
Abstract
Introduction
More recently, we demonstrated that hHcys may generate its detrimental effects by activation of NOD-like receptor protein (NALP3)-centered inflammasomes, an intracellular inflammatory machinery in podocytes. This proteolytic, high-molecular-weight complex is primarily comprised of NALP3, the adaptor protein apoptosis-associated speck-like protein (ASC), and caspase-1, where each component is necessary for inflammasome assembly and release of active caspase-1 (16). The activation of these inflammasomes during hHcys turns on local inflammatory response, inducing kidney senescence and progressive degenerative glomerular dysfunction and sclerosis, where active caspase-1 promotes maturation of interleukin-1β (IL-1β) to induce decreases in nephrin expression in podocytes (33). However, it remains to be determined how the NALP3 inflammasomes are formed and activated during hHcys and how this induces glomerular degenerative pathology, ultimately leading to end-stage renal disease.
Innovation
Hyperhomocysteinemia (hHcys) stimulates the formation and activation of a novel intracellular inflammatory machinery, termed the inflammasomes, leading to podocyte injury and eventual glomerular sclerosis. The present study for the first time demonstrates that hHcys-induced activation of inflammasomes in podocytes is attributed to nicotinamide adenine dinucleotide phosphate (NADPH) oxidase–mediated redox signaling, where the production of reactive oxygen species may not only be involved in inflammation-induced tissue injury by oxidative damage, but may also serve as signaling molecules to regulate this very early activation of the inflammasomes.
In this regard, several mechanisms underlying inflammasome activation have been reported, including lysosome rupture, ion channel gating, and reactive oxygen species (ROS) activation (23). Activation of the NALP3 inflammasome by increased ROS, the most widely accepted and considered to be the most plausible mechanism, suggests that this inflammasome is a general sensor for changes in cellular oxidative stress. ROS activation of inflammasomes within podocytes may be an early initiating mechanism of glomerular injury during hHcys, because the production of ROS has been considered to be one of the major early factors mediating hHcys-induced glomerular injury (24,31). In the kidney, there are many enzymatic systems that contribute to the production of ROS, including the mitochondria, xanthine/xanthine oxidase, and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase. However, NADPH oxidase has been considered as the major source of superoxide in the kidney, and its action relies on an enzymatic complex with five subunits, including gp91 phox , p47 phox , p67 phox , and Rac1/2, all being reported to be expressed in the kidney (6). Further, we have demonstrated the importance of NADPH oxidase in the pathogenesis of hHcys-induced glomerular sclerosis by showing that inhibition of NADPH oxidase and its subunits can attenuate glomerular damage and restore normal renal function (35). Similarly, antioxidants have been shown to ameliorate homocysteine (Hcys)-induced toxicity (14). On the other hand, there are reports that hHcys-induced renal injury or other organ damages are also associated with local inflammatory response and corresponding pathological actions (8,20). It is possible that NADPH oxidase activation at the early stage of hHcys triggers the formation of inflammasomes, resulting in a downstream local inflammatory response.
The present study hypothesized that NADPH oxidase-mediated redox signaling may play an essential role in triggering hHcys-induced inflammasome activation within podocytes, thereby inducing glomerular inflammatory injury such as immune cell infiltration and consequently leading to glomerular sclerosis. To test this hypothesis, we first used cultured murine podocytes to examine whether inhibition of NADPH oxidase attenuates Hcys-induced NALP3 inflammasome formation and activation and also addressed the functional relevance of this early inflammatory event. Using pharmacological inhibitors or mice lacking the gp91 phox gene, we also tested the in vivo role of NADPH oxidase activation in hHcys-induced NALP3 inflammasome formation and activation, glomerular inflammatory pathology, and glomerular sclerosis compared with wild-type littermates.
Results
Inhibition of NADPH oxidase attenuated Hcys-induced inflammasome formation
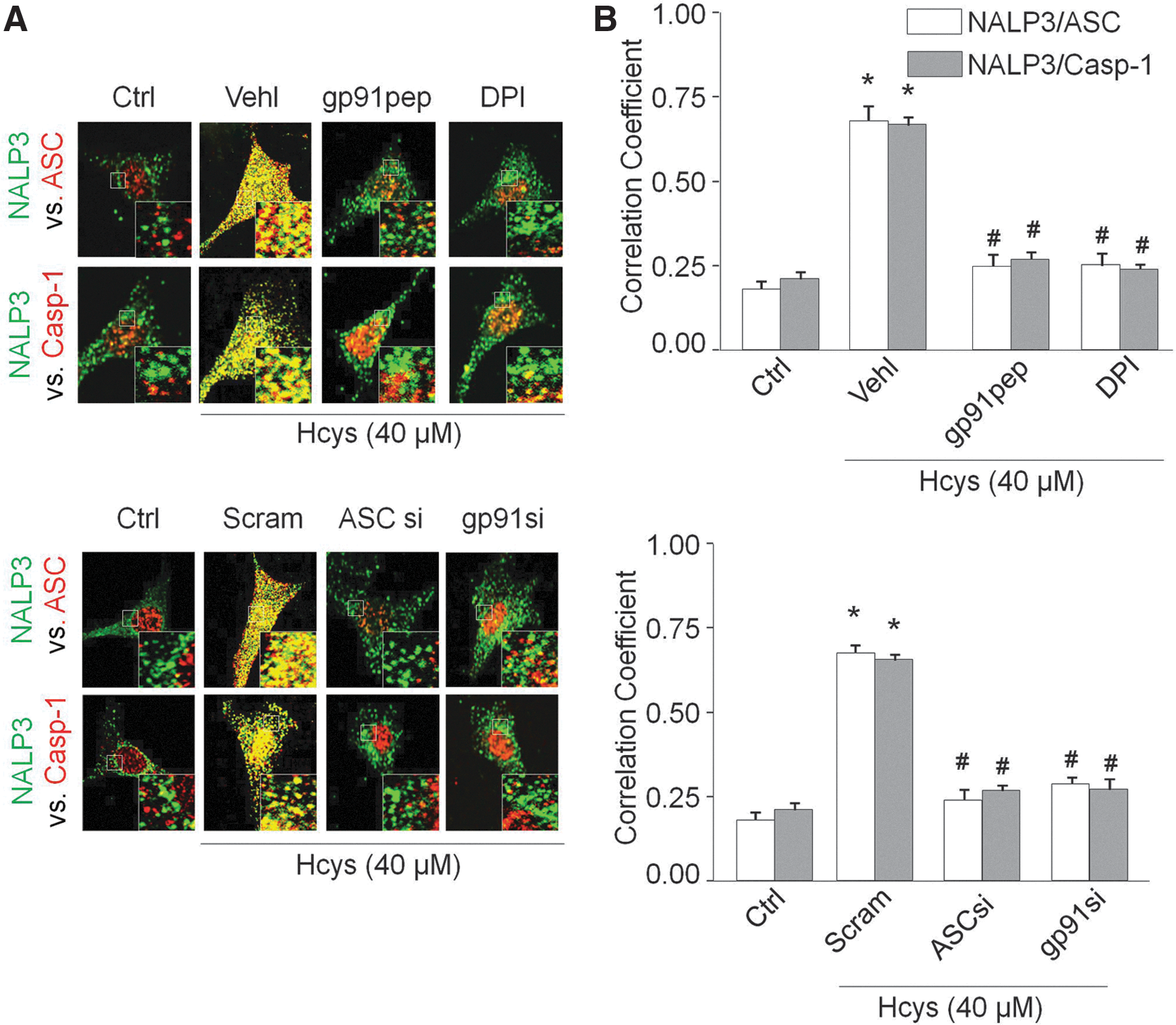
As shown in Figure 1A, confocal microscopic analysis demonstrated that Hcys induced the colocalization of inflammasome markers (NALP3 with ASC and NALP3 with caspase-1) in podocytes compared with control cells. Pretreatment of podocytes with NADPH oxidase inhibitors diphenyleneiodonium (DPI) or gp91ds-tat peptide significantly abolished the Hcys-induced aggregation of NALP3 with ASC and NALP3 with caspase-1, suggesting blockade of inflammasome formation in these cells. Further, small interfering RNA against ASC and gp91
phox
also blocked Hcys-induced inflammasome formation. The Pearson correlation coefficient (PCC) was calculated for each of the groups and summarized in Figure 1B. In addition, coimmunoprecipitation experiments demonstrated that Hcys significantly increased the binding of NALP3 and caspase-1 together with ASC compared with control cells, which is attenuated in the presence of apocynin (APO) and gp91ds-tat peptide (Supplementary Fig. S1; Supplementary Data are available online at
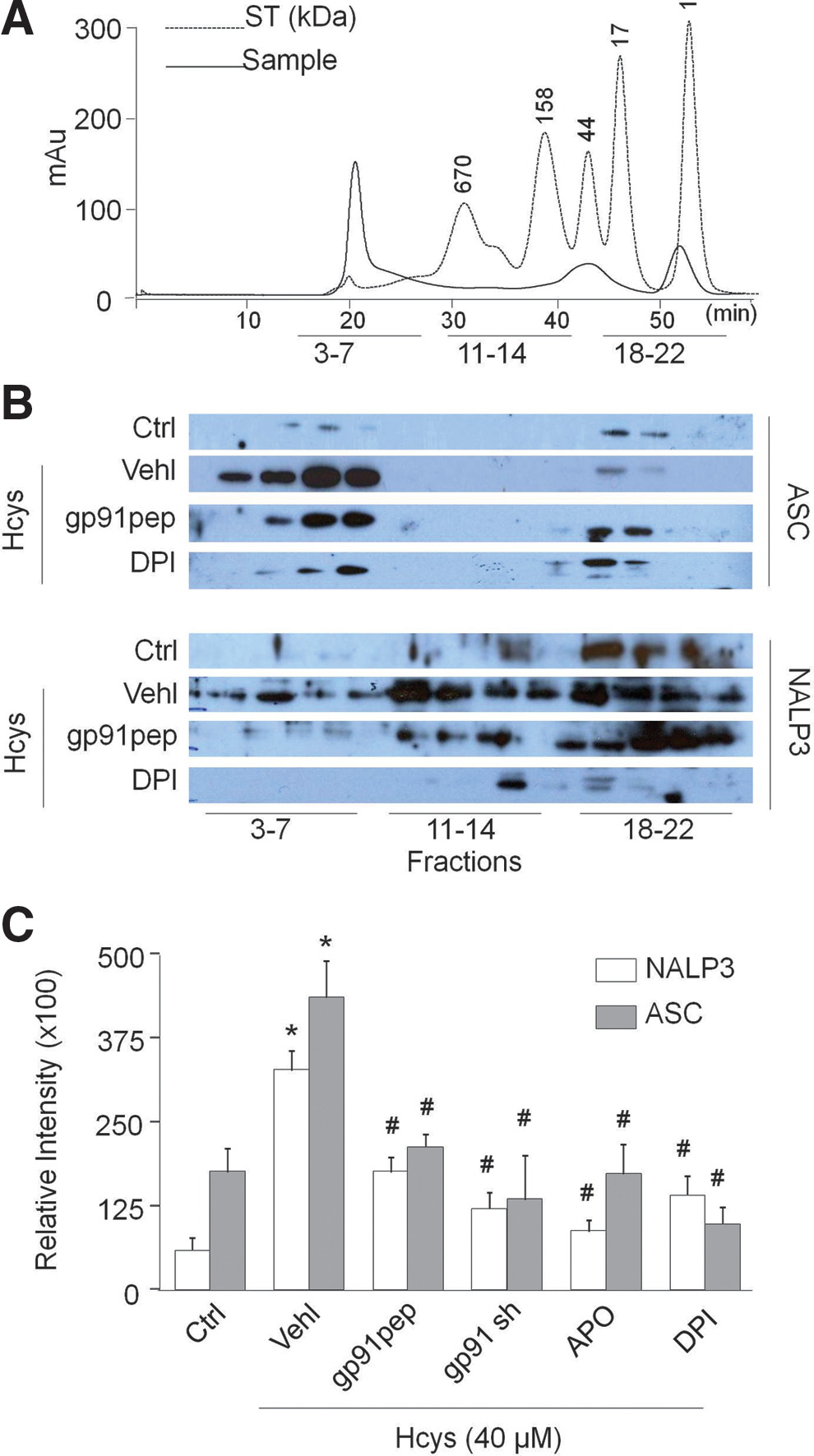
Size-exclusion chromatography (SEC) was employed to further determine the role of NADPH oxidase in the process of Hcys-induced inflammasome complex formation. A representative chromatogram is shown in Figure 2A, illustrating the peaks produced from both a standard and typical protein sample when run and separated by the Sepharose 6 column. As depicted in Figure 2B, under control conditions, the specific bands for NALP3 and ASC were located in the low-molecular-weight fractions (18 –22). Upon stimulation with Hcys for 24 h, the NALP3 and ASC bands markedly shifted to high-molecular-weight fractions (3 –7), termed the inflammasome fractions due to complex formation. However, when NADPH oxidase was inhibited in podocytes by treatment with gp91ds-tat or DPI before the addition of Hcys, a clear decrease in inflammasome protein complex aggregation was observed in the high-molecular-weight fractions. The intensity of these bands was quantified by ImageJ software and summarized in Figure 2C.
Inhibition of NADPH oxidase blocked inflammasome functionality by suppressing caspase-1 activity and IL-1β secretion
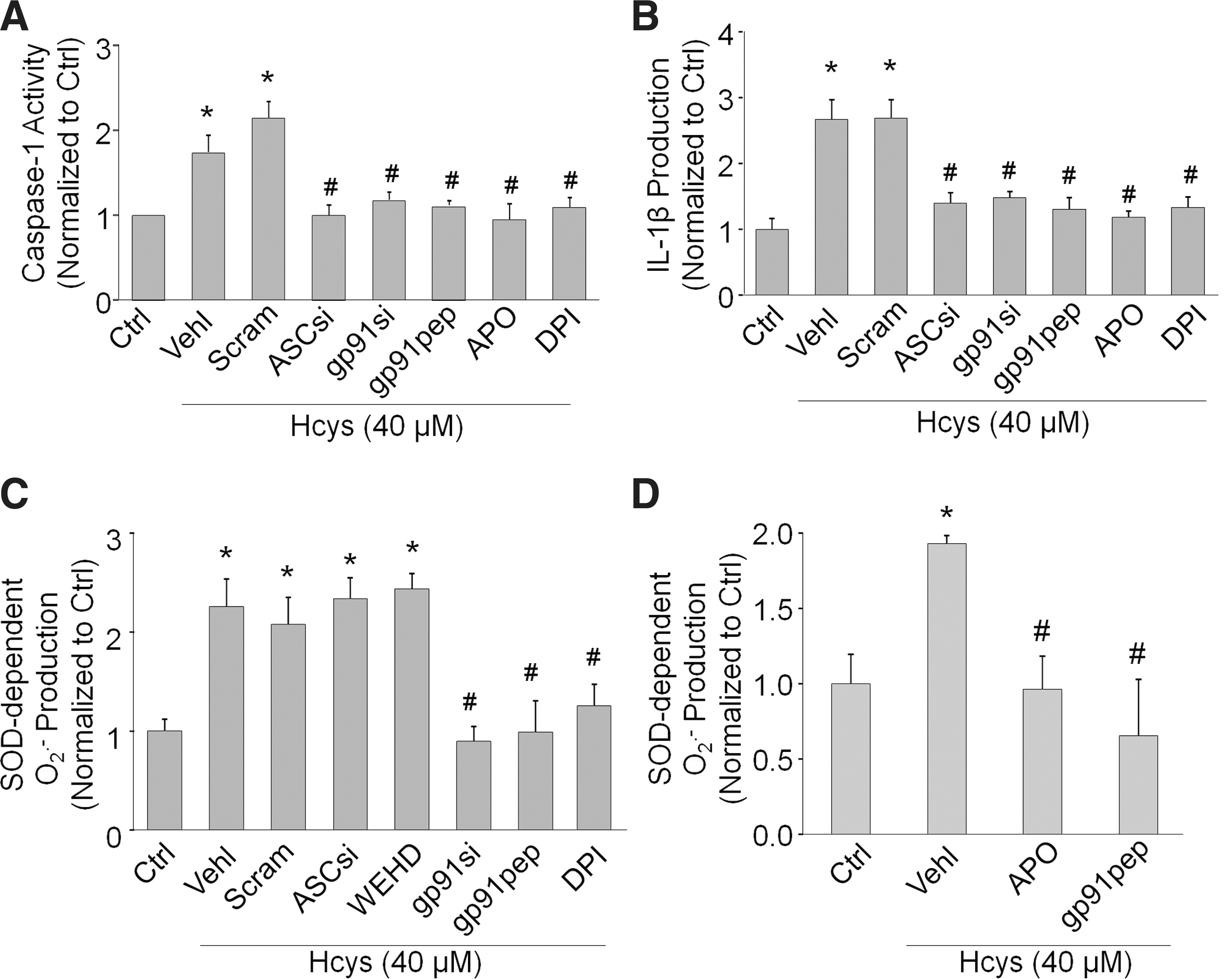
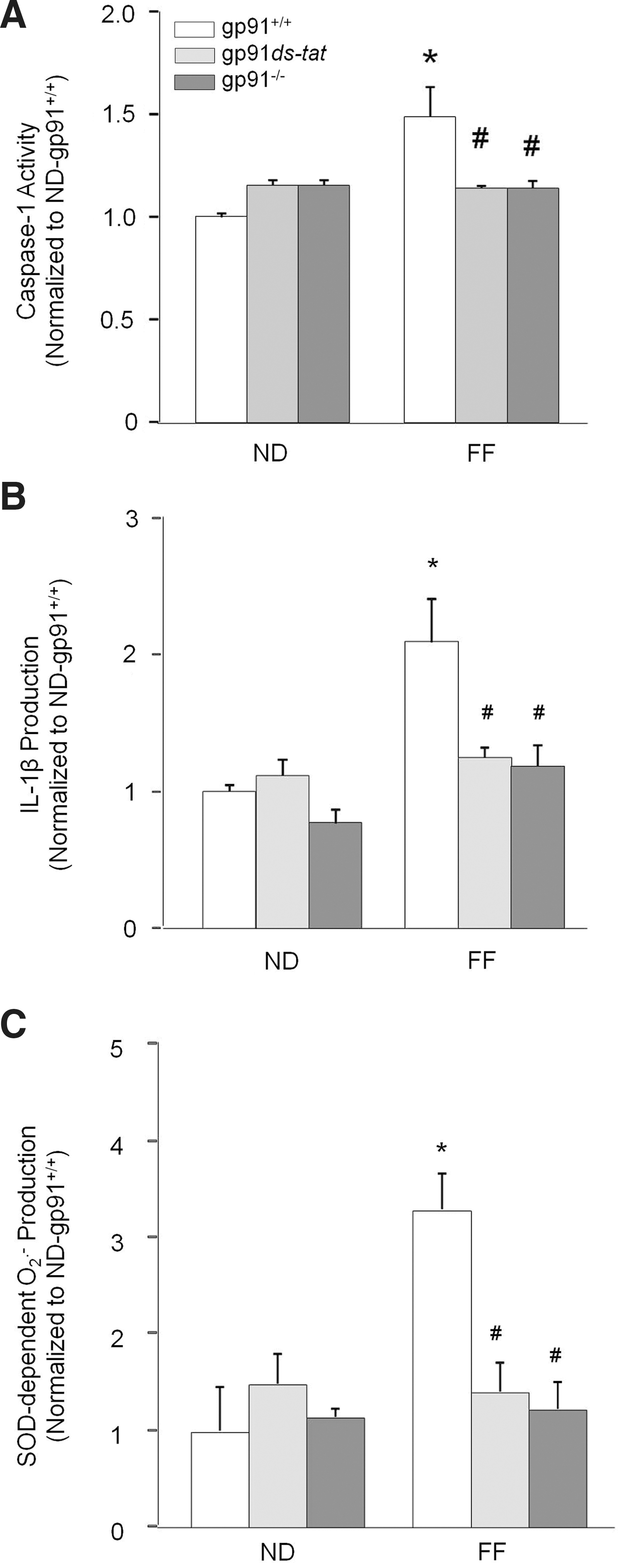
We further tested whether inhibition of NADPH oxidase also attenuates Hcys-induced caspase-1 activity and IL-1β production in podocytes. As shown in Figure 3A, the increase in caspase-1 activity caused by Hcys was markedly inhibited by the NADPH inhibitors APO, DPI, gp91ds-tat, and gp91 phox siRNA, implicating an important role for NADPH oxidase in inflammasome activation. ASC siRNA also produced a similar significant decrease in caspase-1 activity. Figure 3B demonstrated that these decreases in caspase-1 activity also resulted in less IL-1β being converted from the proinflammatory cytokine form to the active form during the simultaneous treatment of Hcys with either NADPH oxidase or inflammasome inhibitors.
Inhibition of the inflammasome failed to inhibit Hcys-induced superoxide production
To determine whether NADPH oxidase-derived superoxide plays a role in inflammasome activation, superoxide levels were measured in podocytes pretreated with NADPH oxidase or inflammasome inhibitors in the presence of Hcys. As expected, treatment of podocytes with DPI, gp91ds-tat, or gp91 phox siRNA produced a distinct decrease in superoxide production when compared to Hcys alone (Fig. 3C). Additionally, APO or gp91ds-tat peptide also significantly attenuated the Hcys-induced superoxide production in the plasma membranes of podocytes (Fig. 3D). However, ASC siRNA or caspase-1 inhibition could not prevent the Hcys-induced increase in superoxide. This suggests that NADPH oxidase activation by Hcys and subsequent production of superoxide are upstream of inflammasome activation, given that inhibition of the inflammasome did not affect the levels of NADPH oxidase-derived superoxide.
Attenuation of Hcys-induced podocyte injury by inhibition of NADPH oxidase or inflammasomes
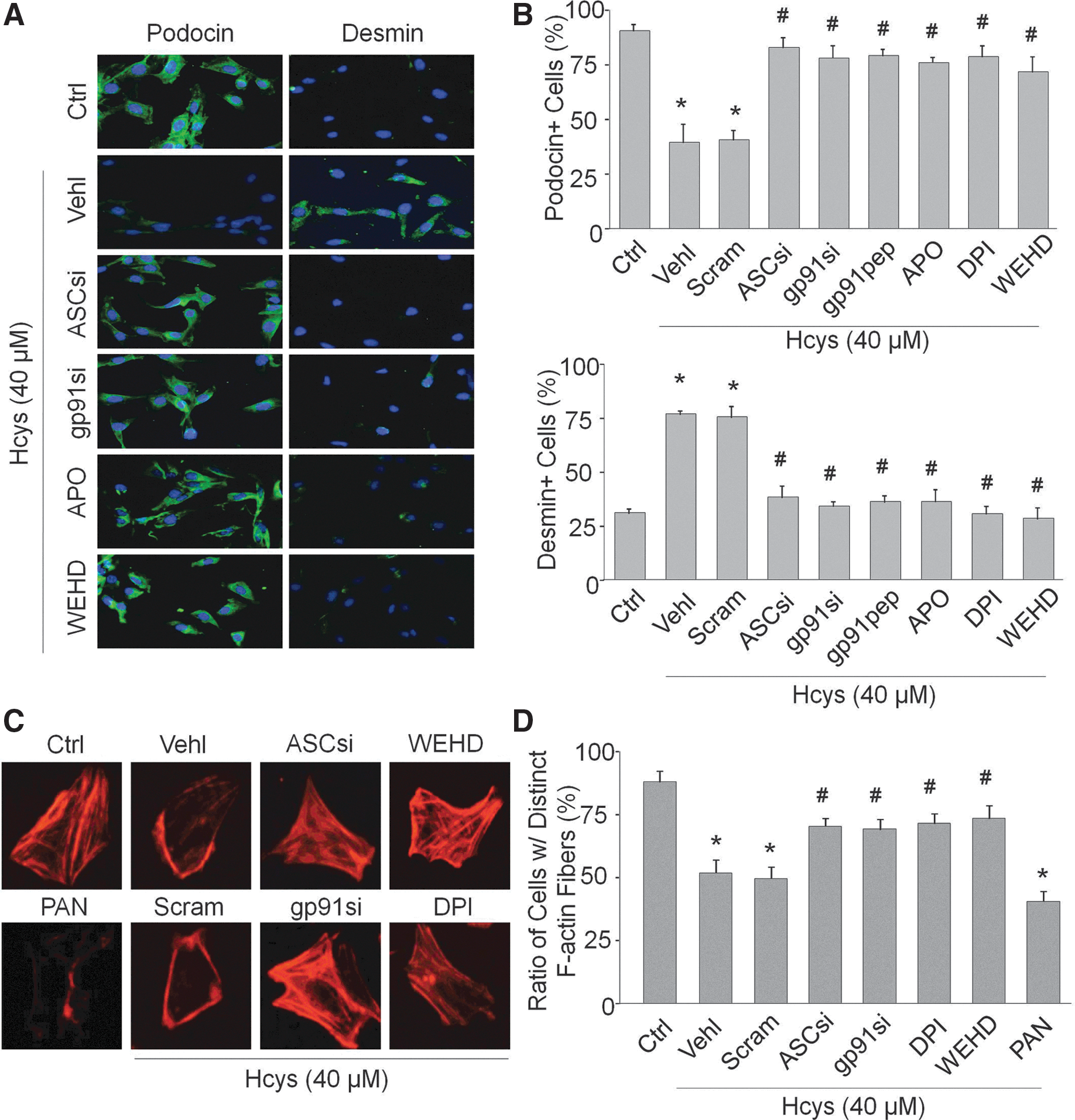
As shown in Figure 4A, immunofluorescent analysis demonstrated that Hcys stimulation increased desmin expression in podocytes compared with untreated cells. Prior treatment with gp91 phox siRNA, ASC siRNA, gp91ds-tat, APO, DPI, or WEHD decreased this Hcys-induced desmin expression in podocytes. Another podocyte marker, podocin, was markedly reduced upon Hcys stimulation in podocytes, and prior treatment with the same inhibitors almost completely attenuated the decrease in podocin expression. Positive cells were counted and summarized in Figure 4B. These results signify the importance of both NADPH oxidase and inflammasome functionality in this injurious process of podocytes when exposed to Hcys. Using rhodamine–phalloidin to stain F-actin, the control condition exhibited well-defined F-actin fibers that run along the longitudinal axis of these podocytes, and as demonstrated by the obvious lack of distinct fibers, Hcys resulted in a significant loss of these longitudinal fibers as they reorganize to the cell border. A classic and specific inducer of podocyte injury, puromycin aminonucleoside (12), served as a positive control (Fig. 4C). However, inhibition of NADPH oxidase or inflammasomes hindered Hcys-induced decrease and rearrangement of F-actin. These changes in F-actin staining are summarized in Figure 4D.
Blockade of hHcys-induced glomerular inflammasome formation and activation in glomeruli of gp91 phox−/− and gp91ds-tat-treated mice
To further confirm the role of NADPH activation in vivo in experimental hHcys mice, double-fluorescent immunostaining of kidney slides was performed. As shown in Figure 5A, under control condition, NALP3, ASC, and caspase-1 were expressed at low levels within the glomeruli, and very few colocalizations of these inflammasome molecules could be detected by confocal microscopy. In gp91 phox+/+ mice, the colocalization of NALP3 with ASC or caspase-1 markedly increased in glomeruli of folate-free (FF) diet-fed hHcys mice. However, the increased colocalization of NALP3 with ASC or caspase-1 was suppressed in glomeruli of hyperhomocysteinemic gp91 phox−/− and gp91ds-tat-treated mice. The summarized data are shown in Figure 5B and C. Scrambled gp91ds-tat was used as a control and was shown in Supplementary Fig. S2 to have no effect on hHcys-induced inflammasome formation. In addition, using podocin and desmin as podocyte markers, we showed that hHcys-induced inflammasome activation in glomeruli was mostly located in podocytes, as demonstrated by the colocalization of podocin with NALP3 or caspase-1 and desmin with NALP3 or caspase-1. This colocalization was substantially blocked in hyperhomocysteinemic gp91 phox−/− mice and gp91ds-tat-treated mice (Fig. 5A and Supplementary Fig. S3). The summarized data are shown in Figure 5D and E.
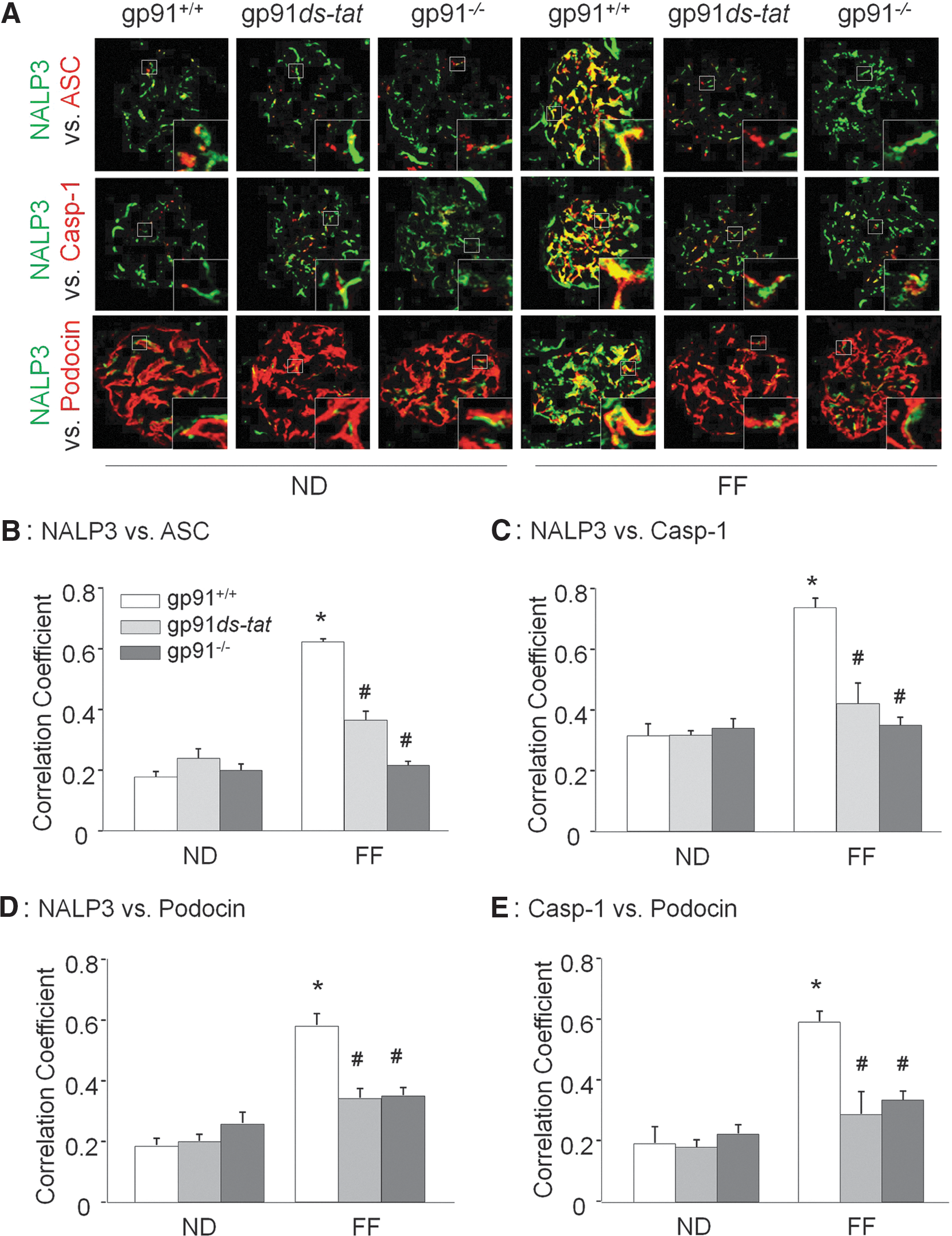
Consistent with decreased aggregation of inflammasome components in the glomeruli, hHcys-enhanced caspase-1 activity and IL-1β production were markedly attenuated in glomeruli of gp91 phox−/− and gp91ds-tat-treated mice (Fig. 6A, B). In addition, hHcys-induced glomerular superoxide production was significantly attenuated in gp91ds-tat-treated and gp91 phox−/− mice compared with their wild-type littermates (Fig. 6C). The plasma Hcys concentration was similar in gp91 phox+/+, gp91 phox−/−, and gp91ds-tat-treated mice on the normal diet (ND). However, the FF diet significantly increased the plasma Hcys concentration in all three groups compared with ND-fed mice (gp91 phox+/+ mice: 12.6±2.0 vs. 4.1±0.41 μM of control; gp91 phox−/− mice: 13.0±1.6 vs. 4.0±0.8 μM of control; gp91ds-tat mice: 11.6±1.0 vs. 5.1±0.41 μM of control).
In vivo inhibition of NADPH oxidase prevented hHcys-induced glomerular inflammation and injury
As shown in Figure 7, immunohistochemical analysis demonstrated that Hcys stimulation induced macrophage (F4/80+) and T-cell (CD43+) infiltration in the glomeruli of hHcys gp91 phox+/+ mice. However, the glomeruli of gp91 phox−/− and gp91ds-tat-treated mice had significantly less macrophage and T-cell recruitment when compared with gp91 phox+/+ mice on the FF diet (Fig. 7). The summarized data are shown in Figure 7B and D. These results suggest that normal NADPH oxidase gene expression and activity are required for inflammasome activation and consequent inflammatory response, which includes macrophage and T-cell recruitment and aggregation in glomeruli of mice during hHcys.

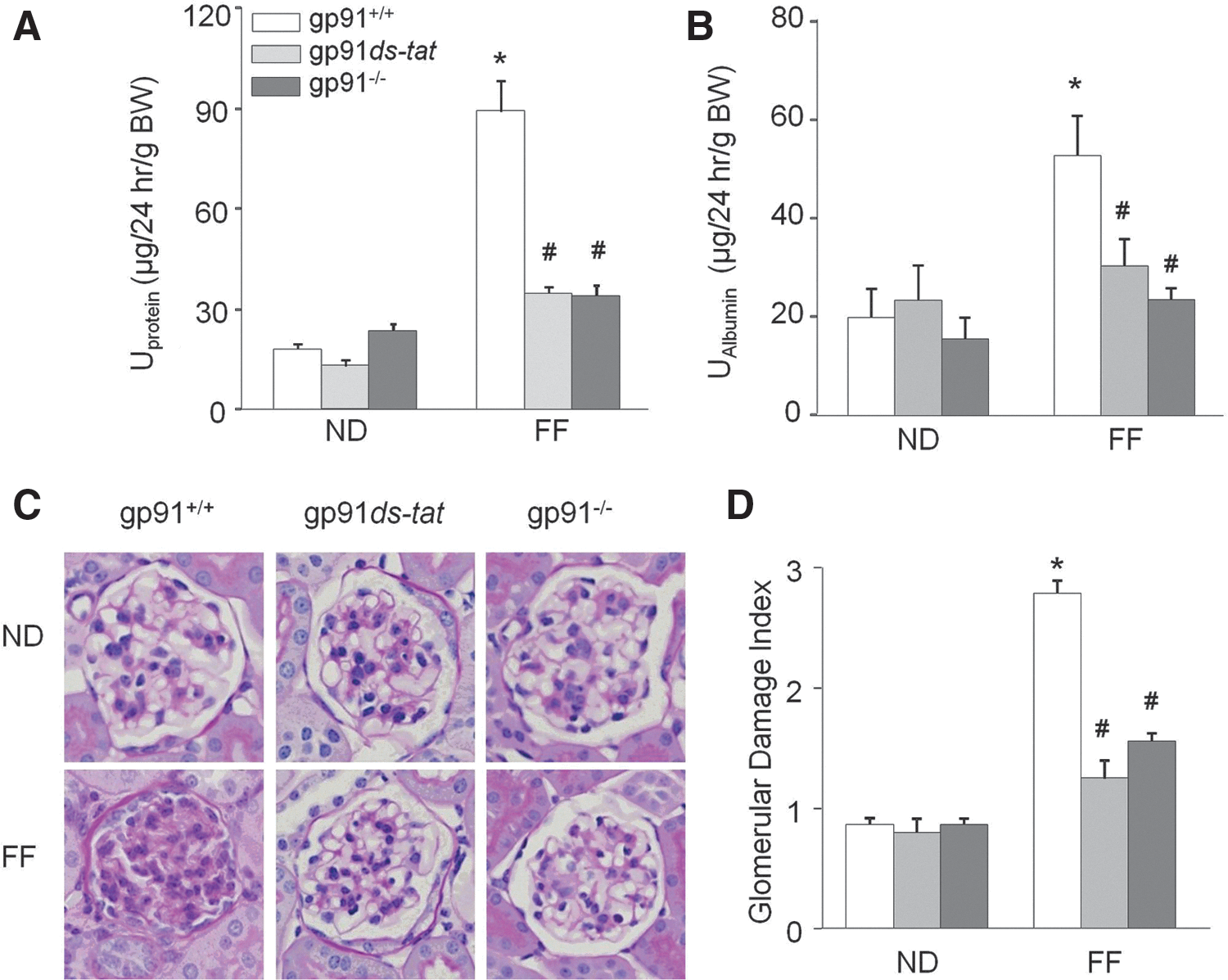
Next, we tested whether gp91 phox contributes to hHcys-induced glomerular injury. As shown in Figure 8, hHcys significantly increased the urinary protein and albumin in gp91 phox+/+ mice compared with ND-fed mice. Mice treated with gp91ds-tat and gp91 phox−/− mice had significantly attenuated hHcys-enhanced urinary protein and albumin excretion in FF-diet-fed mice, but had no effect in ND-fed mice (Fig. 8A, B). Morphological examinations revealed a typical pathological change in glomerular sclerotic damage indicated by capillary collapse, fibrosis, cellular proliferation, and mesangial cell expansion in glomeruli of hHcys gp91 phox+/+ mice (Fig. 8C). The average glomerular damage index was significantly higher in glomeruli of hHcys gp91 phox+/+ mice compared with ND-fed mice. However, in gp91 phox−/− or gp91ds-tat-treated mice, hHcys-induced glomerular injuries were significantly inhibited (Fig. 8D). Additionally, gp91 phox−/− and gp91ds-tat treatment prevented hHcys-induced decreases in WT-1 staining and overall podocyte number (Supplementary Fig. S4).
Discussion
The primary goal of the present study was to reveal whether NADPH oxidase-mediated redox signaling contributes to Hcys-induced inflammasome formation or activation and consequent glomerular injury. In vitro studies using cultured podocytes and an in vivo animal model of hHcys demonstrated that NADPH oxidase is necessary for the formation and activation of NALP3 inflammasomes in podocytes upon Hcys stimulation, thereby leading to podocyte dysfunction, glomerular immune cell recruitment, and ultimately glomerular injury and sclerosis. These results for the first time demonstrate Hcys-induced redox activation of podocyte NALP3 inflammasomes as an early mechanism switching on local inflammatory responses in glomeruli.
hHcys has been known to directly cause deleterious effects in the kidney, promoting a vicious cycle responsible for chronic renal disease, where hHcys decreases renal function and leads to further increased plasma Hcys levels due to decreases in Hcys excretion in the kidney (29). In the human condition, a plasma Hcys concentration <10 μM is considered to be within the normal range, 10–16 μM is clinically defined as mild hHcys, 16–30 μM as moderate hHcys, 30–100 μM as intermediate hHcys, and >100 μM as severe hHcys (29). For mice, it has been extensively studied that similarly to humans, genetic background and strain of the mice play an important role in the levels of plasma Hcys concentration, along with gender, diet, and parental effects (5). However, our most recent work demonstrated thorough concentration- and time-dependent studies that in an in vitro setting, a 40 μM treatment for 24 h in cultured podocytes is sufficient to activate an inflammasome-stimulating response, verifying the effect we see in vivo (33). Indeed, the present study showed significant glomerular damage observed in gp91 phox+/+ mice fed an FF diet for 4 weeks. Coinciding with this glomerular damage characterized by our report and others to include mesangial cell expansion, overall cell proliferation, and capillary collapse (7,35), hHcys caused a significant increase in proteinuria and albuminuria, indicative of an impaired glomerular filtration membrane. In previous studies, these damaging effects of hHcys have been well correlated with its ability to stimulate the inflammatory response. Proinflammatory mediators, such as monocyte chemoattractant protein-1, nuclear factor kappa B, and interleukin-8, and adhesion molecules vascular cell adhesion molecule-1 and E-selectin, have been demonstrated to be upregulated during hHcys (2,25). Further, it has been shown that hHcys results in the recruitment of lymphocytes (18). However, it remains unknown how hHcys or Hcys in vitro activates this response of the innate immune system in glomeruli. We have recently reported that Hcys can activate NALP3 inflammasomes in podocytes, an intracellular molecular switch of inflammation (33). Inflammasome activation has also been implicated in a number of inflammatory and metabolic diseases, including obesity, gout, hypersensitivity, silicosis, and diabetes, where inhibition of the inflammasome proteins NALP3, ASC, or caspase-1 significantly attenuates the downstream inflammatory reaction produced under these conditions (3,17,26,36). Similarly, our report showed that inhibition of either ASC or caspase-1 could prevent podocyte and glomerular damage induced by hHcys, improving renal structural and functional integrity (33), suggesting an important role for inflammasome activation in mediating the deleterious effects of hHcys.
The present study further explored the mechanism by which NALP3 inflammasomes are activated in podocytes in vitro by Hcys and in vivo by experimental hHcys. It has been reported that many endogenous and exogenous danger signals activate the inflammasome, and it has been of great interest as to how all of these very diverse signals can activate the same molecular machinery to turn on inflammation. Of the proposed models of inflammasome activation, the ROS model provides an unifying link, utilizing the common aspect that all these danger signals produce changes in oxidative stress, making NALP3 a more general sensor detecting these changes in oxidative stress (23). Interestingly, we and others have found the damaging effects of hHcys to be strongly associated with increased local oxidative stress, majorly through NADPH oxidase (4,14,31). With reports of gp91 phox /Nox2 being the predominant isoform found in podocytes, our laboratory has also demonstrated that this NADPH oxidase isoform and its activity are essential for hHcys to induce glomerular injury, since knockout of the gp91phox gene in mice protected podocytes and glomeruli from hHcys-induced glomerular sclerosis (35). Therefore, we hypothesized that redox signaling associated with NADPH oxidase in podocytes may be critical in triggering NALP3 inflammasome formation and activation upon Hcys stimulation.
To test this hypothesis, we first performed a series of experiments in cultured podocytes. It was found that pharmacological and genetic interventions to inhibit NADPH oxidase resulted in significant attenuation of NALP3 inflammasome formation, as shown by less aggregation of inflammasome molecules detected by confocal microscopy and SEC. Coinciding with the prevention of inflammasome formation, inhibition of NADPH oxidase gene expression or activity also prevented increases in caspase-1 activity as well as IL-1β production, signifying that NADPH oxidase is necessary for NALP3 inflammasome activation in response to Hcys. In contrast, inhibition of NALP3 inflammasome activation by either ASC siRNA or WEHD, a specific caspase-1 inhibitor, did not affect the ability of NADPH oxidase to produce superoxide, strongly indicating that NADPH oxidase-mediated redox signaling is an upstream event that activates NALP3 inflammasome in podocytes. Although there are reports that NALP3 inflammasomes can be activated by ROS in different cells or tissues (11,23,36), to our knowledge, our results for the first time link NADPH oxidase to the formation and activation of these inflammasomes in podocytes, where Hcys-induced NADPH oxidase activation produces superoxide to conduct redox signaling that triggers the formation of NALP3 inflammasomes and production of inflammatory cytokines such as IL-1β.
Next, we tested the in vivo triggering action of NADPH oxidase activation in the formation of NALP3 inflammasomes using mice lacking the gp91phox gene and in mice treated with NADPH oxidase inhibitory peptide, gp91ds-tat. Although the strategies used to genetically and pharmacologically inhibit NADPH oxidase in mice were not podocyte specific, confocal results demonstrated that this inhibition prevented hHcys-induced NALP3 inflammasome formation as shown by less colocalization of NALP3 with ASC and NALP3 with caspase-1, which mainly occurred in podocytes, given the increased colocalization of NALP3 molecules with podocin and desmin within glomeruli. Correspondingly, both interventions also inhibited hHcys-induced activation of NALP3 inflammasomes, confirmed by decreased caspase-1 activity and IL-1β production. The inflammatory response in glomeruli during hHcys, indicated by recruitment of macrophages and T-cells, was also significantly suppressed by both genetic and pharmacological inhibition of NADPH oxidase. These results from in vivo animal experiments further support the view that NADPH oxidase-mediated redox signaling promotes the formation and activation of NALP3 inflammasomes in podocytes during hHcys. Again, to our knowledge, these findings represent the first in vivo experimental evidence that triggering of NALP3 inflammasomes is attributed to NADPH oxidase-mediated redox signaling, which results in local inflammation in glomeruli during hHcys.
We also performed in vitro and in vivo experiments to address the functional relevance of NADPH oxidase-mediated triggering of NALP3 inflammasomes on Hcys-stimulated podocyte and glomerular injury. Podocytes, the epithelial cells lining the outermost layer of the glomeruli, are essential for proper filtration, and injury to podocytes is indicative of impaired glomerular filtration, in time leading to glomerular sclerosis (13,15). Foot process effacement, considered as the hallmark sign of podocyte injury, is usually accompanied by the destruction of the actin cytoskeleton, increased expression of slit diaphragm molecule and podocyte injury factor desmin, and reduction of the slit diaphragm molecule podocin, which is important for cell polarity and survival. In the presence of Hcys, inhibition, gene deletion, or silencing of either inflammasome component ASC or NADPH oxidase subunit gp91 phox was able to preserve the morphological structure of podocytes by keeping the distinct arrangement of the F-actin fibers intact and was functionally able to maintain podocin expression and prevent desmin expression. In vivo studies showed that gp91 phox−/− and gp91ds-tat-treated mice had less hHcys-induced podocyte injury, indicated by the preservation of podocin staining and podocyte number compared to hyperhomocysteinemic gp91 phox+/+ mice. Although such protection of podocytes observed in in vivo experiments may be due to a suppressed inflammatory response (Fig. 7), it is interesting to note that in in vitro experiments, the direct effects of suppressed inflammasome or NADPH oxidase also protect the functional and structural integrity of podocytes, even before a typical inflammatory response is instigated. This noninflammatory effect or direct action on podocytes has been reported to be associated with IL-1β-induced podocyte dysfunction (22). In addition to the detrimental actions of hHcys-induced inflammatory response, the early effect of activated inflammasome products, such as IL-1β, directly on podocytes may also importantly contribute to podocyte injury. If triggering of such inflammasomes by NADPH oxidase-derived ROS is blocked, both direct and indirect effects of inflammasome activation in the induction of podocytes injury may be blocked, as shown in our results.
Corresponding to protection of podocytes from Hcys-induced injury, inhibition of inflammasome activation by deletion of the gp91phox gene or inhibition of NADPH oxidase activity significantly ameliorated hHcys-induced glomerular injury in mice, as shown by improved proteinuria and albuminuria and decreased sclerotic changes in morphology of glomeruli from mice with hHcys. Inhibition of this inflammasome-triggering mechanism not only protects podocytes from injury, but also prevents glomerular sclerosis, which may be due to decreased podocyte injury as well as suppressed local glomerular inflammatory response. This NADPH oxidase-derived ROS may act as redox-signaling messengers to activate the inflammasome, which serves as the bridging and amplifying mechanism leading to a robust inflammatory response that eventually progresses to glomerular sclerosis. These findings provide evidence that either targeting the inflammasomes directly or targeting the mechanisms leading to their activation may be an effective therapeutic strategy for the prevention and early treatment of glomerular sclerosis and other end-stage organ damage resulted from hHcys.
In summary, the present study demonstrated that in the very early stages of glomerular damage, Hcys in vitro or hHcys in vivo stimulated the formation and activation of the NALP3 inflammasome, which initiated early injurious events in podocytes and glomeruli, leading to more serious glomerular injury and ultimate sclerosis. This NALP3 inflammasome activation, podocyte injury, and the glomerular pathology induced by hHcys could be substantially suppressed by inhibition of NADPH oxidase. These results may establish a new concept that NADPH oxidase-derived ROS upon Hcys stimulation trigger the formation and activation of NALP3 inflammasomes and thereby produce IL-1β and other factors, leading to podocyte and glomerular injury, potentially progressing into glomerular sclerosis.
Materials and Methods
Animals
Eight-week-old, male gp91 phox+/+ and gp91 phox−/− mice (Jackson Laboratories, Bar Harbor, ME) were uninephrectomized to accelerate renal injury, as described previously (32). After allowing a week for recovery after surgery, gp91 phox+/+ and gp91 phox−/− mice were fed either an ND or an FF diet for 4 weeks to induce hHcys (34). Another group of gp91 phox+/+ mice were injected intraperitoneally with gp91ds-tat or scrambled gp91ds-tat at a dose of 5 mg/kg every day, while being maintained on the FF diet for 4 weeks (10). This custom gp91ds-tat peptide constructed with a tat sequence to enable cell membrane penetration and inhibition of NADPH oxidase was synthesized by RS Synthesis (Louisville, KY) with the following amino acid sequence: YGRKKRRQRRRCSTRIRRQL. gp91 phox+/+, gp91 phox−/−, and gp91ds-tat mice were placed in metabolic cages, and urine samples were collected for 24 h before collecting blood samples, sacrificing, and harvesting tissues for analysis. All protocols were approved by the Institutional Animal Care and Use Committee of Virginia Commonwealth University.
Confocal microscopic detection of inflammasome proteins
Indirect immunofluorescent staining was used to determine the colocalization of inflammasome proteins in both podocytes and in glomeruli of the mouse kidney. Detailed double-immunofluorescent staining detection methods are presented in the online Supplementary Data.
Size-exclusion chromatography
SEC was performed in podocytes as described previously (33). The detailed SEC method was presented in the online Supplementary Data.
All other methods are described in the online Supplementary Data.
Footnotes
Acknowledgments
This work was supported by the grants DK54927, HL075316, and HL57244 (to P.L.) and 1F31AG043289-01 (to J.M.A.) from the National Institutes of Health.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.