
Abstract
Introduction
Innovation
Mitochondrial function is altered during hypoxia and following reoxygenation. We report here that sensitivity of the mitochondrial complex I to oxidative assault during ischemia/reperfusion could be determined by the conformational state of complex I. Transition of complex I into the dormant form in the absence of oxygen may make an important contribution to irreversible tissue damage during postischemic reoxygenation and may therefore represent a novel therapeutic target in this setting.
Mitochondrial complex I (EC 1.6.5.3) oxidizes NADH, contributing to the formation of membrane potential and consequently ATP synthesis, and therefore occupies a key position in cellular metabolism. Complex I is also an important source of superoxide and, most likely, it is responsible for the majority of ROS produced by the respiratory chain in vivo (10, 33). Mitochondrial complex I responds rapidly to lack of oxygen and is damaged by subsequent reoxygenation (5, 24, 34, 37, 45). The altered activity of complex I can have a significant effect on mitochondrial ROS generation. Moreover, this enzyme is not only a major source of ROS, but is also susceptible to damage during I/R, including that caused by oxidative and nitrosative stress (6, 12, 51).
Reversible conversion of the active, A-form of complex I into the dormant, D-form has been described in vitro (31), in rat heart ex vivo (39), and recently, in studies of cultured cells (14). If idle at physiological temperatures, the enzyme undergoes conversion into the D-form, which is characterized by a 10,000-fold lower catalytic activity compared to the catalytically competent A-form (53). In contrast to irreversibly inactive enzyme, the D-form is potentially capable of catalyzing a fast reaction and can be converted to the A-form after slow catalytic turnover(s) when substrates become available. Despite recent progress made in the resolution of the bacterial enzyme (9), very little is known about the eukaryotic complex I, so it is not yet possible to suggest the nature of the gross structural changes in the enzyme during activation/deactivation.
Deactivation of the enzyme in the absence of oxygen (14, 39) is an intrinsic property of complex I and it would be expected to play a functional role. However, in the time frame of ischemic conditioning, prolonged accumulation of the D-form may have severe pathophysiological consequences, depending on the duration of exposure, type of tissue, and the presence of natural effectors of the active/deactive (A/D) transition and of the ROS/antioxidant balance.
We have previously investigated the effects of lack of oxygen on the conformational state of mitochondria complex I and its sensitivity to nitric oxide-metabolites in isolated mitochondrial membranes and cultured cells (14, 17). Taking into account the role of complex I in generating ROS in I/R (13, 22, 42) and subsequent damage to oxidative phosphorylation, we sought to characterize changes in complex I states during I/R in an established experimental model. Here we present data showing that reversible deactivation of mitochondrial complex I takes place in situ under ischemic conditions and report the differential sensitivity of the two forms of the enzyme to superoxide anion and peroxide. We found that 50% deactivation of complex I occurs within 10 min of cardiac arrest, while reperfusion resulted in the return of complex I A/D equilibrium to its initial level. The D-form of complex I isolated from the ischemic samples was found to be sensitive to ROS treatment, and that sensitivity was eliminated by activation. Furthermore, oxidative modification of the D-form of the enzyme in vitro resulted in a decrease in the rate of NADH oxidation indicating functional damage of the enzyme. Subunits responsible for functional modification of the D-form of the enzyme have also been identified.
Results
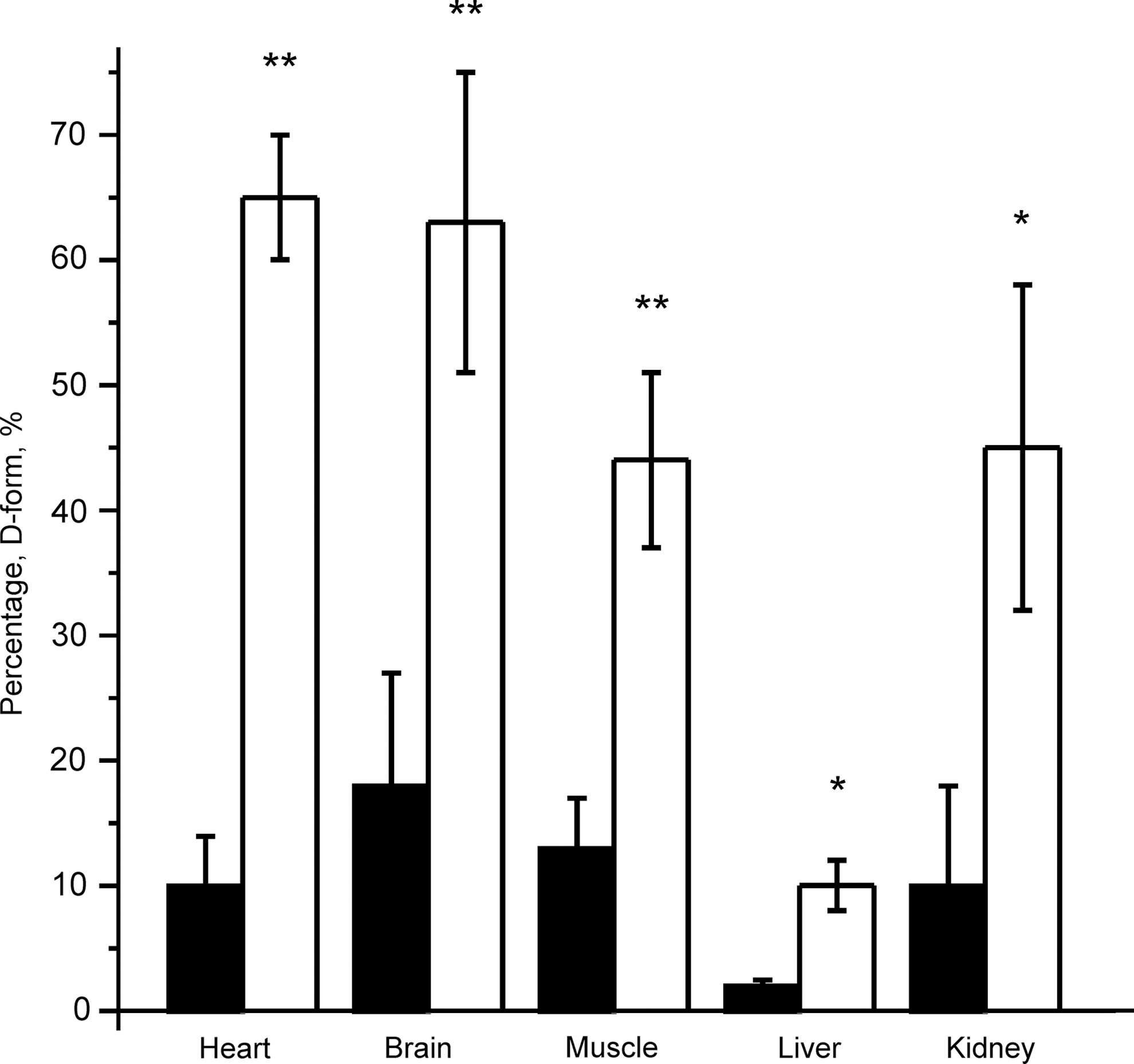
A/D transition in vivo in different tissues
First, the effect of induced cardiac arrest on the A/D ratio of complex I was investigated in different tissues 20 min after cardiac arrest (Fig. 1). The greatest degree of deactivation was observed in highly metabolizing tissues such as the heart and brain.
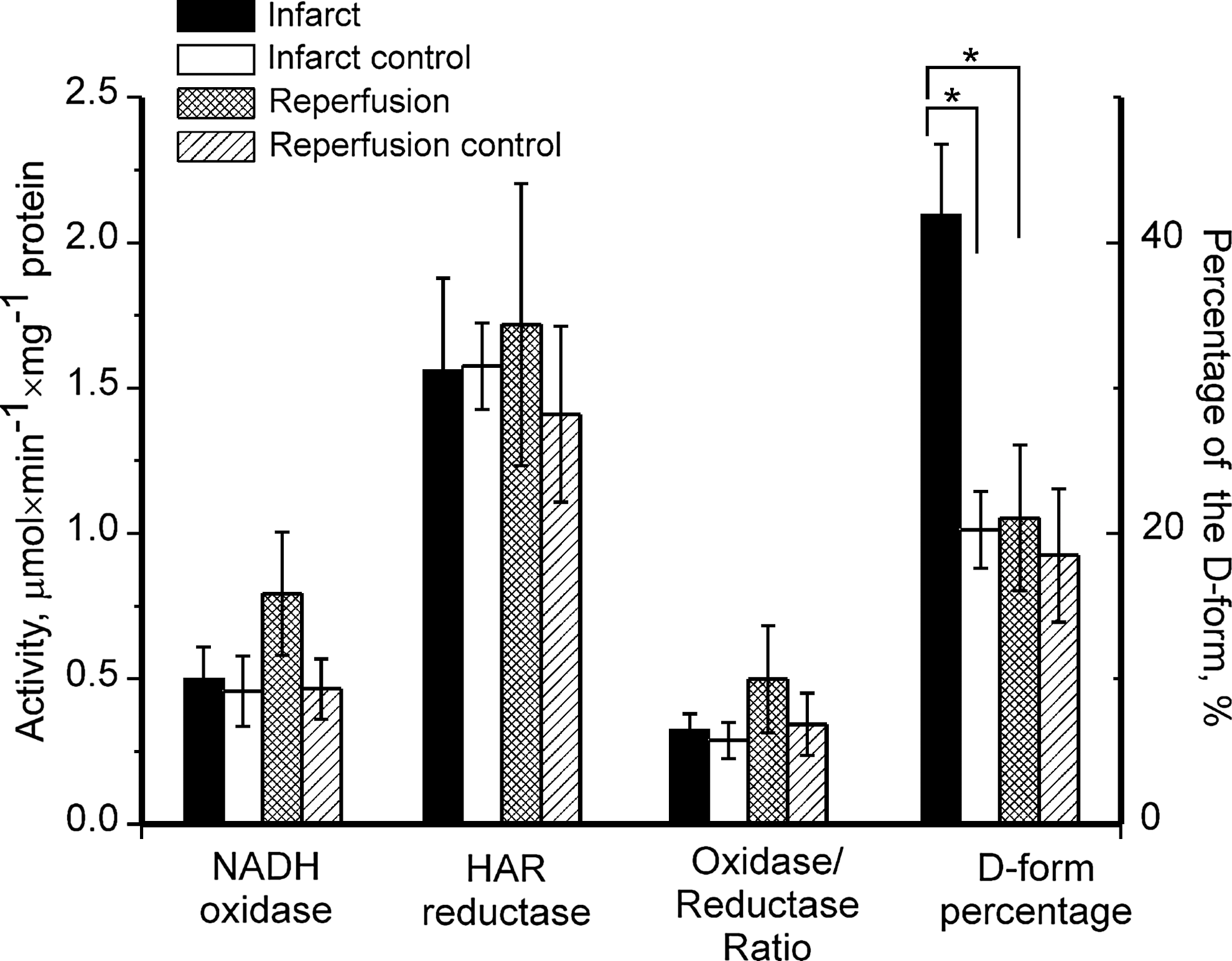
Cardiac I/R and activation state of complex I
Next, reversibility of complex I deactivation was assessed in situ. The effect of local myocardial ischemia with or without reperfusion, on complex I-catalyzed activities and on the A/D ratio is shown in Figure 2. There was almost twice as much complex I in the D-form in samples from ischemic tissues as there was in control tissues. In samples taken after reperfusion of the ischemic area, the D-form content was similar to that in the control samples. As judged by the NADH-oxidase and hexaammineruthenium (III) chloride (HAR)-reductase activity, the complex I activity and content in ischemic and reperfused samples was not different from that in the control samples. There was no significant difference between NADH- and succinate-supported generation of superoxide in any of the samples (data not shown).
Time course of complex I deactivation in heart
To determine the time course of complex I deactivation after cardiac arrest in situ, heart mitochondrial membranes were isolated at different periods after cardiac arrest, taking care to preserve the A/D ratio. The time course of myocardial complex I deactivation after cardiac arrest is shown in Figure 3. The t1/2 of deactivation was around 10 min. However, it should be noted that neither the total NADH-oxidase nor cytochrome c oxidase activity was significantly altered by ischemia.

Myocardial superoxide production by the mitochondrial respiratory chain after cardiac arrest
The percentage of the D-form of complex I in mitochondrial membranes obtained from mouse hearts from the control group and 20 min after cardiac arrest was 10% and 65%, respectively (Table 1). The rate of NADH-supported superoxide generation was significantly higher in mitochondrial samples obtained 20 min after cardiac arrest than in control samples. Turnover-dependent activation eliminated that difference between these samples (data not shown). Rate of succinate-supported superoxide generation was found to be similar in control and ischemic samples (Table 1).
Values represent means of quadruplicate measurements in two experiments.
Activity was measured in the presence of 1 μM antimycin A.
n=3 animals per group, two experiments, quadruplicate measurements p<0.05 versus control.
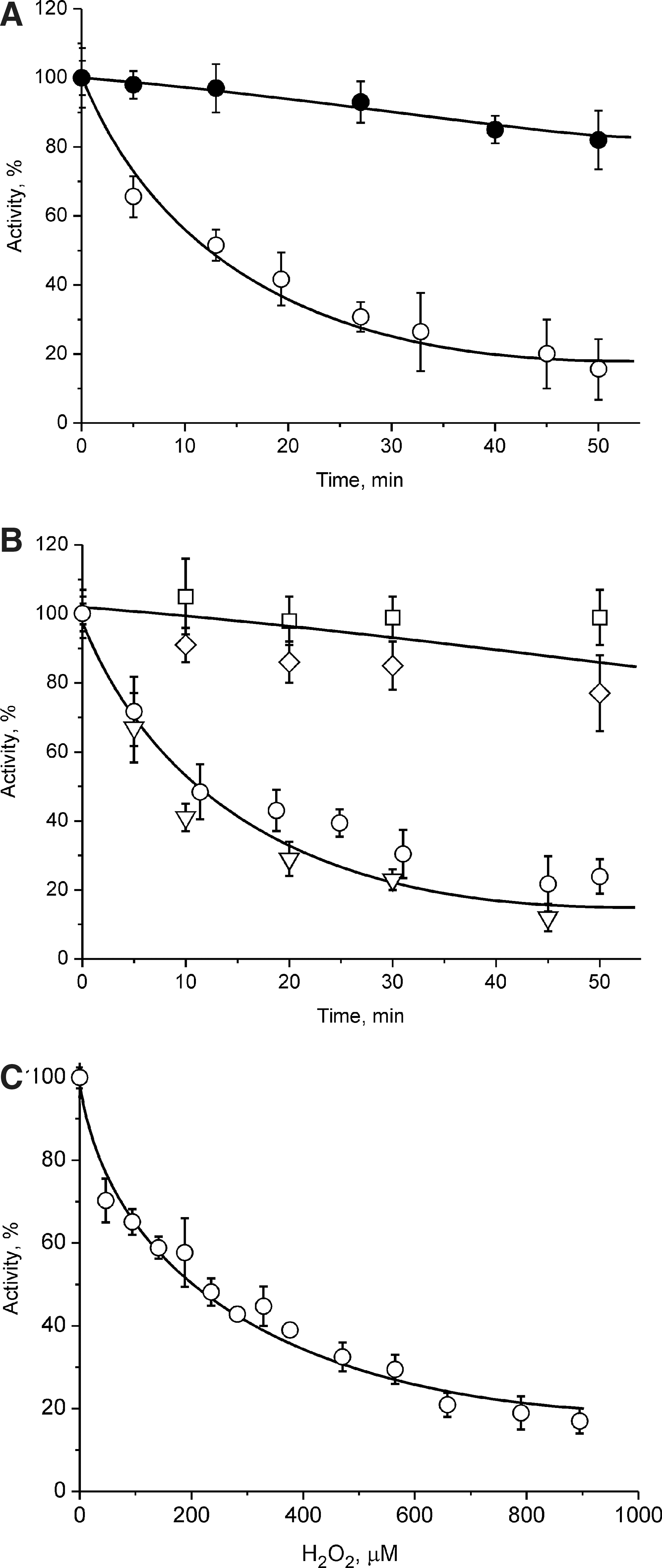
Sensitivity of the A- and the D-forms of complex I to H2O2 and O2·−
Mitochondrial membranes isolated from heart at 0 and 20 min after cardiac arrest (containing 8% and 67% of complex I in the D-form, respectively), were subjected to incubation for 30 min in the presence of the xanthine/xanthine oxidase superoxide generating system. After addition of 50 μM xanthine, the system is able to generate superoxide at an initial rate of 50 nmol/min/mg protein, as assessed by superoxide dismutase (SOD)-sensitive reduction of acetylated cytochrome c. As shown in Table 2, incubation of mitochondria with this O2 · −-generating system resulted in a significant decrease in the NADH-oxidase activity in heart tissue samples obtained 20 min after cardiac arrest. This effect was abolished when all the D-form is converted into the A-form. Addition of 50 U/ml catalase to the incubation medium did not alter the observed effect, while the presence of 50 U/ml SOD completely prevented the inhibitory actions of the O2 · −-generating system in all samples. There was no significant effect on the succinate-oxidase activity of mitochondrial membranes from either sample.
The heart mitochondrial fractions were diluted to 1 mg/ml with a medium containing a 50 mM phosphate buffer pH 7.2, 20 mM KCl, 0.1 mM EDTA, and components of the superoxide-generating system (50 μM hypoxanthine and 5 μg/ml xanthine oxidase from bovine milk). Incubation was carried out at 20°C. Superoxide dismutase 50 U/ml was used to scavenge the superoxide generated. To activate complex I, NADPH was used as described previously (17). All activities are given in μmol substrate×min −1 ×mg −1 .
p<0.05 versus control.
Figure 4A demonstrates a dramatic difference in the sensitivity of the A- and the D-form of complex I to hydrogen peroxide. Incubation in the presence of 1.0 mM H2O2 led to the inhibition of the NADH-oxidase reaction of the D-form, but not the A-form. The time course of inhibition of the NADH:Q1 reductase activity was similar to that of NADH-oxidase, indicating a direct effect on complex I. Presence of 50 U/ml SOD did not affect the time course of inactivation, while 100 U/ml catalase or 1.5 mM reduced glutathione protected the D-form of the enzyme from inactivation (Fig. 4B). Careful titration of the D-form of the enzyme in the presence of catalase inhibitor 3-amino-1,2,4-triazole (Fig. 4C) shows the sensitivity of the D-form to H2O2.
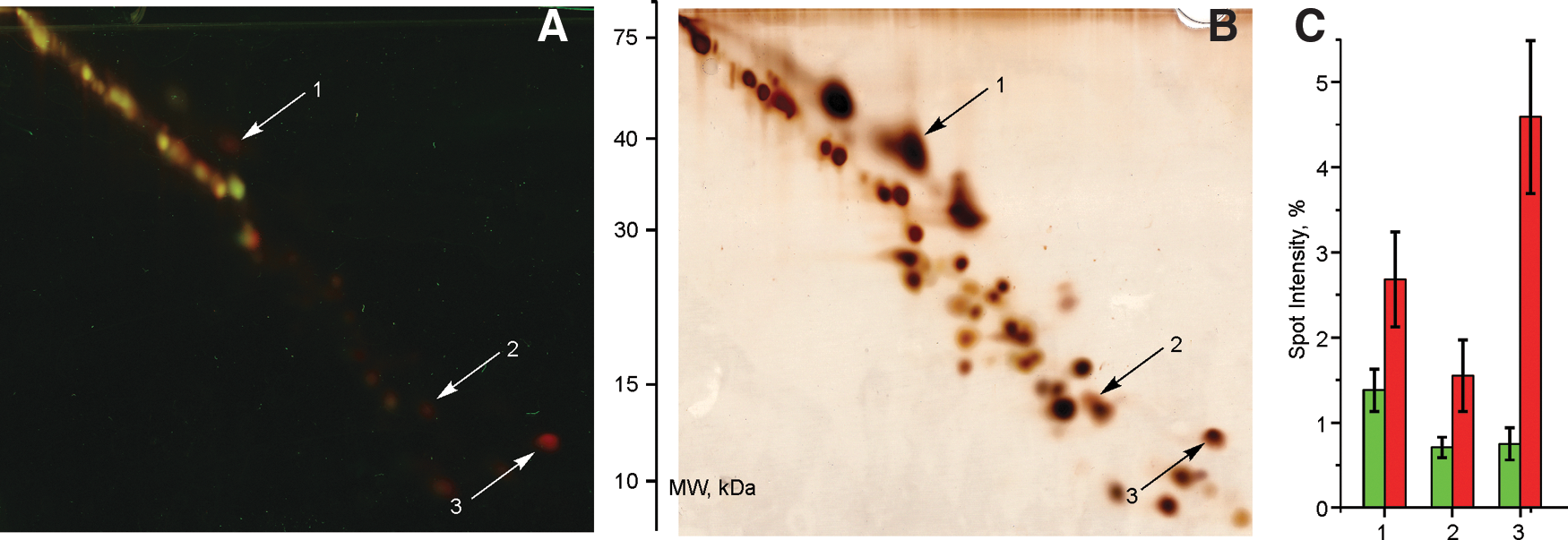
Identification of the subunits involved in oxidative modification of the D-form by redox difference gel electrophoresis
Cysteine-reactive Cy3-N-ethylmaleimide (NEM) and Cy5-NEM dyes were used to label oxidized protein thiols in the A- or the D-form of complex I after H2O2 treatment. Blue native PAGE followed by double SDS-gel separation of complex I subunits allows identification of differentially labeled thiols of complex I in H2O2-treated samples of the A- form and the D-form without interference from other mitochondrial proteins (Fig. 5A). Control samples without H2O2 treatment demonstrated significantly more labeled spots with no difference in the fluorescent signal between the A- and the D-form (not shown). Most of the fluorescent spots had equal Cy3 and Cy5 signal so that the ratio of Cy3 and Cy5 fluorescence intensity was between 0.8–1.2, indicating equal amount of oxidized thiols in corresponding subunits in both samples. The total number of fluorescent spots in H2O2-treated samples was much less than the number of protein spots revealed by silver staining (Fig. 5B). Reciprocal labeling experiments, where the D-form was labeled with Cy3 and the A-form with Cy3 showed the same results, although a slight dye-dependent shift in the electrophoretic mobility of some Cy3- and Cy5-labeled subunits was observed. However, one spot with apparent mass of 40 kDa and two spots of around 13 kDa were significantly more labeled in the D-form than in the A-form (Fig. 5C). Table 3 lists three complex I subunits containing thiols with a significantly altered redox state upon H2O2 treatment of the A and the D samples identified by LC-MS/MS under conditions listed in Supplement. In-gel digestion of the 40-kDa spot produced only a single peptide of the mitochondrial ND4 subunit. However, from the unusual electrophoretic mobility [located at the upper diagonal (46)], it can be concluded that this is indeed ND4. One low molecular weight subunit was identified as B12. The highest difference in the fluorescence intensity between the A and the D samples was found in the third highly hydrophobic spot located above the diagonal. The corresponding protein was expected to have 605.8 Da higher molecular mass due to Cy5-NEM labeling (See Supplementary Data Table S1 [Supplementary Data are available online at
Number of cysteins located in the interhelical region was determined from the high-resolution structure of prokaryotic complex I (9).
A/D, active/deactive.
Discussion
I/R injury has been associated with many types of surgery and vascular interventions. This phenomenon is particularly relevant to cardiac operations in which reperfusion of coronary flow is necessary to resuscitate the myocardium after a period of ischemia, and to percutaneous coronary intervention after myocardial infarction. If performed in a controlled fashion and within a short time postischemia, reperfusion may facilitate cardiomyocyte survival, reduced cardiac damage, and improved post-traumatic recovery. However, the resultant increase in ROS generation in I/R poses significant risks and may mediate irreversible tissue damage. It is therefore essential to understand underlying mechanisms to inform novel therapeutic strategies to prevent I/R injury in such clinical situations.
Two catalytically and structurally distinct forms of complex I have been shown to be present in mitochondrial membranes: one the fully competent, active A-form and the other the dormant, deactivated, D-form.
In the present study, we performed analysis of the A/D ratio of mitochondrial complex I from various tissues after the onset of cardiac arrest. In the absence of oxygen, the respiratory chain is over-reduced, resulting in the lack of second complex I substrate ubiquinone, decrease of complex I catalytic turnover, and eventual deactivation of the enzyme. In highly metabolic tissues such as the brain and heart, 20 min of global ischemia significantly shifted the A/D equilibrium toward formation of the D-form. This form exhibits a much lower catalytic activity than the A-form, but unlike the irreversibly inactivated or denatured enzyme, the D-form can be converted to the A-form during that slow catalytic turnover(s) and is potentially able to catalyze a rapid physiological reaction.
To measure the full complex I activity, enzyme preparations should be activated before the measurements as carried out in the present study and described previously (17). It is important to stress that accumulation of the D-form during the ischemic period observed here is fully reversible, but could easily be mistaken for enzyme inactivation when the NADH-oxidase activity is assessed by conventional methods. Preparations composed of a mixture of the A- and the D-form catalyze oxidation of NADH with a lag phase during continuous assay. This lag phase represents slow activation of the D-fraction of the enzyme during the time of the measurement (14) and can be easily interpreted for a linear initial rate when an assay buffer of pH>7.5 is used or divalent cations are present (53). If the preparation composed mostly of the D-form (i.e., postischemic mitochondrial samples) is assessed without activation, the observed initial rate is low and represents only the contribution of the A-form fraction. In our experiments, total activated NADH-oxidase did not change with the time of ischemic treatment.
The time course of complex I deactivation in situ revealed that the half-time (t1/2) for the heart was 12 min after the onset of global ischemia. Using an in vivo mouse infarction model, we showed that local cardiac ischemia results in accumulation of the D-form, while reactivation occurs in situ after reperfusion. At the same time, the D-form of complex I isolated from the ischemic area can be reactivated in vitro during turnover-dependent reactivation when substrates are added (14, 31, 39). The difference in the D-form content in mitochondria derived from the samples subjected to 20-min global ischemia (<60%) and local infarction (40%) most likely reflects possible oxygenation of the border regions of the ischemic zone during post-treatment surgery as well as some inaccuracy in the excision of the ischemic area of the heart.
In our I/R experiments, the complex I activity and content, estimated from the NADH-oxidase and NADH:HAR reductase activity, was not significantly different in mitochondrial membranes from the control and ischemic samples. A similar oxidase/reductase rates ratio in all of the samples indicates that neither the complex I content nor its turnover were affected by ischemia or following reperfusion. Since the complex I full activity could be restored after turnover-dependent reactivation, the enzyme did not undergo significant covalent modifications affecting its activity. It is therefore likely that our chosen conditions of 20-min ischemia, with or without 10-min reperfusion, were not sufficient to cause the significant damage to complex I observed by others using a longer ischemic treatment (23, 24). Nonetheless, complex I catalyzed activities in mitochondria obtained from control, ischemic, or reperfused samples in our experiments, is in agreement with the findings of several other studies (51).
Oxidative stress is considered as an important mechanism of I/R injury. Complex I-dependent production of ROS is increased during reperfusion, although the underlying mechanism remains unclear (2, 38, 43, 54). We have shown that the complex I-dependent superoxide anion generation was significantly higher in mitochondrial membranes isolated from the ischemic heart, most likely, due to the presence of the D-form accumulated during the ischemic period. This is in agreement with the earlier observation of higher capacity for superoxide generation by the D-form (20).
We also determined the sensitivity of the A- and the D-forms of complex I to superoxide anion and hydrogen peroxide. In heart mitochondrial membranes, exogenously added H2O2 inhibits only the D-form of the enzyme, but not the A-form. This has been further tested on control samples and those obtained 20 min after ischemia, using a superoxide generating system able to produce 0.25 nM/min superoxide. As expected, incubation of mitochondrial membranes from the ischemic tissue in the presence of a low steady-state concentration of superoxide resulted in a dramatic inhibition of the NADH-oxidase activity in comparison with control. Activation of complex I completely abolished that inhibition, rendering the A-form of the enzyme less sensitive to superoxide.
It is not clear what level of sensitivity of the enzyme to ROS may be expected in situ, where the enzyme is exposed to additional factors such as high osmolarity and electrical field, and is surrounded by other proteins [including enzymes mediating H2O2 oxidative action (21)]. Prolonged exposure of the D-form of the enzyme to low steady-state levels of endogenous ROS at physiological temperature would inevitably result in irreversible damage to some fraction of the enzyme population leading to accumulation of irreversibly modified enzymes over time. Therefore, taking into account the high degree of flux control of complex I over oxidative phosphorylation (18, 26, 32), a slight decrease in the NADH:ubiquinone reductase activity, even if this is not sufficient to induce an acute effect on apparent respiration, may lead to a significant decrease of the ATP production by mitochondria (52).
Together, our observations suggest that accumulation of the D-form of the enzyme takes place during ischemia (i), this accumulation increases ROS production (ii); the presence of complex I in the D-form may potentially increase susceptibility of mitochondria to oxidative damage (iii), so could result in the so-called vicious cycle of damage during I/R (5, 8, 14, 48).
The link between inhibition of complex I in I/R and the nature of the oxidative modification of the enzyme is not completely understood, but it is likely to involve oxidation, nitrosation, nitration, glutathionylation, or disulfide formation of cysteine thiols (41). There have been a number of reports on covalent modifications of complex I in I/R: thiol oxidation of NDUFS1, NDUFS2, NDUFV1, NDUFV2, NDUFA6 (51), and tyrosine nitration of NDUFS2, NDUFS3, and NDUFV2 (35) in the heart and nitration of GRIM-19 in brain mitochondria (6). Our redox difference gel electrophoresis (DIGE) results suggest three subunits that may be involved in the functional modification of the D-form by H2O2: ND3, B12, and ND4. However, the subunit B12 (NDUFB3) has no cysteine residues in its primary sequence and can therefore be excluded. The highest difference in Cy-dye labeling of the H2O2-treated A- and D-form makes ND3 the most likely candidate. Moreover, identification of peptide, thiol labeled by Cy-dye identifies cysteine-39 as the exact region of modification. This cysteine is exposed only in the D-form of the enzyme as shown before (16). Most likely, if Cys-39 of the ND3 subunit complex I is modified, the enzyme does not catalyze the physiological NADH:ubiquinone reaction, making it an early mitochondrial target for oxidative/nitrosative stress during I/R.
Modification of the FeS clusters of complex I can be a major factor for complex I inhibition (44, 50). In our experiments, the possibility of FeS cluster damage cannot be excluded, however, in vitro studies indicate that the accessibility of all clusters to the outside environment is the same in the A- and D-form (29).
The combined processes of accumulation of mitochondrial fatty acid (27) and an increase in the matrix Mg2+ and Ca2+ concentration (11, 19, 47) would shift the A/D equilibrium of complex I toward the D-form during an ischemic episode affecting myocardial recovery. Moreover, after reoxygenation, the possible opening of the mitochondrial permeability transition pore could also result in Ca2+ overload (1, 7, 28, 40). Together with the release of cytochrome c (4), this would significantly delay complex I reactivation. At that stage, the exposure of Cys-39 of the ND3 subunit of the D-form of complex I (16, 17) may be an important factor determining specific inhibition of the NADH:ubiquinone reductase activity by an as yet unknown mechanism. As shown in our experimental settings, functional modification of the D-form may give a rise to a population of mitochondria with a decreased respiration rate, an over-reduced pool of matrix NAD(P) nucleotides, and a low ATP-synthesizing capacity. Such a population would delay or significantly retard the functional recovery of the cardiomyocytes in I/R. Our results suggest that the deactivation of mitochondrial complex I and increased susceptibility of the enzyme to ROS or nitric oxide metabolites (17) after the ischemic period may represent one of the important contributory mechanisms involved in cardiac injury during acute I/R. Combined classical therapy and interventions for fine tuning of the A/D ratio in mitochondria during the reperfusion process may provide new avenues for ischemic treatment.
Materials and Methods
Experimental animals
Female C57BL/6J mice (8–12 weeks, Charles River) were employed for all studies and were fasted overnight before experimentation. Animals were housed under constant climatic conditions with free access to food and water. All experiments were performed in accordance with the Guidance on the Operation of the Animals (Scientific Procedures) Act, 1986 (UK).
Cardiac arrest and tissue extraction
Cardiac and respiratory arrest was initiated by cervical dislocation, carcasses were placed in a portable 37°C incubator to maintain physiological body temperature. After specific time periods (2–30 min), organs or tissues were rapidly (within 90 s) extracted, washed in an ice-cold phosphate saline buffer, and snap-frozen in liquid nitrogen.
In vivo ischemia reperfusion
Mice were subjected to acute myocardial ischemia by ligation of the left anterior descending coronary artery under 2% isofluorane/oxygen anesthesia as previously described (36). This procedure has been shown to produce reliable and reproducible myocardial infarcts of ∼40%. Animals were sacrificed by cervical dislocation either after 20 min of ischemia or after a further 10-min period of reperfusion. Hearts were then rapidly excised and separated into ischemic or nonischemic regions (control) before being frozen in liquid nitrogen and stored at −80°C for further analysis.
Mitochondrial membrane isolation
Isolation of the mitochondrial membranes preserving of the A/D ratio was performed essentially as previously described, with minor modifications (39). Pieces of frozen tissue were then placed in a liquid nitrogen precooled metal mortar and pulverized by striking with a mallet. The resulting powder was added to 10 ml of the isolation medium (200 mM Tris-HCl, pH 8.8, 0.5 mM EDTA 1 mM K3Fe(CN)6, and 2 mM potassium malonate) and homogenized in a IKA tissue disruptor (2 min×6000 rpm). Particular care was taken to cool down all the mediums, glassware, and centrifuge rotors. An alkaline buffer and 1 mM ferricyanide were used to allow rapid oxidation of reduced matrix pyridine nucleotides to prevent turnover-dependent complex I activation. Tissue debris was discarded after brief centrifugation for 10 min at 16,000 g at 0°C. The supernatant was diluted to 40 ml with the same medium and the membranes were collected by centrifugation for 25 min at 48,000 g. The membrane pellet was rinsed with 20 mM Tris-HCl (pH 8.0), 0.25 M sucrose, and 0.2 mM EDTA and resuspended in 400 μl of the same buffer. The membranes were then frozen in liquid nitrogen and stored at −80°C until use. The protein content was determined by bicinchoninic acid (BCA) assay (Sigma).
Activity measurements
Oxidation of NADH was determined spectrophotometrically (Varian Cary 4000) as a decrease in absorption at 340 nm with 150 μM NADH in 1 ml of the standard assay medium (0.25 M sucrose, 50 mM Tris-HCl pH 7.0, 0.2 mM EDTA) supplemented with 5 μM cytochrome c and containing 10–25 μg protein/ml mitochondrial membranes. Additional measurements of NADH:Q1 or NADH:HAR oxidoreductase reductase were assayed in the presence of 1 mM cyanide with the addition of 80 μM Q1 or 1 mM HAR, respectively (Sigma). The formation of superoxide radicals was monitored as the SOD-sensitive reduction of acetylated cytochrome c [ɛ550–539nm=21.5, (15)] in the same assay medium, pH 8.0, supplemented with 20 μM acetylated cytochrome c, substrates (50 μM NADH or 5 mM succinate) and containing 0.4–0.5 mg/ml mitochondrial membranes. Acetylated cytochrome c was prepared as described previously (15).
Determination of A/D ratio
The diagnostic test for determination of the A/D ratio is based on the fact that in the presence of divalent cations such as Mg++ or Ca++ and at alkaline pH (8.5), the rate of reactivation of complex I is very slow (39) (see also (30) for the details). The total amount of the enzyme (A+D) was estimated after full activation of complex I by preincubation of the sample in 0.1 ml of a standard medium (pH 7.0) with 20 μM NADH for 30 s before the addition of 0.9 ml of the standard medium (pH 8.8), 5.5 mM MgCl2, and 165 μM NADH. For estimation of the A-form fraction, 20 μM NADH was omitted from the initial preincubation in the pH 7.0 medium. In these conditions, the initial rate of NADH-oxidase accurately corresponds to the activity contributed only by the A-form, since the activation of the D-form is significantly slower than the time of the assay. In all inhibition studies, the D-form was treated with an effector and activity assessed only after activation by NADH.
To prepare SMP in which complex I is present almost entirely in the D-form, an aliquot of frozen membranes was thawed, diluted to 5 mg/ml with a standard assay medium (pH 8.5), and incubated at 35 °C for 1 h. To obtain a fully active enzyme, after thermal deactivation, membranes were incubated aerobically for 10–20 min at room temperature with 1% ethanol, 400 μM NADH, and 0.1 mg/ml alcohol dehydrogenase (17).
Redox DIGE and subunits identification
Redox DIGE was performed essentially as previously published (25). Control and membranes treated with 1 mM H2O2 were washed by centrifugation, 20 mM NEM was added to the suspension, incubated at 30°C for 30 min, and membranes were washed once with the standard buffer. After resuspending the pellet at around 1 mg/ml, 5 mM dithiothreitol (DTT) was added and samples were incubated at 10°C for 15 min. Membranes were pelleted by centrifugation and washed three times with the same buffer, resuspended at 1 mg/ml, and treated with 30 μM Cy3- or Cy5-maleimide. Each sample was treated with both dyes. After 30 min, the reaction was quenched with 10 mM DTT and washed twice before pooling of labeled samples.
Complex I subunits were separated as described previously (16). The gels were scanned using the Fujifilm FLA fluorescent scanner and stained with silver (49). Images were quantitatively analyzed using Aida Image Analyzer software (Raytest).
Spots of interest were excised, the proteins in-gel digested, and identification was performed through mass spectrometric analysis at the BSRC Mass Spectrometry and Proteomics Facility, University of St Andrews (See Supplementary Data). All proteins were identified with >99% confidence (Prot Score>2.0) with False Discovery Rates of Local FDR<5%, Global FDR<1%.
Statistical analysis
All results are expressed as mean±SEM unless specifically indicated and were analyzed by one-way ANOVA to determine statistically significant differences of means among groups.
Footnotes
Acknowledgments
This work was supported by the Medical Research Council UK grant to A.G. (NIRG G1100051). We would like to thank Dr. Vsevolod Belousov for critical reading of the manuscript. The authors are grateful to Matthew Fuszard (St. Andrews University) for the help with mass spectrometry analysis of the samples, to Annie Higgs and Amanda Birch for help in the preparation of this manuscript and to Sir Prof. Salvador Moncada for valuable discussion.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.