M (Fig. 4B). The results show that A1M co-migrates with the major Complex I-containing band and a supercomplex band containing both Complex I and III, whereas no co-migration was seen between A1M and the major Complex III-containing band. Taken together, this supports a specific association between A1M and a Complex I subunit. However, a large fraction of A1M migrated at a position corresponding to ∼200–300 kDa, suggesting that A1M is also associated with other, as yet unidentified, large structures in mitochondria. The blotting intensity of all bands decreased with increasing digitonin concentrations, suggesting that all bands seen in the gels result from noncovalent protein–protein interactions (Supplementary Fig. S1A; Supplementary Data are available online at
A1M protects mitochondrial structure and function
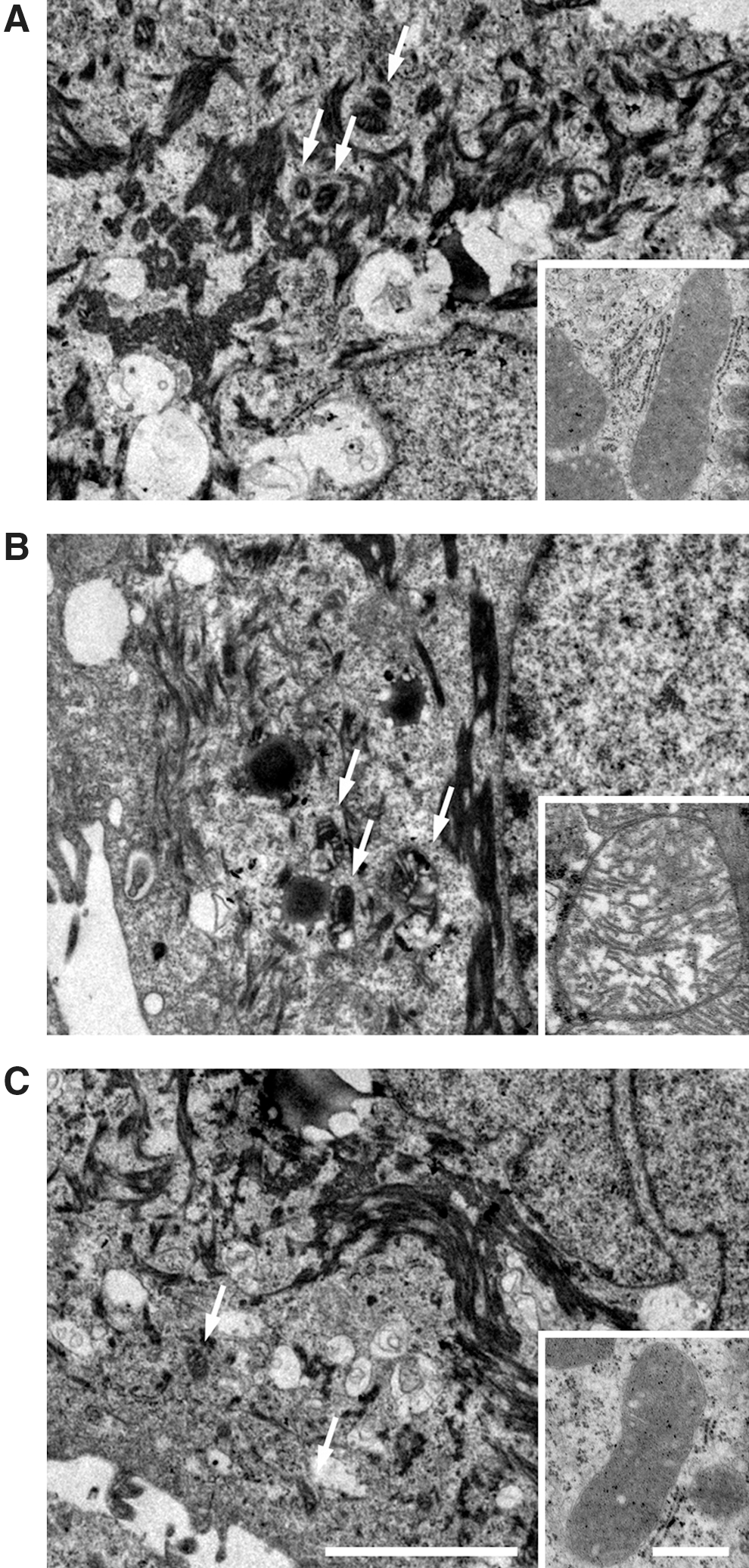
Hypothesizing that the physiological role of mitochondrial-bound A1M is to confer protection of this organelle against oxidative damage, we first employed TEM to investigate the impact of A1M on the structure of mitochondria in cells exposed to oxidative stress (Fig. 5). TEM was performed on cultured human primary keratinocytes. Extensive destructive effects were seen by heme (Fig. 5B) and H2O2 (Supplementary Fig. S2B), that is, vast formation of vacuoles, structural disorganization of keratin fibers, and swelling of the mitochondria (Fig. 5B and Supplementary Fig. S2B, zoom). These effects were counteracted by the addition of A1M, where a particular impact was seen on the mitochondrial swelling (Fig. 5C and Supplementary Fig. S2C, zoom). The results suggest that A1M protects and preserves cellular structures otherwise damaged and disintegrated by heme and ROS.
We finally investigated the effects of A1M on mitochondrial function by measuring ATP production of purified mitochondria exposed to heme- or H2O2-induced oxidative stress (Fig. 6). A significant reduction in the rate of ATP production was seen by 5 and 20 μM heme (Fig. 6A). This reduction was reversed by A1M, and no reduction in the ATP production rate was seen by heme in any of the tested concentrations when A1M was present. Similar results were obtained using H2O2 (Fig. 6B). Thus, H2O2 significantly reduced the rate of ATP production, but the effects were significantly reversed in the presence of A1M.

Discussion
In this work, we have shown that the radical scavenger A1M binds to mitochondria and more specifically to mitochondrial Complex I. Furthermore, it was shown that the protein can protect mitochondria from heme- and ROS-induced swelling and loss of ATP production capacity. On the basis of these findings, we suggest a novel cell housekeeping mechanism: regulation of the redox homeostasis of mitochondria by A1M.
The interactions between A1M and mitochondria were observed in several different cell types, that is, human blood cells of lymphocytic, myelocytic, and leukocyte origin, human keratinocytes, and isolated mouse liver mitochondria, suggesting that the interactions and protective effects studied here may be generalized to all types of cells. However, since different cell types have different metabolic and redox conditions, and therefore may behave differently, additional studies are needed to confirm this.
Induction of cell apoptosis was used to mimic situations characterized by stress-induced cell insults causing damage to the plasma membrane and destruction of the external barrier of the cell. Exogenous A1M was bound intracellularly with high affinity as soon as the cells could internalize PI, suggesting that the uptake mechanism is a passive leakage of A1M from the extracellular compartment. Previous studies have shown that the amount of A1M internalized from the extracellular compartment can range between 4% and 18% of the total amount added, suggesting a specific binding and accumulation of A1M in the cells (36, 37). At present, the exact mechanism for the cell uptake is not known nor is the mechanism of the mitochondrial translocation. Previous studies have also shown that the cell uptake of A1M can be increased by alpha-particle irradiation (36). Thus, it can be speculated that ROS and oxidative challenge to cells stimulate the uptake mechanism and upregulate a tentative cellular and/or mitochondrial receptor.
It is becoming increasingly clear that many forms of in vivo necrotic cell death actually can be described as a regulated series of ATP-dependent cell disintegration events, that is, programmed necrosis following specific pathways called necroptosis (7, 47). Thus, a possible role for A1M is to participate in preservation of mitochondrial function in both apoptotic and necroptotic cells and possibly other types of necrotic cells during nonautophagic cell death, to ascertain availability of ATP for energy-dependent cell degradation processes. The radical-binding properties of A1M suggest that the protection mechanism is based upon its antioxidant effects, but other mechanisms, such as stabilization of Complex I or the mitochondrial membrane, cannot be ruled out. Furthermore, the mitochondrial electron transport chain is a major source of cellular ROS (6, 27) such as superoxide radicals and H2O2 formed by Complexes I and III (39, 48). Therefore, an alternative physiological explanation of the localization of A1M to mitochondria is that the protein participates in protection of surrounding tissue components against ROS generated by the mitochondrial respiratory chain during apoptotic or necrotic events in inflammation, infection, or ischemia/reperfusion. In this context, the role of A1M, bound to Complex I, may be to eliminate the ROS before they can participate in unwanted, destructive reactions with healthy tissue. Possibly, A1M fulfills both of these physiological roles in vivo: (i) preservation of mitochondrial structure and function and (ii) protection of surrounding tissue from mitochondrial activity.
A1M was found to interact with four different proteins: the NDUFAB1 subunit in the hydrophobic portion of the NADH dehydrogenase complex (46), N-acetylglucosamine kinase (18), the snRNA-binding protein LSm5 (41), and a cancer antigen, NY-CO-3 (43). None of the four proteins were a false positive, that is, interacting with the DNA-binding bait fusion protein. Thus, the candidates were regarded as true A1M-interacting proteins in the yeast two-hybrid system. Considering that eight of the 11 clones isolated by the yeast two-hybrid system were the same Complex I subunit, this protein was seen as the most interesting of the proteins and was therefore chosen as a target of further investigations. Since the binding to mitochondria also was supported by alternative methodological approaches and a functional role of the association could be ascribed as compatible with the antioxidation function of A1M, none of the other three A1M-binding proteins have been further investigated in this work. The physiological implications of the binding of A1M to N-acetylglucosamine kinase, the snRNA-binding protein LSm5, and NY-CO-3 may be speculated upon, however. These proteins are located to the cytosol (19), nucleus (40), and plasma membrane (42), respectively, and the role of the binding to these proteins may be to localize internalized A1M to other cell compartments besides mitochondria. Another possibility is that the binding to these three proteins reflects other functions of A1M unrelated to antioxidation and radical scavenging.
A1M is a member of the Lipocalin protein family. The lipocalins constitute a functionally diverse group of ∼50 proteins from bacteria, plants, and animals having an amino acid sequence similarity usually around 20%–25% and share certain structural common features that indicate a common evolutionary origin (11, 12, 1a). Interestingly, two lipocalins found in plants and green algae, violaxanthin de-epoxidase and zeaxtanthin epoxidase, are localized in the thylakoid membranes of chloroplasts, the ATP-producing photosynthetic organelle of plants, where they are associated with the light-harvesting system II and participate in the xanthophyll photoprotection system [reviewed in (14)]. There is an obvious parallel between the violaxanthin protection cycle and the results in this article: the presence of a lipocalin-based protection system in the energy-converting organelles of both plants and animals. In plants, they are believed to have a role in excessive heat dissipation during light harvesting, and in animal cells, the results in this article suggest that a lipocalin (A1M) participates in maintaining the redox homeostasis of respiration by quenching of the side product ROS. Hypothesizing that mitochondria and chloroplasts have a common prokaryotic or eukaryotic ancestor [c.f. (15, 31, 51)], one may speculate that these two lipocalin systems are evolutionarily related.
When a cell is subjected to a fatal insult by, for instance, oxidation, it will ultimately be degraded and its components recycled. Preservation and maintenance of the mitochondrial energy machinery during this process are vital. In this article, we have shown that the reductase and radical scavenger A1M may have a central role in maintaining mitochondrial energy production. It can be speculated that A1M at the same time may counteract and eliminate the ROS generated in the respiratory chain to prevent unwanted, destructive reactions with healthy tissue, which is in line with its previously reported capacity to prevent ROS-induced cell and tissue damage (2, 32 –34, 36, 37, 1c). Thus, these findings suggest a novel mechanism of maintaining mitochondrial redox homeostasis.
Materials and Methods
Proteins and antibodies
Human monomeric plasma A1M was isolated by anti-A1M affinity chromatography and Sephacryl S-300 gel-chromatography, as described previously (49). Recombinant human A1M, containing an N-terminal His-tag, was purified from the culture medium of baculovirus-infected insect cells (49) or expressed in Escherichia coli and purified and refolded as described (21, 33). Human serum AGP and ovalbumin were purchased from Sigma-Aldrich Co., and BSA was from Roche Diagnostics Scandinavia AB. Hemin (Ferriprotoporphyrin IX chloride) was purchased from Porphyrin Products, Inc., and a 10 mM stock solution was prepared fresh by dissolving in DMSO (Sigma-Aldrich). H2O2 was from Acros Organics. Mouse monoclonal antibodies against human A1M (BN11.3 and 23.26) were raised as described (30). Rabbit polyclonal anti-mouse A1M antibodies (Sven; IgG-fraction) were prepared as described (42). The hamster anti-mouse CD3 antibody (145.2C11) was kindly provided by Dr. Rikard Holmdahl, Lund University. Fluorescein isothiocyanate-conjugated goat anti-mouse immunoglobulin (GAM-FITC) and phycoerythrin-conjugated streptavidin (SAPE) were purchased from DAKO A/S, and 7 AAD was from Sigma-Aldrich Co., and annexin V-FITC was from Trevigen, Inc.
Cell culture
A mouse CD4+ T-cell hybridoma cell line (HCQ.4), 70Z/3, K562, and human primary keratinocytes (Cambrex Biologics) were employed for studies on A1M binding to cells and mitochondria. Cells were cultivated as described previously (26, 29, 38) and processed and analyzed as described below.
Induction of apoptosis
Apoptosis was induced in the T-cell hybridoma by three different treatments: Cells were incubated on anti-CD3 antibody-coated plastics (4 μg/ml) (44), or incubated in a medium supplemented with either 5% ethanol or 10% of DMSO (25). Apoptosis was detected as DNA fragmentation by agarose gel electrophoresis (described below), and cell viability was measured by trypan blue exclusion. In the pre-B-cell line, apoptosis was induced by the 3-CPA (Oxigene, Inc.) as described in (26).
Agarose electrophoresis
To detect DNA fragmentation, ∼1×106 cells were lysed, proteinase K- and RNAse A-treated, and analyzed by agarose electrophoresis.
Labeling of A1M
For analysis of A1M binding to cells, A1M was biotinylated, FITC-conjugated, or radiolabeled with 125I as described previously (9, 13, 16).
Flow cytometry
A1M binding to cells was analyzed by flow cytometry. Approximately 1×106 cells were analyzed for A1M binding in one of three different ways: (i) The cells were incubated with 1 mg/ml of plasma or recombinant insect cell A1M, followed by 10 μg/ml of monoclonal mouse anti-A1M (BN 11.3) and GAM-FITC (diluted 20 times). (ii) The cells were incubated with 10 μg/ml biotinylated A1M followed by SAPE (diluted according to the manufacturer's recommendations). (iii) The cells were incubated with 0.1 mg/ml FITC-conjugated A1M. All incubations were performed in phosphate-buffered saline (PBS)+1 mg/ml of BSA for 10 min at room temperature (RT). Between the incubations, the cells were washed 2–3 times in PBS. To detect leaking cells, cells were incubated with PI (Invitrogen, Inc.) or 7AAD (according to manufacturers' instructions). To detect apoptotic 70Z/3 cells, cells were also incubated with FITC-conjugated annexin V in a Ca2+-containing buffer (according to the manufacturers' instructions). A Becton Dickinson FACSorter and the Cell Quest software package were used for all analyses.
Fluorescence and confocal microscopy
K562 cells were washed and resuspended in the culture medium to 0.5–4.0×106 cells/ml and incubated with or without A1M as indicated in the figure legends. Cells were then either incubated with MitoTracker (Invitrogen, Inc.) for 15 min at 37°C and washed in a fresh medium (Fig. 3A) or directly washed in a fresh medium (Fig. 1D). Cells were then resuspended in an ice-cold Na-medium (5.4 mM KCl; 1.2 mM KH2PO4; 0.8 mM MgSO4; 5.6 mM D-glucose; 127 mM NaCl; 10 mM Hepes; 1.8 mM CaCl2; pH 7.3), followed by fixation with 1% BD CellFIX on ice for 15 min and at RT for 45 min. Cells were then washed in a blocking solution (Na-medium; 1% BSA; 5% goat serum) followed by permeabilization in 0.02% Triton-X and blocking in 1% BSA, 5% goat serum, and 0.2% Tween-20 for 1 h at RT. The cells were then incubated overnight with monoclonal mouse anti-A1M (23.26) at 5 μg/ml, at 4°C. Subsequently, goat anti-mouse IgG F(ab`)2 fragments conjugated with Alexa Fluor® 488 (Invitrogen, Inc.) was applied for 1 h at RT. Cells were mounted using ProLong Gold AntiFade Reagent containing 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen, Inc.). Cells and fluorescent markers were analyzed using an epifluorescence microscope (Nikon Eclipse TE300) and a confocal laser-scanning microscope (Zeiss LSM 510 Meta). The epifluorescence microscope was equipped with the appropriate filter combinations to selectively visualize the used fluorophores. A Plan Apochromat 100× lens was used for analyses, and the image data were collected with a Hamamatsu C4742-95 CCD camera. To analyze intracellular labeling and colabeling in subcellular structures, confocal scanning of optical sections was recorded through the cells. For excitation of the fluorophores, the 405-nm laser line was used for DAPI (diode laser 405–30); the 488-nm laser line was used for Alexa Fluor 488 (Argon laser), and the 561-nm laser line was used for MitoTracker (DPSS 561-10). The individual fluorophore emission wavelengths were detected using the following filters: band-pass 420–480 nm for DAPI, band-pass 505–550 nm for Alexa Fluor 488, and long-pass 575 nm for MitoTracker. The pinhole for detection of Alexa Fluor 488 (488-nm excitation) was set to correspond to 1 (one) Airy unit, and the pinholes for the other detection channels were then adjusted to give optical sections of the same thickness, that is, to ensure comparisons of the corresponding confocal volumes. Laser power and detection settings (gain and offset) were optimized for the individual channels, giving a detection range from highly saturated pixels of larger structures to nonsaturated pixels of small structures. The different fluorophores were sequentially scanned, that is, with optimal settings for one fluorophore in each channel, at a 512×512 or 1024×1024 frame size. To determine cellular morphology, differential interference contrast images were obtained using the 405-nm laser as transmitted light. The spatial relation between the Alexa Fluor 488 fluorescence (green) and MitoTracker fluorescence (red) was determined via merging of the optical sections from the individually scanned channels (yellow when colocalized), confirmed via analyses of merged images using LSM Zen software (Profile, data not shown).
Yeast two-hybrid system
A GAL4-based yeast two-hybrid system was used to search for A1M-interacting cellular proteins. DNA encoding the A1M part (amino acids 1–183) of the A1M-bikunin gene (AMBP) was amplified by PCR using a pCR-Script construct as a template. The fragment was completely sequenced and ligated into the yeast two-hybrid vector pBD-GAL4 Cam phagemid vector (Stratagene). The recombinant vector was then transformed into the Saccharomyces cerevisiae yeast host strain (YRG-2; Stratagene). The yeast strains were grown and maintained and two-hybrid assays were performed using standard protocols as recommended by Stratagene and
Mitochondrial preparation from mouse liver tissue
Mouse liver tissue was collected in an ice-cold isolation buffer (320 mM sucrose, 10 mM Trizma Base, and 2 mM EGTA) and subsequently homogenized in 2 ml homogenization buffer (isolation buffer supplemented with 1% BSA). Mitochondria were prepared from homogenates by sequential centrifugation, including density purification on 19% Percoll. The protein concentration of mitochondrial preparations was determined using Nanodrop, and isolated mitochondria were used without freezing.
Competitive cell- and mitochondrion-binding assay
The specificity of A1M binding to cells and mitochondria was investigated by a competitive cell-binding assay as described (3, 50). Apoptosis was induced in HCQ.4 cells by anti-CD3 cross-linking for 15–18 h. The cells were harvested and compared to normal cells in the binding assay. A1M, ovalbumin, BSA, and AGP diluted in PBS+2% BSA were used as unlabeled control proteins. An affinity constant for the binding was calculated using a Scatchard plot of the data.
Immunocapture of Complex I
Complex I was immunoprecipitated on freshly prepared mitochondria using the Complex I Immunocapture Kit (MitoSciences). After the immunoprecipitation, bound proteins were eluted using the SDS buffer and subsequently analyzed using SDS-PAGE and Western blotting.
Isolation of respiratory chain complexes and supercomplexes
Freshly isolated, nonfrozen mitochondria were suspended in PBS supplemented with a Complete Mini-Protease inhibitor. Mitochondria were pelleted for 5 min at 5000 g and subsequently dissolved to a concentration of 5 mg/ml in the MB2 buffer (1.75 M aminocaproic acid, 7.5 mM Bis–Tris, pH 7.0, +2 mM EGTA, pH 8.0). Mitochondrial membrane proteins were solubilized by incubation with 0.5 g digitonin/g protein for 5 min on ice. Samples were centrifuged for 30 min at 13000 g; the supernatant was collected and the protein concentration measured as before. Finally, SBG (750 mM aminocaproic acid and 5% Serva Blue G) was added to a final concentration of 4.5%.
Blue native PAGE, SDS-PAGE, and Western blotting
Five μg mitochondrial membrane proteins was separated on a BN-PAGE 4%–16% Bis–Tris gel and either stained with Coomassie Brilliant Blue or blotted to a polyvinylidene fluoride (PVDF) membrane. Complex I immunoprecipitated proteins were separated on a 12% SDS-PAGE and transferred to a PVDF membrane. After blocking, the membranes were incubated with antibodies detecting Complex I subunit NDUFV1 (Sigma), Complex III subunit Core I (Invitrogen, Inc.), or mouse A1M (Sven). Primary antibodies were detected by incubation with horseradish peroxidase-coupled goat anti-mouse (DAKO) or goat anti-rabbit (DAKO).
Transmission electron microscopy
Human keratinocytes (about 1 million cells), incubated for 20 h at RT with 20 μM heme, with or without 0.25 mg/ml A1M, were pelleted by centrifugation and prepared as previously described (33). Thin sections with gold-labeled anti-A1M (BN11.3) were immunolabeled as described previously (40), with the modification that Aurion-BSA was used as a blocking agent. Specimens were observed in a JEOL JEM 1230 electron microscope operated at an 80-kV accelerating voltage. Images were recorded with a Gatan Multiscan 791 CCD camera.
ATP assay
Cellular ATP production was measured using a luminescence assay kit (Promega), based on the ATP-dependent activity of luciferase. ATP levels were normalized to the corresponding sample protein content.
Statistical analysis
Origin 8 software was used for statistical analysis. Student's t-test was used for statistical evaluation and was considered significant when p<0.05.
Footnotes
Acknowledgments
This work was supported by the Swedish Research Council, governmental ALF research grants to Lund University and Lund University Hospital, the Swedish Cancer Foundation, the Royal Physiographic Society in Lund, the Foundations of Greta and Johan Kock and Alfred Österlund, the Blood and Defence Network, Lund University, and A1M-Pharma AB. The authors wish to acknowledge Maria Baumgarten for outstanding electron microscopy work, Dr. Ulrich von Pawel-Rammingen for kindly donating the human leukocyte library and for fruitful discussions, Drs. Christine Persson and Kurt Schesser for fruitful discussions, and Eva Miller, Kerstin Torrikka, Kerstin Boll, Lisa Palm, Jenny Johansson, and Eva Hansson for excellent technical assistance. We are grateful to Dr. Bo Holmqvist, ImaGene-iT AB (Medicon Village, Lund, Sweden), for help with the confocal microscopy.
The authors MGO and BÅ are cofounders of the company A1M Pharma AB. This does not present any conflict of interest.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.