
Abstract
Introduction
See text for details.
HAS-SNO, S-nitrosylated human serum albumin; SNO-Hb, S-nitrosohemoglobin; NF-κB, nuclear factor-Kappa B; MMP, matrix metalloproteinases.
The thioredoxin system comprising thioredoxin (Trx), selenoenzyme thioredoxin reductase (TrxR), and NADPH is the major protein-disulfide reductase of the cells (36 –40). Trxs are crucial proteins, and the cells' major disulfide reductases with Trx1 in the cytosol and Trx2 in mitochondria (61) regulate the activities of several transcription factors, redox regulation of different metabolic pathways, oxidative stress defense, DNA synthesis by being a hydrogen donor for ribonucleotide reductase, apoptosis, and NO signaling. The active site disulfide form of Trx undergoes NADPH-dependent reduction by TrxR and, in turn, reduces oxidized cysteine groups (disulfide or sulfenic acid) on substrate proteins by utilizing the two cysteinyl residues in its Cys-Gly-Pro-Cys active site. TrxR is a homodimeric, FAD-containing selenoenzyme with three isoforms: TrxR1 (cytosol), TrxR2 (mitochondrial), and TrxR3 (present primarily in testis) (2). Mammalian cytosolic TrxR has a redox center, consisting of Cys59/Cys64 adjacent to the flavin ring of FAD and another redox center consisting of Cys497/SeCys498 close to the C-terminus (71, 120). This selenothiol is the proper active site that can reduce many of the potential substrates of the enzyme, and the essential role SeCys residue makes it the target of several drugs in cancer treatment (2, 77, 120). The structure and function of Trx1 is regulated by structural SH-groups that can be oxidized to an extra disulfide or nitrosylated (33). Trx plays a major role in protein S-denitrosylation but, under certain conditions, it can also trans-S-nitrosylate other proteins (33, 72, 93). Taking account of the recent progress in the field of research, this review focuses on the regulation of cellular processes by reversible S-nitrosylation and Trx-mediated cellular homeostasis of RSNOs and S-nitrosoproteins.
Trx-TrxR System
Trxs are a family of small proteins (12 kDa or larger) catalyzing thiol-disulfide oxidoreductions by using redox-active cysteine residues that are present in a Trp–Cys-Gly-Pro-Cys sequence motif (36
–40, 51). In oxidized Trx (Trx-S2), the active site is a disulfide and in reduced Trx [Trx-(SH)2], it is a corresponding dithiol. Trx-(SH)2 reduces protein disulfides (Reaction 1) and forms Trx-S2, which, in turn, is reduced by TrxR and NADPH (Reaction 2) (38, 39, 51).
TrxR from bacteria, fungi, and plants are FAD-containing homodimers of 70 kDa with active site cysteine residues (2). In contrast, mammalian TrxRs are larger dimeric selenoenzymes (114 kDa or larger) with a catalytically active selenocysteine residue located in the C-terminus (2, 28, 120). The catalytic mechanism of mammalian TrxR homodimer involves the transfer of electrons from NADPH to the enzyme-bound FAD, which reduces the disulfide within the CVNVGC motif in the N-terminal domain that subsequently reduces the selenylsulfide motif to a selenothiol at the C-terminal Sec-containing GCUG motif of the neighboring subunit (2). The selenothiol is the proper active site that can reduce many of the potential substrates of the enzyme.
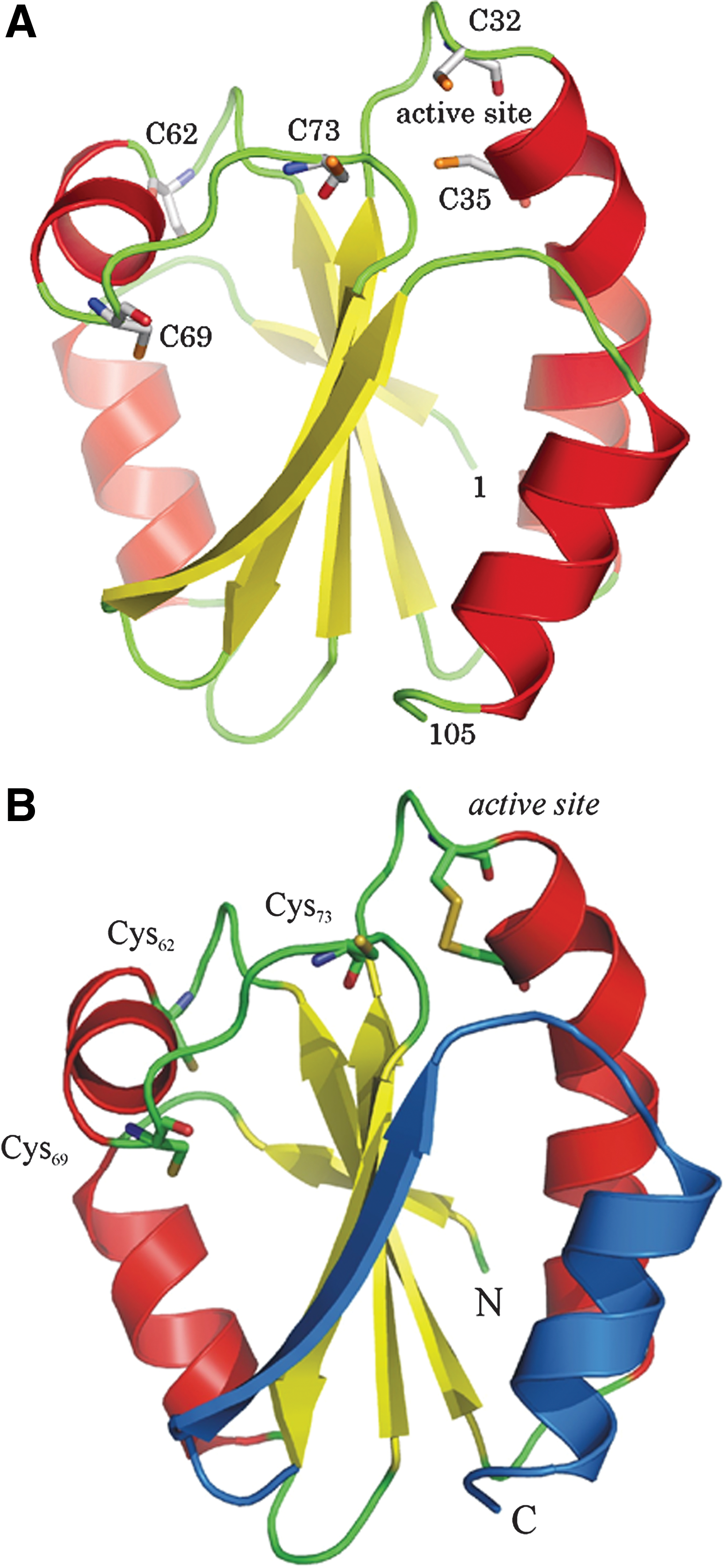
Trxs exist in all living cells and in multiple forms such as approximately 19 isoforms in plants (89). Humans have two classical Trxs, including Trx1 (cytosolic/nuclear) and Trx2 (mitochondrial), both of which are essential and embryonically lethal (61). The structures of Trxs from many species, determined by X-ray crystallography and multidimensional NMR, showed the Trx fold comprising a central core of β-sheet surrounded by α-helices (36 –40, 61, 108) (Fig. 1). The active site of Escherichia coli and human Trx1 comprises Cys32 and Cys35, which are located at the end of the second β-sheet and in the beginning of α helix2. The pKa value of Cys32 is lowered to 7, and this is due to hydrogen bonding within the active site. The pKa value of Cys35 is high in Trx-(SH)2. The mechanism of Trx-catalyzed reduction involves docking and formation of a transient mixed disulfide with the substrate after a nucleophilic attack by the depronated N-terminal Cys residue in the active site (38, 61). Kinetically, Trx is known to be four to six orders of magnitude faster as a dithiol reductant compared with a strong chemical reductant such as dithiothreitol. Recently, the mechanism of Trx-catalysis has been probed by single molecule techniques (109). The Trx fold is found in a large number of proteins, including glutaredoxins, glutathione (GSH) peroxidases, GSH transferases, protein disulfide isomerases (PDIs), and many other proteins (70). The structure and function is regulated by structural SH-groups that can be oxidized to an extra disulfide or nitrosylated. Moreover, depending on the redox states of the active site dithiols, Trx1 catalyzes either trans-S-nitrosylation or S-denitrosylation of its target proteins (33, 93, 110).
Formation of RSNOs and S-Nitrosoproteins
Nitric oxide synthases (NOSs) are the family of enzymes that catalyze the production of NO from L-arginine in the presence of NADPH and O2 (1, 45, 62). In mammalian cells, NOSs are the major sources of endogenous NO and the three mammalian NOS isoforms (inducible NOS [iNOS], endothelial NOS [eNOS], and neuronal NOS [nNOS]) exhibit a similar catalytic profile and composition. However, iNOS was found to generate a much greater extent of NO, to more than 100 fold, compared with that produced by constitutive NOSs (eNOS and nNOS) (55). Moreover, iNOS and nitrosative stress have been implicated in many human diseases, including atherosclerosis, insulin resistance, inflammation, and neurodegenerative diseases (21, 60, 79).
S-nitrosylation, the covalent attachment of the NO function to the thiol group of cysteine, is a ubiquitous redox modification and an important mechanism for post-translational regulation of different proteins. The products of S-nitrosylation, RSNOs, have been suggested to play crucial roles in human health and diseases (25). Several mechanisms, such as trans-S-nitrosylation, acidic S-nitrosylation, and catalysis by metalloproteins, have been proposed for the formation of RSNOs (31, 35). However, some of the pathways can be predominant over others depending on the biological context of cells; such as the acidic S-nitrosylation under inflammatory conditions (31, 83) and metal-catalyzed S-nitrosylation in the presence of ceruloplasmin (101).
NO forms metal-nitrosyl complexes and N2O3, which can act as S-nitrosylating agents in cells (11, 54). Activated mouse macrophage and vascular smooth muscle cells have been reported to produce NO2 − at a rate of 50–70 nmol/mg of protein/h for approximately 20 h (4, 105). The formation of GSNO is expected to follow the production of endogeneous NO due to the presence of GSH, the most abundant intracellular thiol. GSNO has been detected in the brain (54) and liver (98) of healthy rats in amounts of 15–34 pmol/mg protein. Thus, the formation of low-molecular-weight RSNOs could be predicted in both the intra- and extracellular environments, as NO can S-nitrosylate extracellular thiols on diffusion across plasma membranes. In another study, on lipopolysaccharide (LPS) stimulation, RAW 264.7 cells were found to be generated 17.4±1.0 pmol/mg of S-nitrosoproteins, which decayed with a half life of about 3 h (118). Moreover, S-nitrosoalbumin and S-nitrosohemoglobin (SNO-Hb) were found to be increased by ∼3.4- and ∼25-fold, respectively, in response to the LPS challenge in rats (50). The administration of LPS to the experimental animals resulted in an upregulation of iNOS and sustained overproduction of NO that contributed to the increased level of circulating RSNOs. The overproduction of NO has been proposed to contribute to circulatory failure, myocardial dysfunction, organ injury, and multiple organ dysfunction syndrome associated with endotoxic shock (76, 103). It was further demonstrated that plasma albumin became saturated with NO at physiological NO concentrations and catalyzed the RSNO formation involving a micellar catalysis mechanism (79). The albumin-mediated S-nitrosylation and its vasodilatory effect were found to be directly dependent on the concentration of circulating low-molecular-weight thiols. The transmembrane movement of RSNOs from the plasma albumin-SNO reservoir was suggested to be mediated by cysteine, which serves as a carrier (58, 59, 119). The mechanism involves trans-S-nitrosylation of free cysteine from S-nitrosoalbumin (or S-nitrosylated albumin) and stereoselective uptake of L-CysNO by two major members of the ubiquitous amino acid transporter system L (LAT1 and LAT2) (58, 59, 119).
Functional Alteration of S-Nitrosoproteins
Till date, more than a thousand of S-nitrosylated cellular proteins have been identified (25) (Table 1). The S-nitrosylated proteins have been suggested to be involved in a variety of cellular processes/diseases, including signal transduction, insulin resistance, apoptosis, and cancer. There are several proteins containing multiple cysteine residues, while the S-nitrosylation of specific cysteine depends on the concentration of S-nitrosylating species at the modification site, pKa values of thiols, hydrophobic environment, and several other factors (20). The S-nitrosylated human serum albumin (HAS-SNO) was suggested to act as a circulating reservoir for NO produced by endothelial cells (44, 82, 96). HAS-SNO showed potent antibacterial activity and inhibited the proliferation of cultured human vascular smooth muscle cells. Furthermore, antitumor activity of HAS-SNO was examined in murine colon 26 carcinoma cells (C26) and also in C26 tumor-bearing mice (52). SNO-Hb was found in the systemic circulation, at concentrations ∼10 times higher in the arterial than the venous blood (49, 97). SNO-Hb has been suggested to act as an endogenous NO donor and a physiological regulator of blood flow.
Nuclear factor-Kappa B (NF-κB) is a versatile transcription factor that regulates a wide variety of cellular processes, including inflammation and survival. NO has been suggested to play an anti-inflammatory role through the S-nitrosylation of components of the NF-κB pathway. Marshall and Stamler reported the inhibition of NF-κB activity by the S-nitrosylation of a critical thiol in the DNA-interacting p50 subunit (69). Under normoxic condition, HIF-1α protein was found to be S-nitrosylated, which prevented its degradation (56). Similar to many other transcription factors, the GTPases were found to be S-nitrosylated. S-nitrosylation of Ras resulted in an increase of cellular Ras-GTP levels in vivo, which activated the downstream signaling pathways (34). In many cancers, Ras GTPases (H, N, or KRas) are mutated to remain in the active GTP-bound oncogenic state, while eNOS enhances the S-nitrosylation and activation of endogenous wild-type Ras, required throughout tumorigenesis (42, 45, 62). One of the important substrates of S-nitrosylation that regulate cardiac function is the ion channel RyR. It was reported that NO and RSNO-mediated oxidation of critical thiols in both skeletal (RyR1) and cardiac (RyR2) isoforms of the RyR resulted in channel activation and subsequent addition of a sulfhydryl reducing agent, resulting in channel closure (6, 14, 99, 112). Later, knocking out nNOS was reported to reduce S-nitrosylation of the RyR on the sarcoplasmic reticulum, which leads to diastolic Ca2+ leak with a negative impact on cardiac electrical stability and contractility in cardiac myocytes (29). The S-nitrosylation of RyR1 depleted the channel complex of FKBP12 (calstabin-1, for calcium channel stabilizing binding protein) and resulted in “leaky” channels. It was proposed that the sarcoplasmic reticulum Ca2+ leak via RyR1 was due to the hypernitrosylation of the channel, and calstabin1 depletion contributed to muscle weakness in muscular dystrophy (5).
NO is known to regulate apoptosis through the S-nitrosylation of many apoptosis-regulating proteins, including caspases that play an essential role in the initiation and execution of apoptosis. S-nitrosylation of the critical catalytic cysteine residue by basal NO production inhibits caspase activity and apoptosis in resting cells (22, 68). In vivo experiments were presented for the S-nitrosylation-mediated inhibition of caspase-3 (85). In addition to caspase 3, the activities of six other caspases were inhibited through S-nitrosylation in vitro, including caspase 8 and 9 (57). FLIP (FADD-like IL-1β-converting enzyme-inhibitory protein) and B-cell lymphoma-2 (Bcl-2) are the key regulatory proteins in the mitochondrial death receptor pathway. S-nitrosylation of FLIP was found to inhibit its degradation through the ubiquitin pathway (46). Similar to FLIP, S-nitrosylation of Bcl-2 was reported, which resulted in the inhibition of its ubiquitination and subsequent degradation (3, 46). Apoptosis signal-regulating kinase 1 (ASK1) is a serine/threonine protein kinase that functions as an MAPK kinase kinase in the JNK and p38MAPK signaling pathways. The results suggested that NO mediated the interferon-γ-induced inhibition of ASK1 in L929 cells through the S-nitrosylation of Cys869 (77). Furthermore, Hara et al. reported a cell death cascade whereby cellular stressors activated NO formation, leading to the S-nitrosylation of GAPDH, which triggered its binding to Siah1 (an E3 ubiquitin ligase), translocation of GAPDH/Siah complex to the nucleus, and apoptotic cell death (32).
Akt/PKB is a serine/threonine protein kinase that plays a central role in the metabolic functions of insulin, while the genetic disruption of Akt2/PKBβ leads to insulin resistance in mice (15). NO donors induced S-nitrosylation and inactivation of Akt/PKB in vitro and in intact cells (15, 114). S-nitrosylation of Cys224 was identified as a mechanism for the inactivation of Akt/PKB (114). Moreover, it was further identified that S-nitrosylation of Akt/PKB was increased in the skeletal muscle of diabetic mice compared with wild-type mice. Carvalho-Filho et al. demonstrated that insulin resistance was associated with iNOS induction in two different models of obesity, and it was mediated by the S-nitrosylation of insulin receptor β subunit, insulin receptor substrate 1, and Akt (12). Abnormal S-nitrosylation plays an important role in the process of neurodegeneration. MMPs, which are involved in remodeling extracellular matrix, have been suggested in the pathogenesis of neurodegenerative diseases and stroke. During cerebral ischemia, MMPs were found to be colocalized with nNOS, and S-nitrosylation was found to activate MMPs (30). Furthermore, the S-nitrosylation of parkin was reported in vitro, as well as in vivo in the mouse model of PD and in brains of patients with PD (17, 113). Parkin is an E3 ubiquitin ligase which is involved in the ubiquitination of proteins that are important in the survival of dopamine neurons in PD. The S-nitrosylation resulted in the inhibition of parkin's ubiquitin E3 ligase activity, which was suggested to contribute in the degenerative process of the disorder by impairing the ubiquitination of parkin substrates (17, 113).
Here, we have discussed some of the S-nitrosylated proteins with relation to signal transduction, insulin resistance, neurodegeneration, apoptosis, and cancer. It is noteworthy to mention that the Trx system represents a major antioxidant defense line in most of the eukaryotes and prokaryotes, and it is known to be involved in the redox regulation of many transcription factors. Thus, further characterization of the role of Trx will enhance our knowledge about the regulation of S-nitrosylation in different cellular processes. Researchers have documented the protective effects of Trx system on autoimmune diabetes, which suggests its possible therapeutic application (41, 67). Furthermore, Trx was found to be a physiological inhibitor of ASK1, and the anti-apoptotic role of Trx was further correlated with the NO-signaling and S-nitrosylation of protein thiols (86, 102).
Trx-Catalyzed S-Denitrosylation
Many S-nitrosoproteins are denitrosylated by GSH (84), while four enzyme systems have been characterized to catabolize GSNO: alcohol dehydrogenase class III (ADH) (48), carbonyl reductase (5, 95), PDI (94), and Trx (E. coli and human Trx) (75, 93, 100). GSNO was found to be an active substrate of rat ADH class III, and GSH sulphinamide was suggested to be the major stable product of the GSNO processing, with minor yields of GSSG and NH3 (48). Later, a GSH-dependent formaldehyde dehydrogenase (GS-FDH), purified from E. coli, Saccharomyces cerevisiae, and mouse macrophage, was found to catabolize GSNO (65). Peptide-mass analysis of the purified GSNO reductase (GSNOR) from RAW 264.7 identified the protein as mouse GS-FDH or ADH III, the homolog of the bacterial GSNOR. The enzyme was found to be specific for GSNO (65). However, it was found that S-nitroso-N-acetylpenicillamine (SNAP) and HAS-SNO were not the substrates for ADH-catalyzed denitrosylation reaction. Further studies suggested GSNOR as an important regulator in human asthma (16, 27, 47, 64, 73, 80, 81, 83, 111). Carbonyl reductase was also reported to reduce GSNO; however, the catalytic efficency (K cat) value was found to be lower than that of GS-FDH (5). S-denitrosylation activity of recombinant human PDI was previously characterized, and a mechanism of PDI-catalyzed GSNO denitrosylation, involving a nitroxyl (HNO) disulfide intermediate stabilized by imidazole residues of the subunits, has been proposed (94). It is noteworthy to mention that most PDIs contain more than one active site, and a combination of active and inactive Trx-like domains (19).
Trx is a ubiquitous protein, while ADH and PDI are expressed mainly in the liver and kidney (66) and in the lumen of the endoplasmic reticulum (8), respectively, (Human Protein Reference Database,
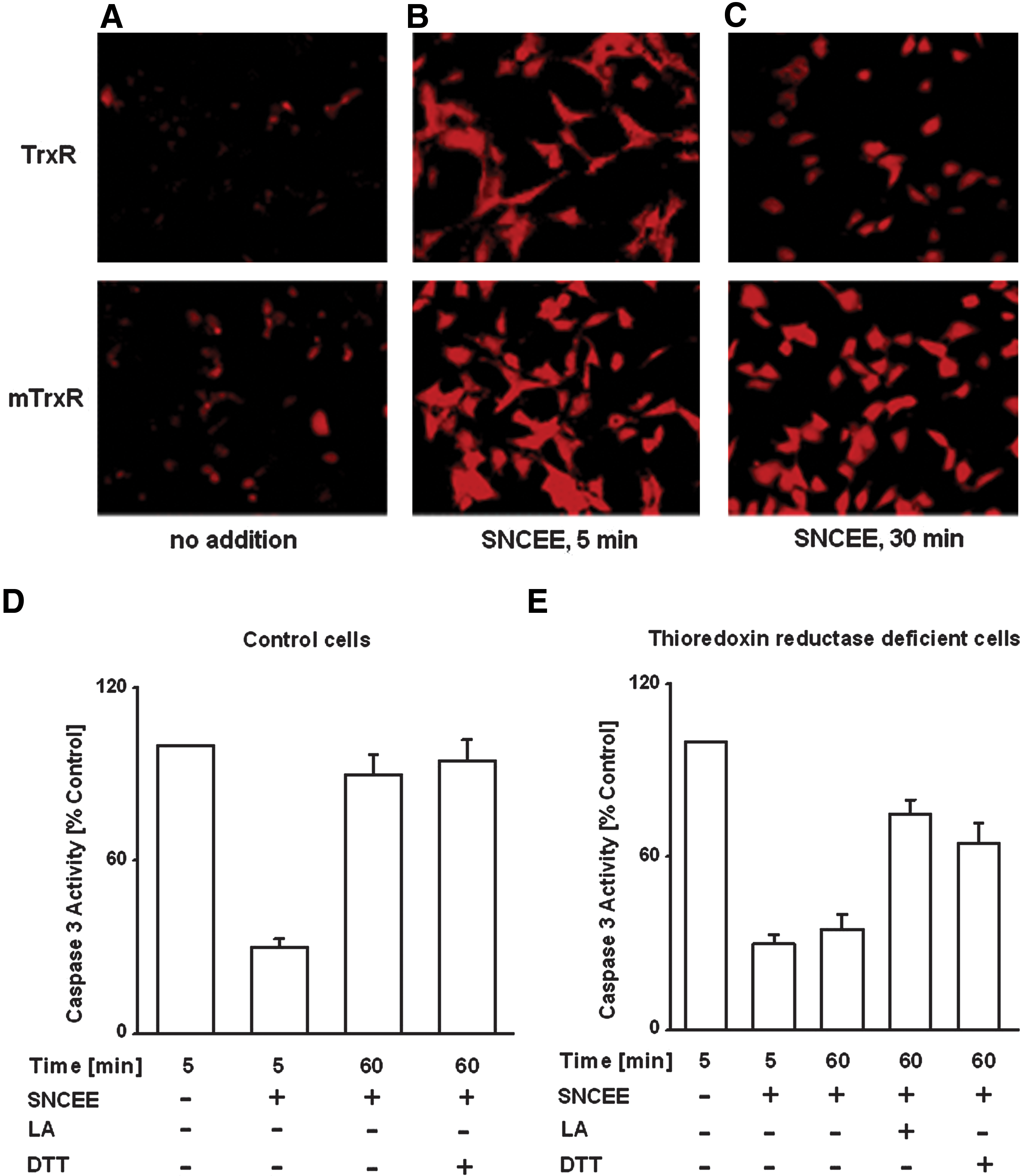
The interaction of Trx with intracellular RSNOs was assessed using Trx/TrxR wild-type HeLa cells (TrxR) and HeLa cells transfected with a cysteine-to-serine mutant expressing plasmid (mTrxR) that rendered cells with dysfunctional TrxR and impaired Trx activity (100). S-nitroso-L-cysteine ethyl ester (SNCEE) treatment resulted in a marked accumulation of RSNOs in cells (Fig. 3B, C; red stains). Clearance of intracellular RSNOs (Fig. 3; red stains) occurred at an increased rate and more completely in the TrxR versus mTrxR HeLa cells. The background intracellular level of RSNOs in wild-type TrxnR HeLa cells was approximately two fold lower than in mTrxR cells (Fig. 3C), indicating that decreased Trx activity was associated with less effective catabolism of intracellular RSNOs. Similarly, in HepG2 cells, it was observed that NO donor treatment resulted in the inhibition of caspase-3 and caspase-8 activities (Fig. 3D, E) (90, 91, 93). However, a subsequent incubation of the cells in RSNO-free medium resulted in the reconstitution of caspase activities, due to endogeneous denitrosylation of the S-nitrosocaspases (90 –93). The latter process was markedly inhibited in TrxR-deficient HepG2 cells, suggesting that the Trx/TrxR system tends to maintain intracellular caspases in a reduced state. It was further observed that lipoic acid (LA) markedly reactivated the caspases in TrxR-deficient HepG2 cells (80 –93) (Fig. 3). LA or (R)-5-(1,2-dithiolan-3-yl)pentanoic acid, a cyclic disulfide, is an essential prosthetic group in the dihydrolipoyl transacetylase component of the α-keto acid dehydrogenase complex in mitochondria. In cells, LA is reduced to dihydrolipoic acid (6,8-dimercaptooctanoic acid) by lipoamide dehydrogenase (LD) with consumption of NADH and by NADPH-dependent reductases. The LA/LD/NADH system was shown to catalyze the denitrosylation of both GSNO and S-nitrosylated caspase 3 (100). These results suggested the possible role of low-molecular-mass dithiols that mimic the activity of Trx in reactions of S-denitrosylation of proteins.
The S-nitrosylated proteins are expected to be denitrosylated by GSH, as it is the most abundant intracellular thiol. Chase experiments conducted in rat spinal cord slices were preincubated with GSNO (84), and the results revealed the intracellular GSH-mediated denitrosylation of protein RSNOs in intact cells by rapid trans-S-nitrosylation reactions. GSH-dependent S-denitrosylation resulted in an ∼75% decrease in high-molecular-mass RSNOs from Jurkat cells. It is noteworthy that S-nitrosocaspase 3, which was relatively stable in the presence of 5–10 mM GSH (117), was readily denitrosylated by the Trx/TrxR system (90, 93). Recently, using Jurkat cells, Stamler and coworkers identified 46 proteins as the substrates of the Trx 1-catalyzed denitrosylation reaction (9). The substrates are involved in a wide range of cellular functions, including cytoskeletal organization, cellular metabolism, signal transduction, and redox homeostasis. The results pointed to Trx1 as a major protein denitrosylase in mammalian cells. In another study, Trx2 has been found to mediate the denitrosylation of mitochondria-associated S-nitrosocaspase (7). However, further studies are needed to better understand the Trx-dependent catabolism of RSNOs in cells.
Role of S-Nitrosothioredoxin
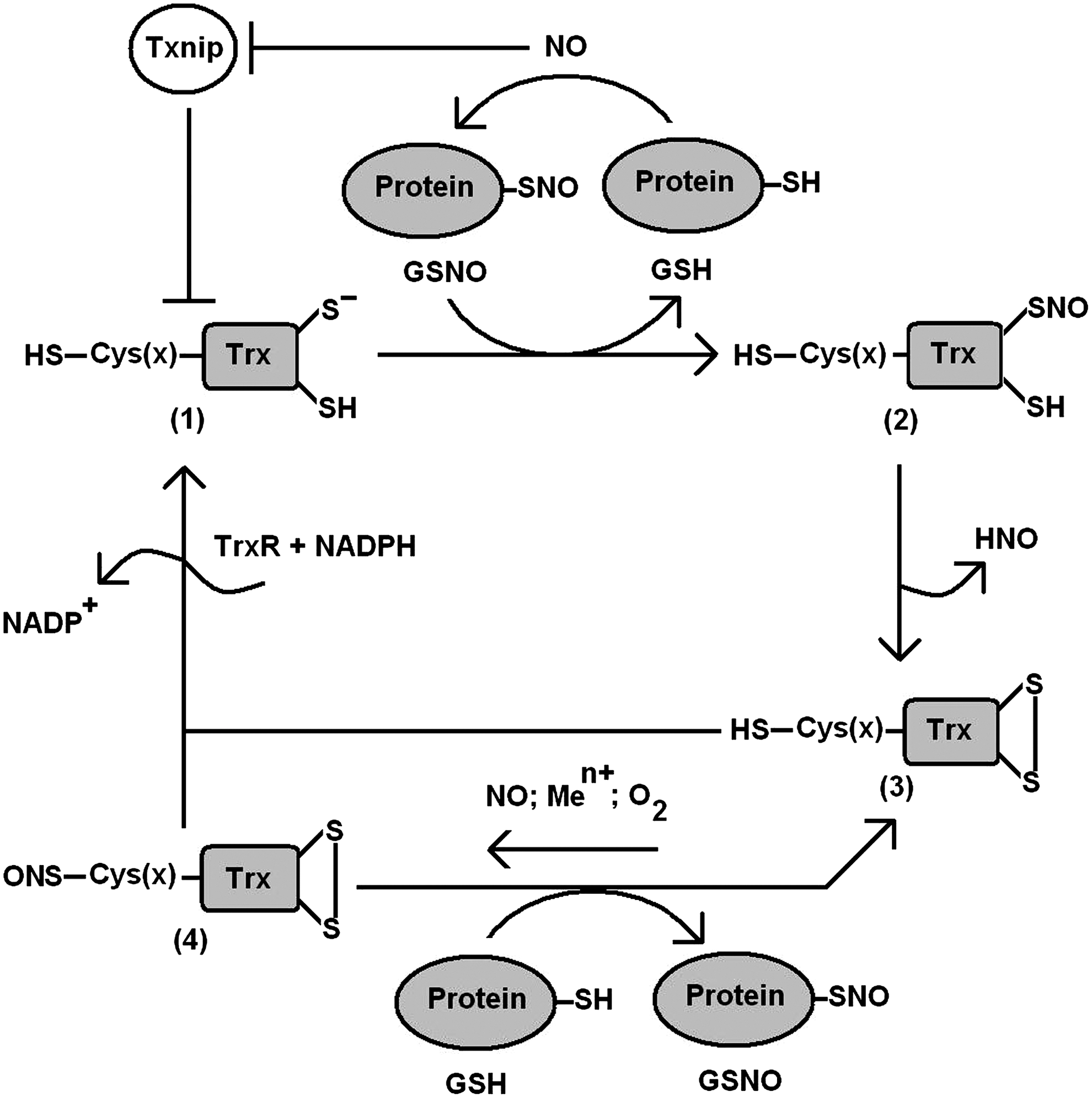
Unlike Trxs from lower species, mammalian Trx1 contains additional cysteines at 62, 69, and 73 positions (107). The nitrosylation of either Cys69 or Cys73 leads to the formation of S-nitrosothioredoxin (ONS-Trx-S2), which has been suggested to impede apoptosis via trans-S-nitrosylation of caspase 3 (33, 72, 110) (Fig. 2). However, experimental evidence was provided for the denitrosylation of ONS-Trx-S2 in the presence of either reduced Trx [Trx(SH)2] or GSH (93). The results suggest that both S-nitrosothioredoxin and S-nitrosocaspase 3, if formed in cells, will undergo GSH and Trx/TrxR system-dependent denitrosylation (Fig. 2). Recently, an optimized mass spectrometric method has been used to demonstrate the GSNO-mediated nitrosylation at Cys73 of Trx after the formation of a Cys32/Cys35 disulfide bond, on which Trx1 disulfide reductase and denitrosylase activities were attenuated (110). Moreover, the overexpression of Trx1C32S/C35S mutant in HeLa cells promoted the S-nitrosylation of specific target proteins, and 47 novel Trx1 trans-S-nitrosylation target proteins were identified. The results demonstrated that Trx1 can catalyze either trans-S-nitrosylation or S-denitrosylation reactions depending on the redox state of the cell. The subcellular localization and stability of the S-nitrosothioredoxin should be tightly regulated by different reductants (reduced Trx1 or GSH) (93, 110). Thus, redox states of the active site dithiols of Trx are critical regulators that determine the S-nitrosylation status of the additional cysteines (33, 93, 110). On inhibition of disulfide reduction or S-denitrosylation activity, ONS-Trx-S2 can catalyze trans-S-nitrosylation of target proteins (Fig. 2).
Trx-Interacting Protein
Trx-interacting protein (Txnip; also called “vitamin-D3-upregulated protein-1” and “thioredoxin-binding protein 2”), a 50 kDa ubiquitous protein, binds directly to the active site Cys of Trx and inhibits the reducing activity of Trx (78, 106) (Fig. 2). Studies suggest major physiological roles for Txnip in glucose metabolism and cell differentiation (18, 106). The expression of Txnip was induced by a variety of stresses, including UV light, γ-rays, heat shock, and H2O2, as well as glucose treatments. Furthermore, overexpression of Txnip renders cells more susceptible to oxidative stress and promotes apoptosis (13, 116). Schulze et al. reported that the expression of the gene encoding Txnip was suppressed by NO in rat pulmonary artery smooth muscle cells (88). The net effect of these transcriptional effects was suggested to increase the Trx activity. Another study implicated Txnip as a regulator of S-nitrosylation by interfering with the Trx-catalyzed denitrosylation (Fig. 2) (24). Endogenously synthesized NO was suggested to repress Txnip to facilitate Trx-mediated denitrosylation. In the study, HEK293 cells transfected with iNOS exhibited increased intracellular levels of S-nitrosylated proteins, while the cotransfection of Txnip with iNOS further resulted in an increase in protein S-nitrosylation by ∼40%. This increase corresponds to the groups of S-nitrosylated proteins that are the substrates for Trx1-catalyzeded denitrosylation. Further characterization of the Trx-Txnip system will enhance our knowledge of the development of drugs and therapies.
Conclusion
Delivery of Trx1 as a new therapeutic agent has been documented (74) and recently, it has been shown that the impairment of angiogenesis and myocardial dysfunction can be regulated by Trx1 gene therapy in diabetic rats (87). On the other hand, the involvement of Trx system in the regulation of cellular RSNOs and RSNO signaling has been studied by several researchers (74). However, the dynamics of protein S-nitrosylation and cellular homeostasis of RSNOs remain poorly understood. Further investigations on the role of the Trx system in relation to biologically relevant RSNOs and their functions will facilitate the development of drugs and therapies.