
Abstract
Mitochondrially generated reactive oxygen species are involved in a myriad of signaling and damaging pathways in different tissues. In addition, mitochondria are an important target of reactive oxygen and nitrogen species. Here, we discuss basic mechanisms of mitochondrial oxidant generation and removal and the main factors affecting mitochondrial redox balance. We also discuss the interaction between mitochondrial reactive oxygen and nitrogen species, and the involvement of these oxidants in mitochondrial diseases, cancer, neurological, and cardiovascular disorders. Antioxid. Redox Signal. 18, 2029–2074.
I. Introduction
A. The respiratory chain: energy and free radicals
Mitochondrial energy metabolism is also recognized as the main source of cellular reactive oxygen species (ROS) in most eukaryotic cells (50, 51, 515). However, mitochondria also have the highest antioxidant capacity, making them a player not only as a superoxide anion (O2 •;−) source but also as a cellular redox sink (138, 281, 413). The initial concept that mitochondrial ROS were essentially an undesirable metabolic byproduct generated by the mitochondrial respiratory chain has changed. Based on a large body of experimental evidence, it is now recognized that, under physiological conditions, mitochondrial ROS generation is a continuous and tightly adjusted process required for the regulation of many cellular processes (138, 222, 506). As an example, H2O2, which is a reasonably stable and diffusible molecule (78), is a cellular signal that regulates multiple vital processes within and outside mitochondria such as cell cycle, stress response, energy metabolism, redox balance, and oncogenic transformation (222). Under normal conditions, cells maintain their redox balance through the generation and elimination of ROS using different antioxidant systems, as will be discussed next (222, 341, 381).
Our current view about cellular ROS signaling has been greatly expanded in the last decade. Hamannaka and Chandel (222) described how multiple inputs such as hypoxia, PI3K, TNFα, and oncogenes regulate the generation of mitochondrial ROS and how these ROS activate multiple outputs, including phosphatases and redox-regulated transcription factors, such as NF-κB, that, in turn, regulate the expression of pro-inflammatory genes and kinases. In hypoxia, for example, the stimulation of mitochondrial H2O2 generation can promote cellular protection via adaptive transcriptional programs regulated by hypoxia inducible transcription factors (HIFs); this adaptive program consists of a changed expression of genes regulating erythropoiesis, glycolysis, angiogenesis, cell cycle, and survival, as well as lowered energy turnover (93, 94, 122, 158, 547, 548). A physiological example of redox-related processes is that increased mitochondrial ROS in response to hypoxia are a signal for pulmonary vasoconstriction, which, in turn, improves ventilation-perfusion matching (548).
It has also been proposed (222) that different classes of signaling actions may be regulated by different levels of ROS: (i) low rates of mitochondrial O2 •;− generation are required for cellular processes such as proliferation and differentiation; (ii) moderate cellular stress induces O2 •;− generation at levels that activate adaptive programs, including the transcriptional up-regulation of antioxidant genes; (iii) at higher levels, ROS signal for the initiation of senescence and cell death.
The process of O2 •;− generation by the respiratory chain seems to be highly regulated, and ROS can function both adversely and beneficially. However, there is only little evidence regarding differential signaling events behind the generation of either beneficial or detrimental levels of mitochondrial ROS. In addition to the physiological processes controlled by mitochondrial ROS, a large body of evidence indicates that mitochondrial oxidative imbalance is responsible for the development and progression of a series of abnormalities such as cancer, diabetes, inflammatory diseases, hypertension, neurodegenerative and ischemia-related diseases, as well as aging (138, 222, 341, 381). In this regard, recent evidence indicates the existence of a cross-talk between mitochondria and NADPH oxidases in which mitochondrial ROS activate O2 •;− and H2O2 production by NADPH oxidases that, in turn, stimulate mitochondrial ROS formation, or alter NAPDH oxidase responses to angiotensin (555). Under some circunstances, this may generate a feed-forward vicious cycle of ROS generation, which may contribute to the development of cardiovascular diseases (138, 555). Indeed, scavenging mitochondrial O2 •;− with mitochondria-targeted antioxidants interrupts this vicious cycle and down-regulates NADPH oxidase activity (140). An example of these interactions is the activation of the PKC-dependent phagocytic NADPH oxidase by increased mitochondrial O2 •;− levels mediated by matrix Ca2+ (139).
In this comprehensive review, we will approach mechanisms of mitochondrial free radicals generation and discuss their fate. The role of mitochondrial Ca2+ influx and of reactive oxygen and nitrogen species as signaling agents regulating cellular processess in health and disease will also be covered. Then, we will focus on selected degenerative diseases in which mitochondrial redox homeostasis is compromised and/or involved in their pathogenesis.
B. Superoxide production by the respiratory chain
Superoxide can be generated in at least five sites within the respiratory chain (52): the ubiquinone (UQ)-binding sites in complex I and III, the flavin prosthetic group in complex I, the electron transferring flavoprotein (ETF), UQ oxidoreductase, and glycerol 3-phosphate dehydrogenase. Three of these sites are relatively well characterized with regard to the mechanism of O2 •;− generation (UQo in Complex III and UQ and flavin in Complex I). For the others, controversies in the literature remain. The topology and relative capacity to produce O2 •;− of each site is important, and determines whether the radical is released toward the matrix side or in the intermembrane space; as well as their contribution toward total mitochondrial O2 •;− production (78).
Complex I is believed to produce O2 •;− at two sites: the flavin mononucleotide (FMN) binding site (323) and the UQ-binding site (516). When respiring on NADH-linked substrates feeding complex I (forward electron transport), mitochondria produce O2 •;− at a relatively low rate; rotenone, a UQ-binding site inhibitor, known to block the electron flow at complex I, fully reduce the upstream redox centers, and increase O2 •;− production (230). This suggests that complex I produces O2 •;− at a site that is proximal to rotenone block during forward electron flow from NADH. It has been proposed that fully reduced flavin in the nucleotide-free binding site of complex I could react with O2 to produce O2 •;−. In addition, reduced FMN is considered an important electron donor to O2 to produce O2 •;− at complex I (187). These complex I sites of O2 •;− generation during forward electron transfer may comprise a mechanism explaining the link between the mitochondrial NAD pool redox state and O2 •;− production. With regard to reverse electron transfer back to NADH, we emphasize that succinate, glycerol 3-phosphate, and acyl-CoAs are physiological substrates which can reduce the UQ pool and generate protonmotive force through complexes III and IV. In isolated mitochondria under such conditions, electrons can be driven uphill from reduced UQ to complex I, a process known as reverse electron transfer. If rotenone is added to mitochondria undergoing reverse electron transfer, a lower production of O2 •;− is observed at respiratory chain steps that are proximal to rotenone block (the QH2-binding site) (295). Despite the biochemical characterization of these two sites of O2 •;− production within complex I (52), there are controversies about the relative contribution of these two sites to overall O2 •;− production and even about the existence of two separate sites. A recent study shows that O2 •;− is produced in complex I only by fully reduced flavin during both forward and reverse electron transfer in submitochondrial particles (i.e., inside-out preparation of mitochondria) (423). Moreover, the last N2 FeS cluster of complex I, a structure working distal to FMN during forward electron transfer, has also been proposed as an electron donor to O2 either directly or indirectly via semiquinone (311).
The production of O2 •;− at the level of complex III is related to its peculiar mechanism of electron transfer: the Q-cycle mechanism (119). Electrons delivered to the respiratory chain entry sites flow along the lipid-soluble UQ. Complex III catalyzes the transfer of these electrons from reduced ubiquinone (UQH2) to water-soluble cytochrome c on the outer surface of the inner membrane, as complex III pumps H+ out of the inner mitochondrial membrane. In the “classical” Q cycle, UQH2 delivers sequentially the first electron at center o (outer positive side) to the Rieske iron-sulphur protein (then to cytochrome c1 and c), along with the release of 2 H+ outside the inner membrane and the formation of an unstable semi-ubiquinone anion (UQ•−). This radical is quickly oxidized to UQ by cytochrome bL (cytosolic side of the membrane). The second electron is then delivered to cytochrome bH (matrix side); cytochrome bH is then reoxidized by UQ at center i (inner negative side), forming another UQ•−. The cycle is completed by the oxidation of a second UQH2 providing one electron to cytochrome c and one electron to UQ•− at center i. Since the electron transfer from cytochrome bL to cytochrome bH is slowed down by the electrical gradient acrros inner mitochondrial membrane, the lifetime of UQ•− is prolonged at site o, allowing the reduction of O2 by UQ•− to form O2 •;− (256). Thus, according to this classical Q-cycle model, the main one-electron donor to O2 is located in center o (external side of the membrane) and could be the UQ•− (353). Indeed, complex III produces O2 •;− at a rate that depends on the half-life of UQ•−. Inhibitors that increase the half-life of UQ•− at center o (e.g., antimycin) or high protonmotive force across inner mitochondrial membrane result in higher O2 •;− production rates. Indeed, inhibitors that block UQH2 (e.g., myxothiazol) access to center o, thus decreasing electron delivery to complex III, lower the rate of O2 •;− production. Opposingly, it has been shown that UQ•− is formed very transiently at center o and never accumulates to a significant level in the functionnal complex III (76), as the bifurcated UQH2 oxidation at center o occurs in a quasiconcerted reaction (119). There are also other conflicting data regarding the mechanisms of O2 •;− production by complex III: (i) It was recently shown that O2 •;− production at center o of the membrane-bound or purified complex III was stimulated by the presence of oxidized UQ, indicating that one electron is transferred to O2 in a reverse reaction from reduced cyt bL via UQ acting as redox mediator (148); (ii) a heme b knock-out mutant complex III shows little electron transfer activity but produces O2 •;− at a higher rate, indicating that O2 •;− can also be formed by a route other than the reaction involving the heme bL (560). Thus, the precise mechanism of O2 •;− generation by complex III still remains elusive. Despite complex III seeming to release O2 •;− in an equal amount on both sides of the membrane (363), due to the strong antioxidant capacity of mitochondrial matrix, the net amount of ROS could be greater in the inter-membrane space side than in the matrix side.
Superoxide generation by glycerol 3-phosphate dehydrogenase, an entry site for respiratory chain electrons, occurs toward both sides of the membrane (147, 355). The flavin site appears to be in the intermembrane space, while the UQ-binding site is in the membrane and is proposed to be the main site of O2 •;− production. Its capacity seems to be lower than the capacities of UQ sites at complexes I and III. The ETF:UQ oxido-reductase could produce O2 •;− into the matrix, but its mechanism is still poorly characterized (486).
In addition to the respiratory chain, three other mitochondrial O2 •;− production sites have also been uncovered: the enzymes 2-oxoglutarate (α-ketoglutarate) and pyruvate dehydrogenases inside the matrix and monoaminoxidase in the outer mitochondrial membrane; this later source will be discussed in the subsection on redox imbalance in heart failure (section VIII-C). Although the mechanisms are not fully understood, O2 •;− production by 2-oxoglutarate and pyruvate dehydrogenases is enhanced when the physiological electron acceptor NAD+ is unavailable (11, 489, 500, 509). For example, lower NAD+ availability (i.e., high NADH:NAD+ ratio) is observed in the liver after alcohol consumption due to the metabolism of alcohol and acetaldehyde that reduces NAD+ (562). On the other hand, caloric restriction promotes increased NAD+ availability and lowers the generation of H2O2 through 2-oxoglutarate dehydrogenase in yeast (500).
Although it is currently known that mitochondria can produce O2 •;− at several sites, we do not precisely know at which relative rates it occurs under the supply of various substrates and different energy demands, even in the simplest system that is, isolated mitochondria (501). It is a methodological challenge to measure mitochondrial O2 •;− in different types of mitochondria, and even more so in vivo. Methods available include the determination of specific hydroethidine derivatives by HPLC (140, 568). However, it should be noted that the accumulation of hydroethidine and hydroethidine conjugated to a triphenylphosphonium (Mito-HE or Mito-SOX) in mitochondria is affected by mitochondrial membrane potentials (67, 78), which are in themselves strong regulators of O2 •;− production. A circularly permuted yellow fluorescent protein used as a mitochondrial O2 •;− biosensor (545) has been recently criticized as being sensitive to pH (464). As a result, many studies that aim at determining rates of mitochondrial O2 •;− production do so by indirect measurements such as the release of O2 •;− derived H2O2 from isolated mitochondria (78). Overall, it seems that a large amount of mitochondrial O2 •;− is produced when NADH/NAD+ is high (e.g., low ATP demand or lack of O2) and/or when protonmotive force is high (low ATP demand) (365).
A puzzling observation is the increase of mitochondrial ROS production during hypoxia in cultured cells (213) and in Langendorff perfused hearts during global ischemia, especially in its late phase, where O2 tension is expected to be the lowest (264, 434). Conversely, isolated mitochondrial ROS production is quite independent of O2 concentrations varying between 5 and 250 μM, bearing in mind that O2 concentrations at 5 μM already limit mitochondrial respiration by ∼40% under the tested conditions (233). This suggests an indirect effect of hypoxia on ROS production, requiring additional factors in hypoxic cells or tissues. Indeed, such hypoxic bursts of ROS production seem to be involved in hypoxic adaptive signaling mediated by the transcription factor HIF1A (233, 421, 544).
An often-repeated quote is that O2 •;− formation accounts for 1%–2% of mitochondrial oxygen consumption (11). This information derives from an extrapolation of early measurements performed in isolated mitochondria under nonphysiological conditions (as clearly stated by the authors): saturating substrate and O2 concentrations in the presence of antimycin [a complex III inhibitor that greatly stimulates mitochondrial O2 •;− formation (92)] and is, therefore, not physiologically correct. Knowing how much O2 •;− is produced by mitochondria in vivo is necessary in order to evaluate its significance in oxidative damage and redox signaling. However, extrapolations of ROS production by isolated mitochondria to in vivo conditions are questionable because of several factors, including substrate nature and concentration, local O2 concentrations, and respiratory states, which may affect mitochondrial O2 •;− formation and fate. Thus, even more realistic extrapolations (0.15% of the oxygen consumption) are not reliable (365). Furthermore, intracellular measurements of mitochondrial ROS production are prone to artifacts and not quantitative (78).
C. Fate of mitochondrial O2 •;−
Irrespective of the rate of O2 •;− production in vivo, its formation implies the existence of metabolizing pathways. Superoxide, the primary ROS produced by mitochondria, gives rise to many ROS and reactive nitrogen species (RNS) through many distinct reactions (256, 281). Superoxide is converted to H2O2 by the metal-dependant enzyme superoxide dismutase (Mn-SOD in the matrix and Cu/Zn-SOD in the intermembrane space and in the cytosol, rescepctivelly). However, a part of O2 •;− is under the form of its conjugated acid HO2 •, a very reactive species, and some O2 •;− can react with nitric oxide (NO•) to form peroxynitrite (ONOO−). H2O2 is poorly reactive, can permeate membranes, and is removed by catalase in the cytosol and by glutathione peroxidase (GPx) and peroxiredoxins (Prx), at the expense of glutathione (GSH) and thioredoxin-2 (Trx) in the mitochondrial matrix. The oxidized form of glutathione and Trx are then reduced back by glutathione reductase and thioredoxin reductase (TrxR), respectively, at the expense of NADPH as reducing power. NADPH is, therefore, of central importance in the removal of mitochondrial H2O2. NADPH is, in turn, regenerated by the protonmotive force-dependent NAD(P)H transhydrogenase (NNT) and by isocitrate dehydrogenase (IDH2). Given these properties of mitochondrial ROS metabolism, conditions that affect protonmotive force or reducing power supply can alter both O2 •;− production and H2O2 elimination.
The most reactive oxygen or nitrogen species can damage proteins (71), DNA (396), and lipids (486). Lipid peroxidation itself is a source of new radicals, as it forms carbon-centered radicals in the unsaturated fatty acid chains of phospholipids. Reactions of carbon-centered radicals with O2 generate peroxyl radicals (ROO•), which react with the side chain of polyunsaturated fatty acids, yielding phospholipid hydroperoxides (PL-ROOH) and new carbon-centered radicals. PL-ROOH are cleaved by phospholipase A2 to free fatty acid hydroperoxides (FAOOH). In the presence of Fe2+, alkoxyradicals (RO•) are formed and react with polyunsaturated fatty acids in their vicinity; if not, they generate unsaturated aldehydes. Interestingly, 4-hydroxy-2-nonenal (4-HNE) is formed by spontaneous cleavage of PL-ROOH (164).
To date, there is little evidence for free-radical chain reactions occurring under physiological conditions, especially in mitochondria, as an abundance of scavengers abrogates propagation reactions. However, under pathological conditions, the disruption of intracellular redox signaling facilitates the auto-oxidative deterioration of polyunsaturated fatty acids, allowing the formation of secondary end products of lipid peroxidation (440). With the advent of mass spectometry-based lipidomics, new perspectives in the understanding of lipid peroxidation processes in pathophysiology has emerged.
A characterization of the lipid composition of rat liver subcellular membranes found that the inner mitochondrial membrane has a high content of unsaturated fatty acid acyl chains (440). This unsaturated feature of the mitochondrial inner membrane renders it susceptible to attacks by free radicals under pathological conditions, resulting in 4-HNE generation. 4-HNE is a highly reactive product of peroxidation of arachadonic, linoleic, and linolenic acid acyl chains (164). Considered a strong electrophile and the most cytotoxic aldehyde produced by lipid peroxidation, 4-HNE has the ability to irreversibly modify cellular targets such as proteins, DNA, and phospholipids (440).
Another process that might contribute to mitochondrial 4-HNE generation is the oxidation of mitochondrial cardiolipin. Mitochondria are rich in cardiolipin, a phospholid located in the inner mitochondrial membrane that contains three glycerol backbones and four acyl chains. Due to its interaction with electron transport chain complexes and high polyunsaturated fatty acid content, cardiolipin is considered a likely target for oxidants generated in the mitochondrion. In fact, stimulating lipid peroxidation in the rat heart and brain reduces both cardiolipin content and cytochrome c oxidase activity (406, 469). In addition, cardiolipin has been shown to be oxidized by cytochrome c in the presence of H2O2, resulting in the release of cytochrome c and the initiation of mitochondria-mediated apoptosis (260).
RNS can also contribute to oxidative imbalance and/or damage proteins. NO• can reversibly inhibit cytochrome c oxidase, as discussed later in section IV-B of this review. This inhibition may increase the reduced state of electron carriers in the respiratory chain and, consequently, O2 •;− production. Nitrogen dioxide radical (NO2 •), a product of NO•oxidation, can oxidize or nitrate a wide range of biomolecules. Peroxynitrite can oxidize thiol groups, DNA bases, and tyrosine residues. In mitochondria, excessive ONOO− levels can impair oxidative phosphorylation by inhibiting Complex I, Complex IV and ATP synthase, and MnSOD activity and calcium homeostasis (60).
To deal with this cascade of potentially damaging ROS and RNS, cells employ two strategies: pathways able to metabolize or scavenge these harmful species, and/or systems that regulate the generation of their common source, O2 •;−.
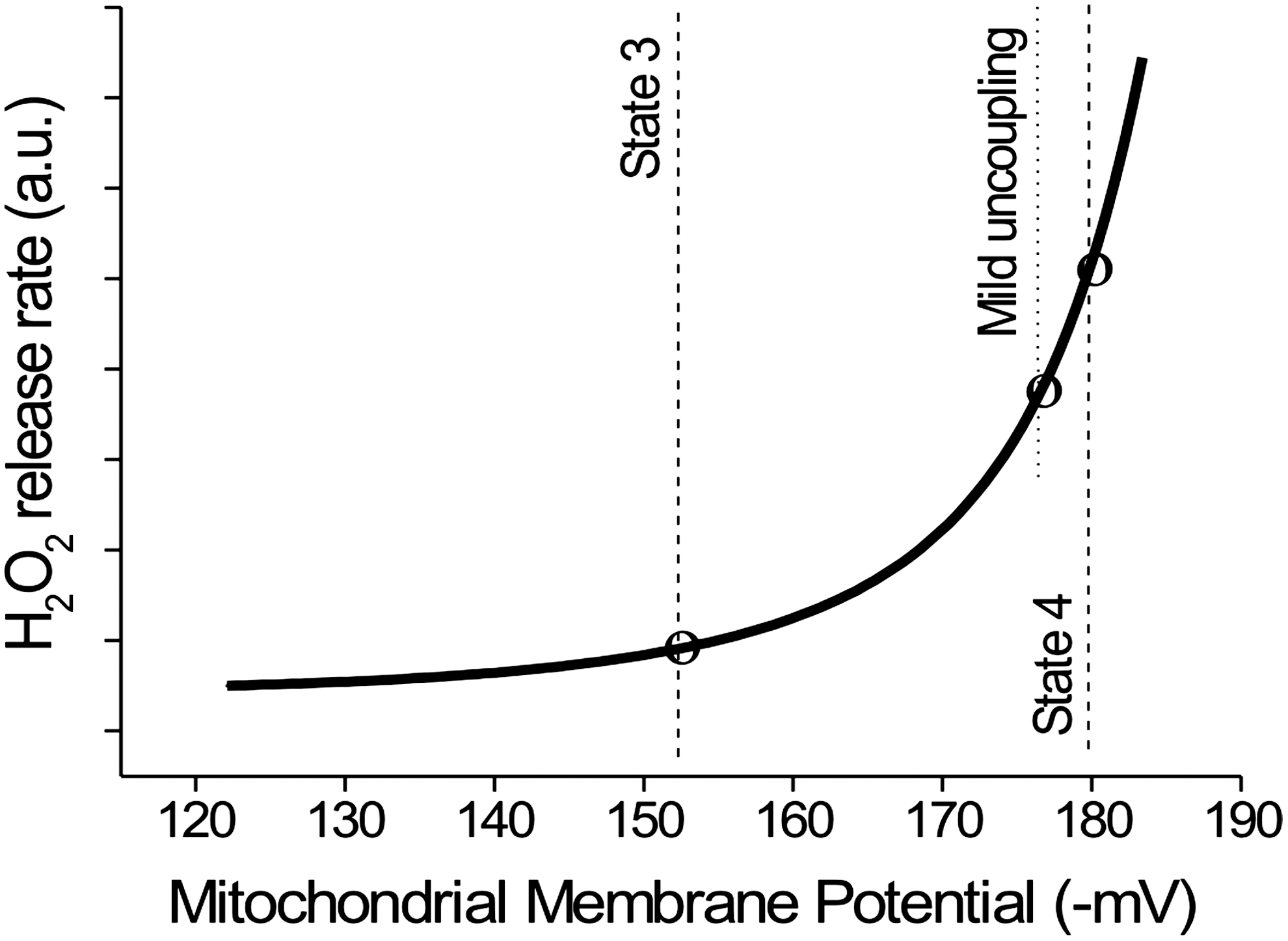
Clearly, a tight control of O2 •;− generation is of paramount importance in order to maintain the ROS production within a range that is compatible with redox signaling and homeostasis. High protomotive force is mechanistically linked to a high level of O2 •;− production; thus, every process that decreases protonmotive force through its dissipation is likely to reduce O2 •;− generation. For instance, less ROS are produced during phosphorylating respiration (state 3) compared with resting respiration (state 4), paralleling the changes in protonmotive force. Since a nonlinear relationship (Fig. 1) links ROS production and the protonmotive force (a small increase or decrease of protonmotive force induces a large increase or decrease in ROS) (276, 488), a large prevention in ROS production is achievable without a dramatic decrease in the efficiency of oxidative phosphorylation (i.e., oxidation-phosphorylation coupling). This “mild uncoupling” (uncoupling is a process that decreases the efficiency of ADP phosphorylation through partial dissipation of protonmotive force) can be achieved by several processes. A “futile” H+ cycle across inner mitochondrial membrane associated with cation homeostasis [e.g., K+ channel plus K+/H+ exchanger (165)] and the H+ re-entry mediated by specialized uncoupling proteins (UCPs) can, theoretically, promote this mild uncoupling.
II. UCP Activity and Regulation
UCPs are members of the Mitochondrial Anion Carrier Family, which transports a large variety of anion metabolites across the inner mitochondrial membrane (154). There are around 40 mitochondrial anion carriers, including UCPs, that are widespread among eukaryotes (479). Despite incompletely understood biochemical mechasnims or relevance for UCP1 analogues, the general activation of UCPs results in the re-entry of H+ from the intermembrane space back to the mitochondrial matrix. In doing so, these proteins uncouple mitochondrial oxidative metabolism from ADP phosphorylation by mitochondrial ATP synthase, thus lowering the ADP:O ratio (i.e., phosphorylation efficiency) (191). Although free fatty acid anions (FFAs) are considered UCP activators of/and purine nucleotides are recognized as inhibitors of UCPs (480), the exact mechanisms by which these proteins mediate the control of the H+ leak across the inner mitochondrial membrane are still controversial (480). Since the inhibition by purine nucleotide is considered diagnostic of UCP activity, UCP1 analogues described after the discovery of the plant UCPs in 1995 (530) were initially considered a distinct class of mitochondrial carriers due to the fact that they lacked purine nucleotides sensitivity (529). However, studies measuring the ADP:O ratio in phosphorylating mitochondria (254) revealed FFA-induced uncoupling that was sensitive to purine nucleotides (i.e., a putative UCP activity) in mitochondria expressing UCP1 analogues (252 –254, 373, 479). Interestingly, all UCPs in mitochondria from protists, fungi, plants, and mammals show this susceptibility to nucleotide inhibition of FFA-induced uncoupling during phosphorylating respiration (252 –254, 373, 479).
A. Redox regulation of UCPs
ROS can either activate or signal for increases in UCP expression. The direct activation of UCPs by ROS or lipid peroxidation products such as 4-HNE and free polyunsaturated FAOOH may comprise an immediate response to oxidative imbalance (151, 152, 248, 432). This “short-term or fast response” downregulates O2 •;− production without significantly decreasing oxidative phosphorylation efficiency (Figs. 1 and 2). The second type of activation occurs through UCP protein expression through ROS signaling (14, 375). This adaptation increases the uncoupling capacity of UCPs and represents a long-term response (Fig. 2) aimed at preventing increased O2 •;− production in chronic pathological conditions. The concept that UCPs are activated by ROS is in line with the role of UCPs in the regulation of O2 •;− production, but the mechanisms of UCP activation by oxidants remain under debate.
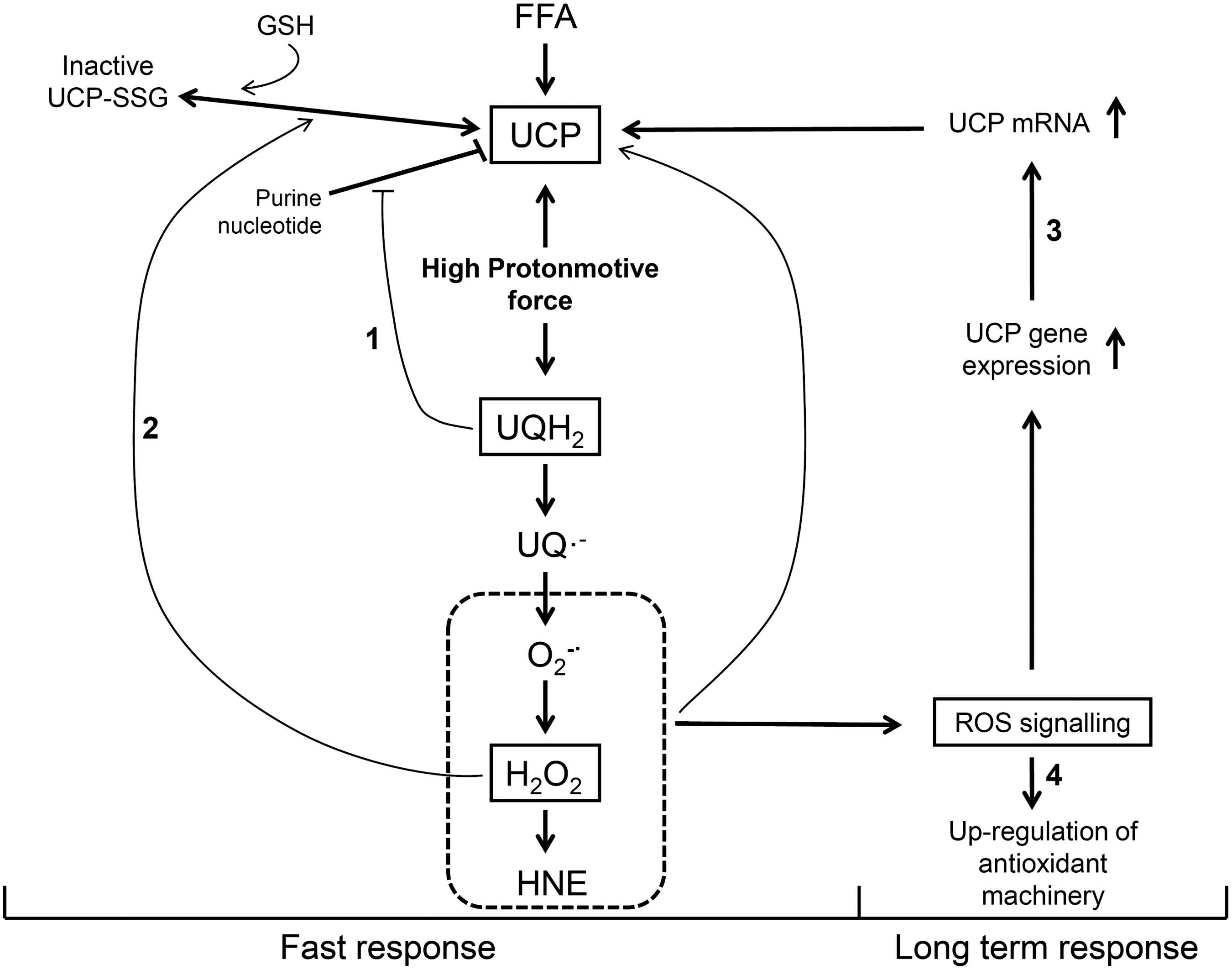
GSH is an important redox sensor (377) that may regulate the activity of UCP2 and UCP3 through reversible S-glutathionylation of thiol residues of these UCPs (334, 335). Glutathionylation, a process that is favored by high matrix GSH concentrations and basic pH (242), decreases UCP2/3-mediated H+ leak, thus increasing protonmotive force and O2 •;− formation (334). Oxidative imbalance decreases GSH availability and promotes deglutathionylation, which activates UCP2/3-mediated H+ leak (334, 335). Of note, UCP1 seems not to be succeptible to this type of regulation (335). These findings suggest that oxidative imbalance, H2O2 detoxifying systems, and UCP activation are mechanistically connected.
Evidence has also been provided that UCP1 analogues operate as anion carriers promoting the electrophoretic extrusion of fatty-acid hydroperoxide anions from the matrix, thus protecting the organelle from these harmiful molecules (205). This concept has been strongly supported by experiments with isolated skeletal muscle mitochondria from UCP3-null mice and their wild-type littermates (325) and suggests that UCP3 is not only involved in mitochondrial uncoupling but also involved in the protection of mitochondria against lipid hydroperoxides.
B. UCPs: purine nucleotide inhibition and UQ redox state
The affinity of UCPs reconstituted into proteoliposomes to purine nucleotides is in the micromolar range (48, 152, 249, 255, 270, 561), suggesting that UCPs are either fully inhibited in vivo (milimolar range of purine nucleotides) or that this inhibition is subjected to some kind of modulation. In this regard, it has been demonstrated in phosphorylating mitochondria that the inhibition of UCP3 by GTP decreases progressively as the reduced state UQ within respiratory chain increases (254). This was achieved by modulating the UQ redox state with malonate during phosphorylating respiration (state 3), while keeping the protonmotive force unchanged (254). According to these data, UCP3 would be maximally inhibited by GTP during high metabolic demand (i.e., state 3 respiration), when UQ is in a more oxidized state. On the other hand, resting-state mitochondria (i.e., highly reduced UQ) do not display GTP-sensitive FFA-induced uncoupling. Based on these results, muscle UCP3 inhibition by GTP was proposed to be dependent on UQ redox state, a metabolic sensor that modulates the inhibition by puridine nucleotides of FFA-induced UCP activity (480). This hypothesis has been supported by independent studies with many different UCPs (145, 478, 496). In vivo GTP concentrations would, therefore, not allow UCPs to operate when energy demand is high.
Overall, purine nucleotides, UQ redox state, and oxidative imbalance are modulators of UCPs activity (150), but many questions remain open with regard to their molecular mechanisms (334).
C. Connecting UQ redox state, O2 •;− production, UCP activity, and energy conservation
The data described earlier provides evidence that the main physiological roles of UCP1 analogues would be the regulation of protonmotive force in order to maintain an optimal compromise between oxidative phosphorylation efficiency and O2 •;− production (480). Indeed, simple in vitro experiments have clearly demonstrated that activation of UCP3 by FFA is protective against O2 •;− overproduction during anoxia/reoxygenation cycles (374). Anoxia leads to a high state of reduction of UQ, and reoxygenation leads to its fast oxidation accompanied by a burst in O2 •;− production. UCP3 activation by FFA atenuated O2 •;− overproduction in mitochondrial anoxia/reoxygenation, which was accompanied by a lower degree of mitochondrial permeability transition (MPT) pore opening and preservation of phosphorylation efficiency. MPT is a redox-sensitive process that can trigger cell death (many aspects of MPT will be approached in the section below). Of note, adenine nucleotide translocator and ATP-sensitive K+ channel also show similar protective effects against anoxia/reoxygenation (80, 165). Figure 3 sumarizes these findings linking UCP activity, O2 •;− generation, and cytoprotection during the process of anoxia/reoxygenation. The role of UCPs in cell protection is a concept that has progressively gained acceptance (55).

With regard to different mechanisms by which UCPs may be involved in ROS homeostasis and cytoprotection, Figure 2 shows a model integrating the roles of ROS and UCP activity as a defense mechanism. Four successive lines of defense are described and connected: Two lines deal with fast regulations against acute ROS production, and two lines are concerned with long-term regulation against chronic ROS production. The first line of defense is the release of purine nucleotide inhibition of UCP activity when UQ reduction levels are high (i.e., high protonmotive force, low ATP demand, and high reducing power), and the second line of defense is the ROS-induced deglutathionylation that promotes UCP activation. The third line is ROS-induced up-regulation of the expression of UCP proteins, and the fourth line is the up-regulation of the expression of enzymes implicated in ROS elimination.
III. Ca2+ Signaling, Mitochondrial ROS Generation, and the Permeability Transition
There is compelling evidence that mitochondrial Ca2+ influx signals for the control of both oxidative phosphorylation (200) and ROS generation (281). In addition, dysregulation in cell Ca2+ homeostasis leading to sustained Ca2+ elevation in the mitochondrial microenvironment is followed by excessive mitochondrial Ca2+ accumulation that may lead to cell death via dysregulation in ATP and redox homeostasis (202, 231, 523, 559).
It is well established that Ca2+ is a signaling agent in biological systems due to (i) its binding properties to complex molecules, (ii) differences in its free concentrations in the extra-cellular environment and cytosol as well as between the cytosol and intracellular organelles, and (iii) the existence of a complex membrane Ca2+ transport system that orchestrates Ca2+ flux across plasma and intracellular membranes in response to cellular and extra- or sub-cellular signals (41). The development by Pozzan and Rizzutto (439) of highly sensitive, genetically encoded, Ca2+ probes specifically targeted to different cellular domains allowed for the demonstration that transient increases in intracellular free Ca2+ concentrations promote a variety of specific responses at different sites. These Ca2+ movements are driven directly or indirectly by ATP hydrolysis, rendering its signaling functions highly dependent on the energy state of the cell (200). Therefore, deficiencies in mechanisms responsible for cellular ATP supply lead to deregulation in Ca2+ signaling that may compromise cell function and survival (231). Experimentally, it is difficult to differentiate between acute ATP depletion and increased ROS generation as the primary cause of cell death, because reduced ATP and increased ROS levels occur concomitantly with increased cytosolic Ca2+ concentrations and amplify each other (58, 202). In this regard, distinguishing between the relative contributions of these events in diseases such as stroke or cardiac ischemia, for example, will allow for improved strategies for their prevention and/or treatment.
Here, we focus on how cellular, and in particular mitochondrial, Ca2+ homeostasis is related to the integrity of this organelle. Mitochondrial Ca2+ overload is known to affect mitochondrial redox state and promote membrane protein thiol cross-linking, causing mitochondrial permeability transition (MPT) (83, 208, 279, 333). MPT is a condition which is characterized by the opening of a high conductance, nonspecific proteinaceous pore (241) that leads to mitochondrial dysfunction (280, 282) and cell death by either apoptosis or necrosis (39, 268, 309, 341). Activation of MPT, a process first described by Hunter et al. (241), is considered a major cause of cell death under a variety of pathophysiological conditions, including ischemia/reperfusion, neurodegenerative disease, traumatic brain injury, muscular dystrophy, and drug toxicity (39, 219, 286, 322, 349, 381, 445, 487, 523). The importance of the MPT process in mammalian physiology is beginning to be properly uncovered (157). Inhibition of MPT pore opening due to cyclophilin D (CypD) ablation impairs mice heart mitochondrial Ca2+ exchange and results in abnormal heart adaptation to overload stress (157).
Ca2+ signaling for mitochondrial ROS generation occurs inside the organelle; so the understanding of the mechanisms of mitochondrial Ca2+ transport is of central importance. Three different mechanisms have been described for the influx of Ca2+ into the matrix and two for the efflux of this cation from mitochondria (210). A uniporter located in the inner membrane mediates an electrophoretic transport of Ca2+ down the electrochemical gradient across inner mitochondrial membrane without coupling Ca2+ transport to the transport of another ion (Fig. 4). Although this mechanism was discovered in the 1960s (128, 524), the molecular nature of the channel was only recently identified (34, 125), as a result of the progress in genome sequencing and knowledge of uniporter distribution in different eukaryotes, that is occurrence in vertebrates (210) and kinetoplastids (144) but absence in the yeast S. cerevisiae (210). Using RNAi techniques, the authors linked the proteins MCU (mitochondrial calcium uniporter) and MICU1 (mitochondrial Ca2+ uptake 1) to Ca2+ transport. MCU is a pore-forming subunit containing two transmembrane helices that are separated by a highly conserved linker facing the intermembrane space. MICU1, an EF-hand-containing protein, constitutes a peripheral regulatory partner of MCU. In addition, recent data reveal a previously unknown role of MICU1 as a gatekeeper to limit MCU-mediated Ca2+ uptake, thus preventing mitochondrial Ca2+ overload and associated stimulation of O2 •;− generation under resting conditions (339). Taken together, MCU and MICU1 explain all biochemical and physiological properties of the putative Ca2+ uniporter (34).
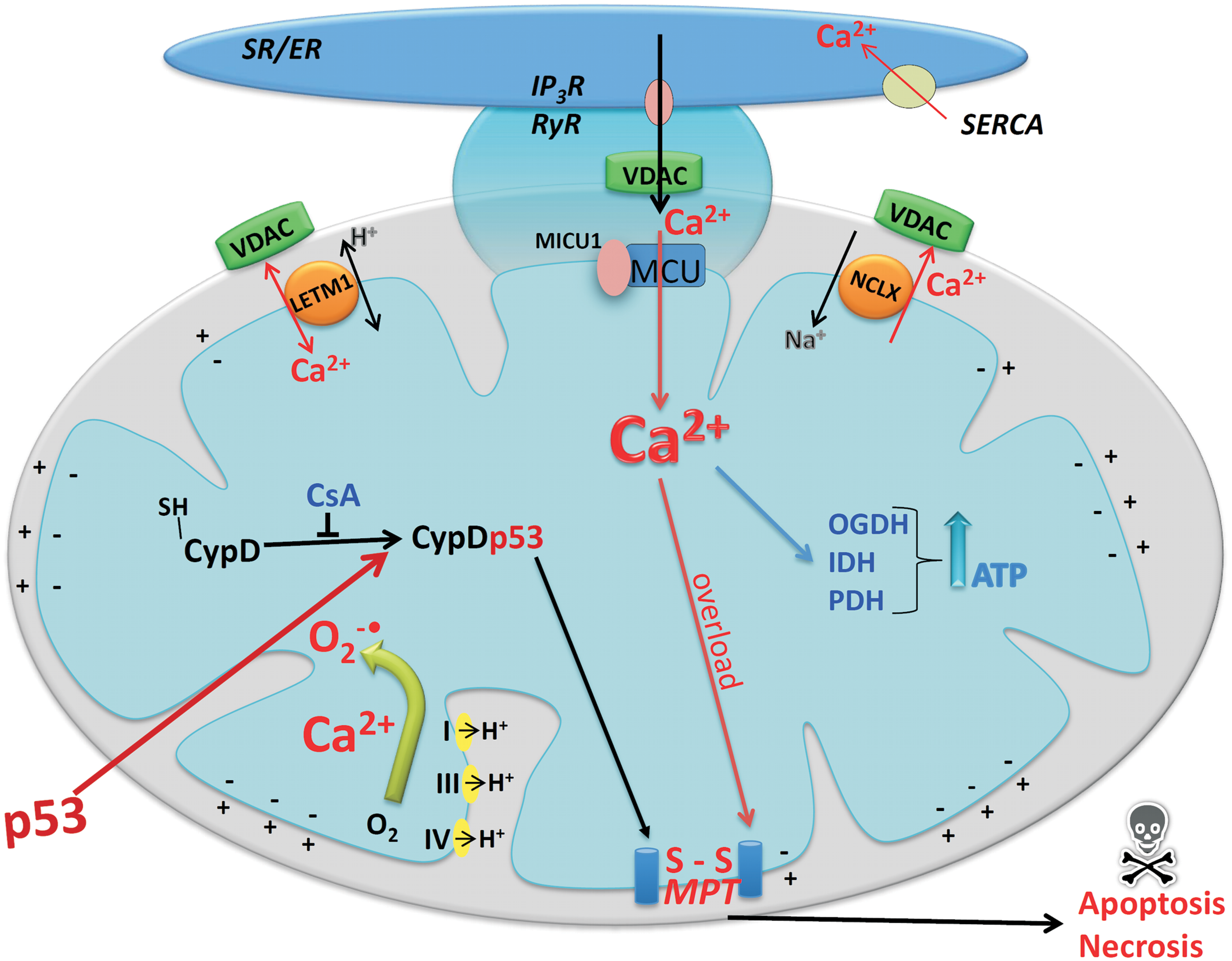
In addition to the uniporter, mitochondria possess two other systems that mediate Ca2+ influx: a mode of uptake called the rapid mode or RaM (484) and a Ca2+ uptake mechanism mediated by a ryanodine receptor, identified in excitable cells (43).
The ability of isolated mitochondria to take up Ca2+ via the uniporter and to release it through the recently identified Ca2+/Na+ exchanger (403) and the putative Ca2+/H+ antiporter was functionally characterized decades earlier (77). Nevertheless, the low affinity of the Ca2+ uniporter (an apparent Km of 20–30 μM) led to the general idea that mitochondria would not participate in cell Ca2+ homeostasis under physiological conditions (77). However, the use of intracellular Ca2+ probes (435) demonstrated that endoplasmic reticulum (ER) calcium release generates sufficient levels of Ca2+ concentrations in the mitochondrial microdomains to permit Ca2+ accumulation by mitochondria under in situ conditions (174, 189, 435 –438, 497). Such microdomais were directly demonstrated in selected regions of contact between the ER and mitochondria (121, 196).
It is now evident that under physiological conditions, the uptake of Ca2+ by mitochondria transfers the signal brought by cytosolic Ca2+ transients to the matrix (200). For example, tricarboxilic acid cycle activation by Ca2+ increases the rate of oxidative phosphorylation by using a transient increase in intracellular and intramitochondrial Ca2+ concentrations (200). This provides reducing equivalents to the respiratory chain under increased demand of ATP production. On the other hand, the close physical contact between mitochondria and plasma membrane Ca2+ channels (407, 532) and the ER (217, 218, 438) allows rapid import of large amounts of Ca2+ from these microdomains that may promote MPT (see Fig. 4) and cell death (286). Therefore, intramitochondrial Ca2+ can regulate both the rate of ATP generation, required for cell function and survival, and MPT, which can lead to cell death.
The MPT pore opening is triggered by a synergic combination of high levels of Ca2+ in the mitochondrial matrix and oxidative imbalance (280) in isolated mitochondria, intact cells, or in vivo (523). Cyclosporin A potently prevents MPT pore opening by binding to CypD and displacing it from the putative complex of proteins assembling the MPT pore [see Kroemer et al. (286)].
The relevance of MPT was initially questioned due to the nonphysiological experimental conditions required to trigger the phenomenon in isolated mitochondria (39, 531). These included mainly high Ca2+ loading in the matrix (241, 306), the irreversibility of the mitochondrial alterations associated with large amplitude swelling (527), and loss of matrix components, including low-molecular-weight proteins (120). However, the better understanding of the factors controlling the opening and closing of the MPT pore and numerous observations that MPT blockers, such as cyclosporin A, prevent cell death under many pathological conditions (133, 225, 322, 517) confirmed the participation of MPT in the pathogenesis of many diseases.
Despite extensive research since MPT was first described (241), the structure of the putative pore remains unresolved and controversial. Literature data suggest that the pore is composed of an assembly of matrix, inner and outer membrane proteins such as the adenine nucleotide transporter (ANT), the voltage-dependent anion channel (VDAC), CypD, aspartate-glutamate and phosphate carriers, hexokinase, and possibly other proteins (309, 341). However, other studies demonstrate that some of these proteins are not essential components of the pore, as they can also occur in inverted submitochondrial particles, devoid of matrix and outer membrane (166); in mitoplasts, devoid of outer membrane (441); and in mitochondria genetically deficient in ANT, VDAC, or CypD (24, 273, 284). In potato tuber mitochondria, MPT was showed to be insensitive to cyclosporin A, despite the fact that cyclosporin A inhibited the isomerase activity of CypD (181). The results show that although there is a pore opening under these conditions, some of its properties are altered. For example, in ANT-deficient mitochondria, atractyloside, a classical ANT ligand, does not promote pore opening; whereas in CypD-deficient mitochondria, MPT requires larger Ca2+ loads and is not blocked by cyclosporin A (24). These results suggest that if the pore is indeed composed of various proteins, it is conceivable that it may still be formed, with slightly different assembly properties, in the absence of one or more components.
An aspect that deserves consideration is the understanding of how Ca2+ and ROS act synergistically in the process of pore assembly (280). There is a general idea that Ca2+ is essential and has multiple roles in the process of pore formation (39), while ROS and other “pore-inducing agents” such as inorganic phosphate (Pi) and thiol oxidants have a facilitating role and lead to MPT irreversibility (84, 225). Indeed, it is very well known that Ca2+ alone can induce MPT, but it is also true that Ca2+ itself stimulates ROS generation by mitochondria (84, 208, 278, 279, 333). In addition, mitochondria are more susceptible to Ca2+ when their antioxidant systems, represented mainly by NADPH and GSH, are exhausted [reviewed in (280)]. The first indication for the redox nature of the MPT arose from experiments showing that isolated liver and heart mitochondria could not retain accumulated Ca2+ when the endogenous pool of pyridine nucleotides was shifted to the oxidized state (306). Further investigations on this mechanism demonstrated that oxidation of mitochondrial NADPH decreased the mitochondrial antioxidant capacity, leading to oxidative imbalance associated with a progressive polymerization of inner mitochondrial membrane proteins via thiol crosslinking (166). Later, a paper by Bernardi's group (117) proposed a modulation of the MPT by the redox state of pyridine nucleotides and thiols at two separate mitochondrial sites (117). In addition, many prooxidants act as MPT inducers, while many antioxidants prevent or even reverse MPT (280).
A recent study (523) identified the mitochondrial p53-CypD complex as an important contributor to oxidative imbalance-induced necrosis and implicated the participation of this protein complex in brain ischemia/reperfusion injury. The authors provided evidence that p53 accumulates in the mitochondrial matrix in response to oxidative imbalance and triggers MPT pore opening and necrosis by interacting with the MPT regulator CypD. In contrast, reduction of p53 levels or cyclosporin A pretreatment of mice prevents formation of this complex and effectively protects against stroke.
Literature data provide evidence that Ca2+ stimulates ROS generation by various mechanisms, including (i) stimulation of the tricarboxilic acid cycle (58); (ii) activation of ROS-generating enzymes such as glycerol phosphate and α-ketoglutarate dehydrogenase (508, 510); (iii) inhibition of respiration by Ca2+-induced NO• generation (195); (iv) opening of the MPT pore (224, 333); and (v) alterations in lipid organization of the inner mitochondrial membrane caused by Ca2+ binding to cardiolipin, leading to lateral phase separation (208). In addition, (vi) Pi, one of the earliest MPT inducers known (210), and Ca2+ cooperate in promoting oxidative imbalance, MPT, and membrane lipid peroxidation (278). This occurs via generation of triplet state intermediates from lipid peroxidation, a process probably catalyzed by cytochrome c (83). Moreover, in in vitro model systems consisting of phosphatidylcholine/diethyl phosphate liposomes, phosphate and Ca2+ cooperate to promote the propagation of radical reactions initiated by triplet acetone-generating systems (278). Finally, (vii) experiments performed with isolated mitochondria demonstrate that high Ca2+ loads promote mobilization of intramitochondrial Fe2+ followed by oxidative imbalance and MPT sensitive to the Fe2+ chelator o-phenantroline, dithiothreitol or exogenous catalase (83). Despite the slow kinetics of the Fenton reaction (221) and the lack of hydroxyl radical specificity, these experimental data suggest that under these conditions, a form of irreversible MPT would be formed via the attack of protein thiols by the hydroxyl radical. Since mitochondria are sites of iron uptake and storage (369), as well as incorporation into heme and electron transfer protein-sulfur iron clusters, this metal is a strong candidate for MPT stimulation under pathological conditions.
Overall, the above data regarding the redox nature of MPT provide the basis for a MPT model in which membrane proteins aggregate to assemble a pore with a hydrophilic core that confers the high conductance of the MPT (166). Although this form of MPT may occur under certain in vitro conditions, it does not contemplate other in situ regulators recently discovered such as the p53-CypD complex (523).
IV. Mitochondria and Nitric Oxide
Recent in vivo data indicate that NO• plays a role in the regulation of cellular metabolic phenotypes (319, 385) and mitochondrial energy transduction (56, 112, 303, 385, 386, 400), generating a strong interest in understanding the relationship between mitochondria and NO•. On the other hand, whether mitochondria possess a nitric oxide synthase isoform, the mtNOS, is a disputed point (54, 525, 526). Many aspects related to the identification, cellular sublocalization, regulation, and pathophysiology of this putative mtNOS have been approached in reviews that we indicate the readers to consult (54, 193, 227, 533). Irrespective of mtNOS existence, there is strong experimental evidence indicating that NO• and its derivatives affect mitochondrial function (4, 57, 59, 60, 110, 112, 420, 422, 470, 472) and biogenesis (111, 319, 384, 386). Recent studies (22, 23, 299, 300, 302, 303) have shown that the supplementation of humans with dietary amounts of inorganic nitrate (which can be converted into NO• in the body) significantly improves energy metabolism during exercise. A mitochondrial mechanism has been suggested for this effect (301).
A. Mitochondrial generation of NO•
Nitric oxide synthases catalyze the conversion of L-arginine into citrulline and NO•:
L-arginine+NADPH+O2+H+→citrulline+NADP++NO•
This reaction is Ca2+ dependent (except for iNOS) and requires FAD, FMN, BH4, heme, and calmodulin.
Several studies (155, 194, 199, 263, 291, 372, 502, 520) show that mitochondrial fractions obtained by standard differential centrifugation techniques possess nitric oxide synthase (NOS) activity, sensitive to specific inhibitors, as identified by the following assays: (i) radio-labeled L-arginine-citrulline conversion; (ii) spectrophotometric changes of oxymyoglobin; and (iii) inhibition of mitochondrial respiration. In addition to these functional approaches, there are many reports showing immunoreactivity of mitochondrial fractions with antibodies against all three known NOS isoforms [nNOS, eNOS, and iNOS, (291, 292)].
A key point to understand the debate and controversies on the existence of mtNOS is that mitochondrial isolation techniques generally yield a fraction also containing other cellular constituents, which may remain as contaminants even when additional purification steps are conducted (431). Therefore, classical biochemical assays or proteomic approaches for the identification of a putative mitochondrial protein are limited by sample preparation, and the NOS activity identified may not be mitochondrial. Some studies attempted to purify mitochondria free of other cellular constituents and still recovered NOS activity in mitochondrial suspensions (199, 291), although some degree of nonmitochondrial contamination remained (199). Nonetheless, it may be possible that an NOS is present in common preparations of isolated mitochondria, not necessarily comprising an evidence of the putative mtNOS.
An isoform of NOS was purified from isolated rat liver mitochondria, and its biochemical properties were described by Giulivi's group (155, 502). These studies show that mtNOS is a neuronal NOS (nNOSα) isoform with post-translational modifications, and that its purified activity required the following substrates and cofactors: L-arginine, NADPH, Ca2+, calmodulin, BH4, and FAD. The high Ca2+ and BH4 requirement for mtNOS activity raised some skepticism in the literature (54, 292, 525, 526). On the other hand, Mann's group employed a new approach to evaluate the mitochondrial proteome (180) that purportedly did not rely on the purification of these organelles. In this proteomic study of white and brown adipose tissue mitochondria, the authors identified the product of the NOS3 gene (i.e., eNOS) as a true mitochondrial protein (180). It is worth noting that this group did not find evidence of mtNOS in proteomic studies of mitochondria from other tissues such as heart, liver, and skeletal muscle, in which they employed a more standard approach relying on isolated and purified mitochondrial samples (179, 359). Overall, comprehensive studies employing robust techniques to approach mtNOS occurrence, properties, and its tissue distribution are still necessary.
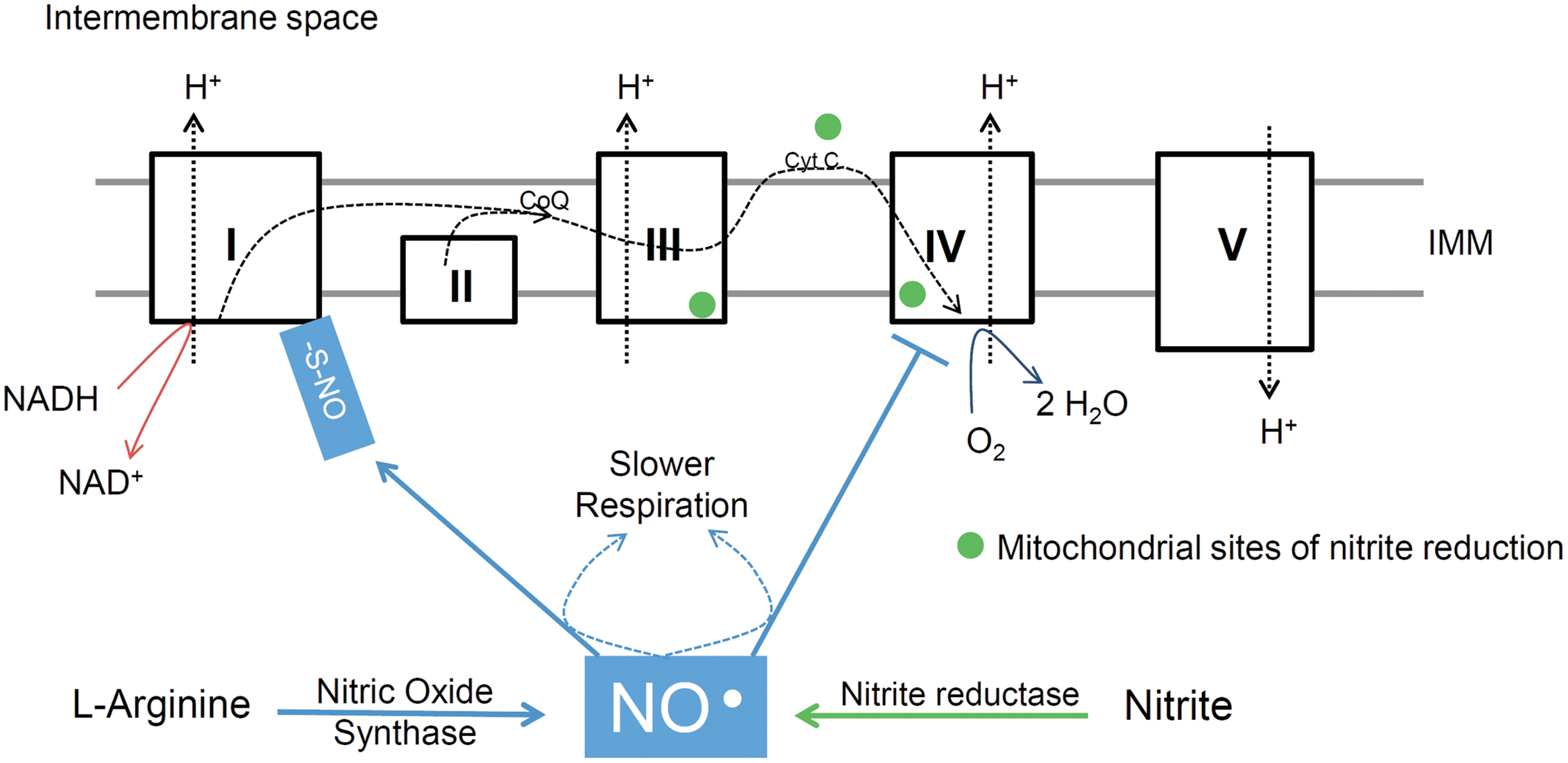
In addition to being generated by NOS isoforms, NO• and other bioactive nitrogen molecules have been shown to be metabolites of dietary inorganic nitrite (NO2 −) and nitrate (NO3 −). Thus, in mammals, both NO2 − and NO3 − are products of NO• degradation and substrates for NO• generation (Fig. 5). NO• enters the cycle when generated by NOS, while NO2 − and NO3 − are acquired through the diet (vegetables such as spinach and beetroot are rich in NO3 −). The amount of dietary NO3 − has been estimated to exceed the amount of endogenously produced NO• through NOS isoforms (331, 332). The reduction of NO3 − into NO2 − involves enterosalivary circulation and is mediated by commensal bacteria in the oral cavity. In turn, NO2 − can be reduced to bioactive NO• through a variety of enzymatic and nonenzymatic reactions. Notably, deoxyhemoglobin, deoxymyoglobin, and the mitochondrial respiratory chain possess nitrite reductase activities (32, 116, 229, 251, 331, 332, 471). The bioactivity of newly synthesized NO• is terminated by its oxidation into NO2 − and NO3 −. An important feature of this nitrogen oxide cycle is that hypoxia and/or acidosis greatly enhance NO• formation from NO2 −, comprising a possible mechanism for hypoxia-induced vasodilation (116, 123, 229, 301).

In vitro studies have demonstrated that mitochondria are able to metabolize NO2 − into NO• (32, 283). Specifically, cytochrome c and respiratory chain Complexes III and IV possess nitrite reductase activities that can be stimulated under hypoxic or acidic conditions (32, 283, 390) (Fig. 6). At these three sites, electrons are transferred from the electron transport chain to NO2 −, promoting reduction into NO•. A recent human study indicates that exogenous NO2 − is inert to mitochondria (isolated from human skeletal muscle and incubated in vitro) at a resting physiological pH of 7.2. However, at a pH of 6.7 (which is promoted intracellularly under conditions such as physical exertion), exogenous NO2 − elicits biochemical effects attributable to NO• (301).
Oxygen desaturated heme proteins are well-characterized nitrite reductases in mammals (116, 127, 229, 251, 471). Myoglobin can catalyze NO• formation from NO2 − in the close vicinity to mitochondria within the skeletal muscle. Thus, NO3 − and NO2 − seem to be a major sources of NO•. Indeed, there is evidence that increased NO2 − bioavailability exerts beneficial effects in vivo (22, 23, 79, 116, 127, 149, 229, 301, 303, 415, 472, 522). NO2 − reduction may even effectively occur in the absence of acidosis, as acute intravenous administration of NO2 − to healthy humans under resting conditions elicits vasodilation (127).
B. NO• effects on mitochondrial electron transport and energy transfer
Inhibition of mitochondrial respiration is the best-known interaction between NO• and mitochondria. Data from a variety of experimental models, ranging from isolated mitochondrial preparations and intact cells to exercising dogs and humans, support the idea that respiratory rates are controlled by NO• (23, 56, 203, 258, 301 –303, 400, 470). Nanomolar levels of NO• inhibit cytochrome c oxidase (mitochondrial complex IV) activity reversibly and competitively with O2 (56, 110, 420, 465) (Fig. 6). Indeed, studies employing isolated mitochondria and intact cells clearly show that NO• inhibition of mitochondrial respiration increases nonlinearly as oxygen availability decreases (4, 56, 203). Although cytochrome c oxidase has an apparent excess capacity with regard to electron flow through the respiratory chain, this enzyme may be operating much closer to its turnover limit if vicinal nM levels of NO• are taken into account (12), thus offering an explanation as to why changes in NO• concentration modulate the rate of mitochondrial oxygen consumption at physiologically relevant intracellular PO2 [∼3–35 mmHg, depending on energy turnover rates and ambient atmospheric pressures; (4, 433)]. In general, NO• inhibition increases at high mitochondrial respiratory rates and/or low PO2 (56, 203). With regard to NO• interaction with cytochrome c oxidase, the oxidized form of this enzyme may play a role in mitochondrial NO• actions and fate, as oxidized cytochrome c oxidase promotes NO• oxidation into NO2 − at considerably fast rates (13, 113).
In vitro studies also indicate that other electron transport chain sites are sensitive to NO• or its derivatives (59). Irreversible inhibition of the activities of mitochondrial respiratory chain complexes has been observed on exposure of submitochondrial particles and mitochondria to millimolar levels of NO• or ONOO− (47, 81, 427). Inhibitory mechanisms include degradation of protein structures, S-nitrosylation, and oxidation (47, 59, 60, 81, 427). However, some of these findings still require demonstrations of biological relevance or in vivo confirmations. Mitochondrial complex I protein thiol modification by NO• (i.e., S-nitrosylation) has been reported to occur in beating perfused hearts under biologically relevant conditions (492). By using exogenous S-nitrosothiol targeted to mitochondria, Prime et al. (422) demonstrated that S-nitrosylation of complex I is associated with slower mitochondrial respiration. These authors also found that this NO• effect only occurs when mitochondria are energized with complex I-linked substrates, thus indicating a specific inhibition of respiration at the level of complex I (422). Importantly, S-nitrosylation of complex I has been associated with improved outcomes from heart ischemia reperfusion in animal models (229, 314, 422, 472).
Boveris' group (420) perfused ex-vivo beating hearts with solutions containing NO• donors and found that myocardial oxygen uptake was decreased up to 50% on increased NO• availability. Developed left ventricular pressure, an index of heart mechanical work, remained unchanged (420). These results are one of the first evidences that the oxygen cost of mechanical work can be fine-tuned by NO• availability. If on one hand increased NO• promotes higher mechanical efficiency (i.e., higher mechanical work to oxygen uptake ratio) (420), decreased NO• availability, by means of pharmacological NOS inhibition, augmented muscle oxygen uptake in exercising dogs (470). Further studies reinforced this concept by showing that pharmacological NOS inhibition accelerated the increase in human skeletal muscle oxidative metabolism during resting-exercise transitions (258).
More recently, a series of work by two independent research groups uncovered interesting metabolic effects of dietary NO3 − during exercise (22, 258, 300 –303, 522). The oxygen cost of exercise was found to be decreased by acute inorganic nitrate intake. Since mechanical efficiency is a determinant of exercise performance, nitrate intake improved performance after nitrate supplementation (22, 299). The acute intake of a dietary amount of NO3 − corresponding to ∼0.5 L of beetroot juice was enough to increase plasma NO3 − and NO2 −, to decrease arterial blood pressure, and to elicit the exercise responses described (299 –301, 303, 522). While NO3 − intake lowers ATP turnover during muscle contraction (22), Larsen et al. (301) found that mitochondrial ADP/O ratios in the skeletal muscles of healthy humans are improved by NO3 − supplementation. Mitochondrial ANT expression was also decreased, and oxygen dependence of mitochondrial respiration was increased. These two findings may provide mechanistic support for higher ADP/O ratios: First, ANT can mediate uncoupling of oxidative phosphorylation; second, a slight inhibition of mitochondrial respiration at the level of cytochrome c oxidase may promote positive effects on energy conservation (112, 204). Cytochrome c oxidase activity can be limited by very low oxygen tensions or by decreased oxygen affinity due to the action of NO• (112). In fact, in vitro analyses of isolated mitochondria show that partial inhibition of mitochondrial respiration by either NO• or cyanide leads to higher oxidative phosphorylation efficiency (112). Based on previous studies modulating NO• bioavailability (209, 388) and on the evidence that NO2 − and NO3 − are sources of bioactive NO• in living mammals (331, 332), effects of NO3 − supplementation on exercise metabolism and mitochondrial energy transfer seem to be mediated by enhanced NO• bioavailability (331, 332).
C. MPT and NO•-mediated cytoprotection
Cell damage or death following MPT opening participates in the pathophysiology of ischemic diseases, as discussed earlier. There is also evidence that pharmacological MPT inhibition is of therapeutic value in brain trauma and muscle dystrophy (198). Adding to the previously demonstrated redox sensitivity of MPT discussed earlier (280, 306, 528), NO• has been shown to modulate mitochondrial suscebility to calcium-induced MPT (57, 307, 380). Initial data showed that incubation of isolated mitochondria with exogenous NO• donors elicited partial inhibition of calcium-induced MPT (57). However, parallel measurements of mitochondrial calcium uptake and membrane potential suggested that NO• inhibition of MPT was secondary to membrane depolarization and calcium uptake failure (57), simply the result of NO• inhibition of mitochondrial respiration. Since the oxidation of membrane protein thiols mediates MPT opening (166), S-nitrosylation of thiols groups by NO• is presumably a mechanism of protection against thiol oxidation, MPT opening, and cell damage (Fig. 7). In fact, cytoprotective effects of ischemic preconditioning are associated with increased S-nitrosylation of mitochondrial proteins along with lower oxidation of thiol groups (272).

Our group has recently demonstrated that co-incubation of isolated mitochondria with NOS inhibitors rendered mitochondria more prone to Ca2+-induced MPT (307). MPT stimulation was associated with a decrease in mitochondrial S-nitrosothiol content (307), building on the concept that reversible modification of thiol groups by NO• plays an important role in the redox regulation of MPT. A very recent study has demonstrated that cysteine 203 of CypD is a target of S-nitrosylation and is of central importance for MPT regulation (380) (Fig. 7). This work showed that mutation of cysteine 203 of CypD to a serine residue increased mitochondrial Ca2+ retention capacity to the level presented by CypD null or cyclosporin A-treated mitochondria. Moreover, exogenous S-nitrosoglutathione presumably promotes S-nitrosylation of CypD and attenuates H2O2-induced MPT opening and cell death (380). These is strong data revealing the role of protein thiol S-nitrosylation as a mechanism for protection against MPT. The peptidyl-prolyl cis-trans isomerase activity of CypD is not significantly altered by the mutation mentioned earlier (317). CypD isomerase activity is inhibited by cyclosporin A and also seems to be modulated by acetylation/deacetylation through mitochondrial Sirt3 activity (474). Sirt3 promotes deacetylation of CypD and atenuates Ca2+-induced MPT (215). Deacetylated CypD was shown to possess lower isomerase activity (474). Adding to the insights provided from the mutated CypD studies (317, 380), a previous study by our group showed that cyclosporin A inhibits the isomerase activity of cyclophilin in isolated mitochondria from potato tubers, while Ca2+-induced MPT in this plant was insensitive to cyclosporin A (181). Therefore, the isomerase activity of CypD is not always associated with MPT regulation.
In vivo animal studies demonstrate that S-NO or NO2 − promotes protection in ischemia reperfusion (131, 149, 259, 422). Improved neurological and cardiac function was observed when mice undergoing cardiac arrest were treated with NO2 − (131). Interestingly, lower mitochondrial oxidative imbalance and a partial inhibition of complex I were also documented in these animals (131). Partial complex I inhibition is likely due to S-nitrosylation, as indicated by a study that pharmacologically targeted S-NO to mitochondria (422). Mitochondria-targeted S-NO is accumulated within mitochondria, promoting S-nitrosylation and inhibition of complex I (422). Similar to NO2 − and other S-NO donors, mitochondrially targeted S-NO reduced the extent of tissue damage in animal models of ischemia reperfusion (422). It is worth noting that these protective effects are probably not only confined to NO• interaction with specific mitochondrial proteins (e.g., CypD or complex I), as S-nitrosylation of other proteins, such as the transcription factor hypoxia inducible factor-1α (HIF-1α), seems to be of central importance for tissue response to hypoxia (314).
S-nitrosylation of complex I and other mitochondrial proteins has been observed after ischemic preconditioning (259, 492). Although the exact mechanisms by which S-nitrosated proteins protect against ischemia reperfusion are still unclear, the advantage of inhibited mitochondrial complex I due to S-nitrosylation may be a decrease in the likelihood of mitochondrial ROS formation, as discussed earlier. In addition, lower NADH oxidation by mitochondrial complex I may help maintain a higher NADH/NAD+ ratio, favoring NADP+ reduction at the expense of NADH (306, 528).
Converse to the cytoprotective roles of NO• discussed earlier, high levels of this oxidant may promote nitrosative stress and cellular injury (47). Indeed, exposure of mitochondria to high levels of NO• or ONOO− potentiates Ca2+-induced MPT (57, 186, 452).
D. NO•-mediated mitochondrial biogenesis
Peroxisome proliferator-activated receptor coactivator 1 α (PGC-1α) is a regulator of mitochondrial biogenesis in metabolically active tissues such as heart, skeletal muscle, and brown adipose tissue, controlling the expression of transcription factors that regulate the expression of a variety of nuclear and mitochondrial genes encoding mitochondrial proteins. Genetically modified mice overexpressing skeletal muscle PGC-1α display a marked increase in mitochondrial density and exercise capacity (315). NO• is an upstream activator of PGC-1α (385), promoting enhanced mitochondrial biogenesis and increases in the abundance of mitochondrial proteins (319, 385, 386) in a manner that is dependent on cGMP (385). Recently, Lira et al. (319) demonstrated that NO• interacts with AMPK both in myotubes and in contracting skeletal muscle, activating NOS and regulating PGC-1α expression.
Mice lacking eNOS have provided important evidence of NO•-mediated mitochondrial biogenesis and metabolic health (79, 385, 386). These mice present lower liver and skeletal muscle mitochondrial density and features of metabolic alterations, despite the expression of other NOS isoforms. Interestingly, dietary supplementation with NO3 −, which can be metabolized into bioactive NO• (331), can reverse metabolic abnormalities in eNOS-deficient mice (79).
Mice deficient in eNOS also present a decrease in mitochondrial biogenesis promoted by the limitation of calorie ingestion (539). Indeed, we have demonstrated that caloric restriction increases eNOS activity and mitochondrial biogenesis in a mechanism involving enhanced insulin receptor activation mediated by adiponectin (86, 87, 89). Furthermore, enhanced NO• and mitochondrial biogenesis may be sufficient to promote some beneficial effects of calorie restriction such as increased neuronal survival in vitro (87). Interestingly, calorie restriction increases mitochondrial content (88, 109, 326, 387), but decreases ROS release, possibly due to mitochondrial uncoupling (88, 277, 296).
V. Mitochondrial Disorders
Mitochondrial dysfunction can arise from more than 1200 different gene mutations, toxic agents, or spontaneously during aging (456, 539). Impairments in mitochondrial oxidative phosphorylation have recently been recognized as the most common cause of inborn errors of metabolism (118). That is somewhat expected, considering that oxidative phosphorylation involves at least 90 proteins and more than 300 auxiliary proteins that help them assemble into mature complexes. On the other hand, in addition to ATP synthesis, cellular metabolism depends on mitochondria and the proper exchange of metabolites across organelle membranes for the biosynthesis of amino acids, vitamins, lipids, prosthetic groups, and many other intermediates, which are also required for oxidative phosphorylation and cell viability. Deficiencies in any part of this intricate metabolism may cause disease. Uncovering of the specific gene mutation involved in mitochondrial disorders has shed light on the biochemical function of many human open reading frames and received a great deal of attention in the last years (37, 136, 141, 288, 402, 511, 513). In the next years, sequencing of a large number of patients will hopefully overcome most diagnostic limitations, and the specific gene mutation in a given patient will be more promptly identified. Since the oxidative phosphorylation apparatus involves proteins encoded in the nucleus, in addition to 13 subunits encoded in mitochondrial DNA (mtDNA) and translated in mitochondria (Fig. 8), this section summarizes our current knowledge of the features of mitochondrial dysfunction and redox imbalance caused by mutations in nuclear genes that affect oxidative phosphorylation (Tables 1 and 2) and by mutations in the mitochondrial genome.
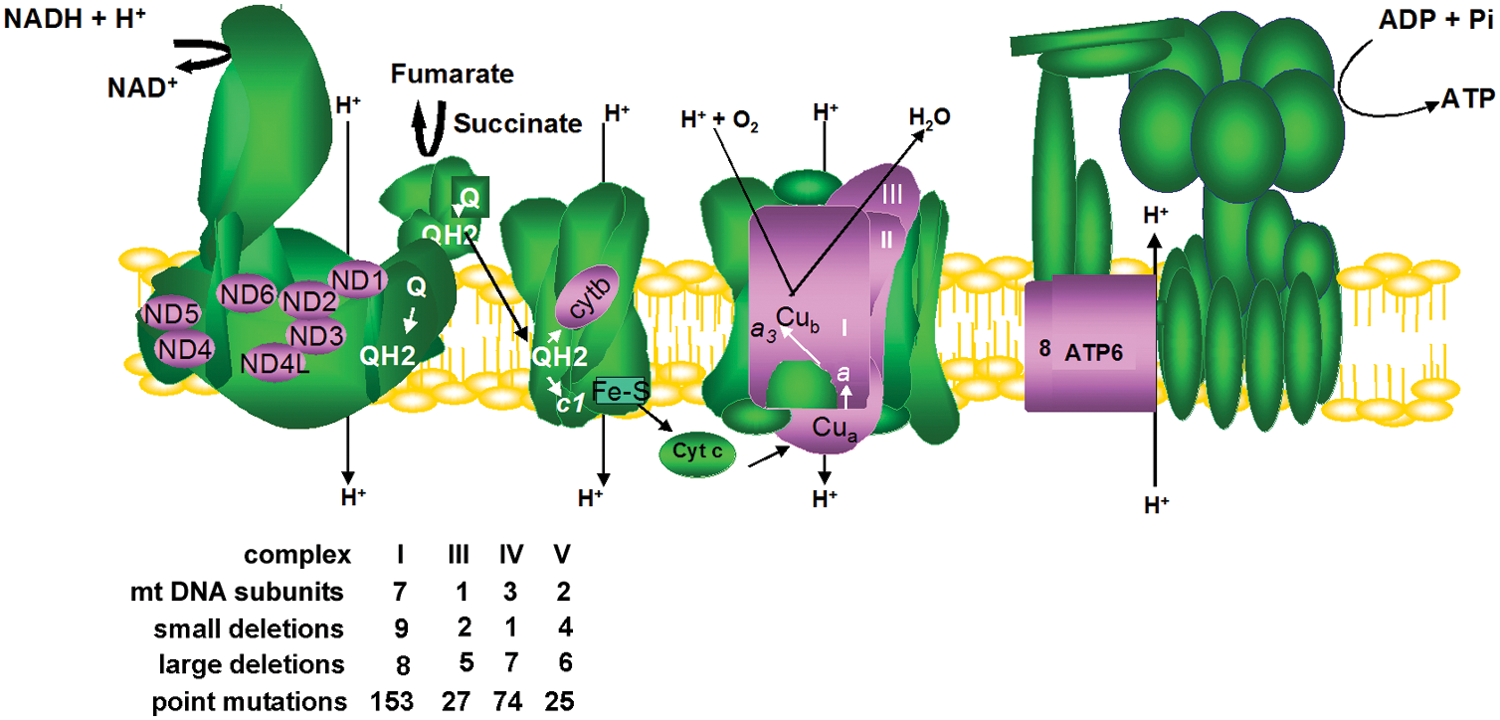
CoQ, Coenzyme Q.
MNGIE, mitochondrial neuro gastrointestinal encephalomyopathy; MLASA, myopathy lactic acidosis sideroblastica anaemia; mtDNA, mitochondrial DNA.
A. Nuclear genes in mitochondrial disorders
The severity and clinical outcome of a particular loss of biochemical function may be different in individuals expressing the same mutant gene, a typical consequence of gene interactions in different genetic backgrounds. In addition, mutations in different genes may elicit the same clinical presentation and, finally, a particular deficiency can have different prognosis depending on the tissue and organ it is being expressed in. It is, thus, not unexpectedly remarkable that tissues with a higher energetic demand are more promptly affected by lower ATP synthesis due to deficient mitochondrial metabolism (521). Altogether, there are 502 clinical categories of mitochondrial disorders already described from 174 gene mutations (456), and neuromuscular-associated pathologies are the most frequent clinical presentation (456). Differently from all other cell enzymatic complexes, biogenesis of mitochondrial respiratory complexes depends on two genomes that create an enormous regulatory complexity required to bring together polypeptides coming from inside and outside the organelle.
Mitochondrial dysfunction, in turn, leads to different biochemical effects that modulate nuclear gene expression, such as the over-expression of ROS scavenging enzymes in fibroblasts of patients with progressive external ophtalmoplegia (329). The amount of ROS produced and the differential ability of tissues to handle ROS excess are certainly components that exacerbate the clinical heterogeneity of mitochondrial diseases (46). ROS and RNS are components of the cross-talk between mitochondria and nucleus. Elevated levels of ROS/RNS were shown to increase the mtDNA copy number and mitochondria abundance, a feedback response to compensate impaired respiration, an adpatation that also occurs in aged tissues (305). Nevertheless, it is a tautological harmful cycle, as mitochondria are both generators and targets of ROS. Elevated ROS can directly damage and mutate mtDNA, a process 10–20 times more likely than in nuclear DNA (61, 540, 542), as mtDNA is located close to the electron transport chain, a major source of intracellular ROS, as described earlier. For instance, high ROS production in mitochondrial diseases leads to augmented 8-hydroxydeoxy guanosine (8-OHdG) and lipid peroxidation levels (418), which are, respectively, related to the process of mtDNA mutagenesis and apoptosis.
Given the important role of ROS in apoptosis, the excess of ROS generated in mitochondrial diseases can trigger apoptosis through MPT, or by means of cardiolipin oxidation (163, 192). Apoptosis participates in the pathogenic process of mitochondrial myopathies and encephalomyopathies (16, 352), as well as in the degenerative process in hereditary optic neuropathies (310). As cells are progressively lost through apoptosis, tissue function declines, leading to the recrudescence of clinical symptoms. Overall, oxidative imbalance resulting from mitochondrial dysfunction plays an important role in the pathogenesis and progression of mitochondrial diseases.
mtDNA abnormalities also stem from direct deficiencies in replication and repair, which depend on nuclear-encoded proteins. Due to this dependence, mtDNA inheritance and maintenance are also transmitted as mendelian traits.
Pathogenic mutations in DNA polymerase γ (POLG1 and POLG2) and twinkle DNA-helicase are the major cause for the multitude of mtDNA deletions in many cases of progressive external ophtalmoplegia (183). POLG1 codes for the catalytic core of the mtDNA pol γ, and POLG2 codes for the accessory β-subunit. mtDNA stability is also considerably affected by deoxynucleotide supply in the organelle. Patients harboring mutations in genes involved in this process usually present depletion of mtDNA (232).
The clinical categories of patients with mtDNA deletions and depletion are very heterogeneous. Their symptoms range from severe encephalopathy and cardiomyopathy in childhood to late-onset progressive external ophtalmoplegia. There is also evidence of mtDNA depletion associated with sporadic Parkinson's disease, Alzheimer's, and other neurodegenerative diseases (540).
mtDNA instability can also originate in mitochodria with defective fusion and fission dynamics (38), which has also been associated with cell death in neurodegenerative diseases (102) as well in the pathophysiology of obesity and type-2 diabetes (570). Organelle dynamics is an essential process, influencing mitochondria morphology, biogenesis, distribution, and metabolism. Morphology modulation of mitochondrial filaments, for instance, allows mitochondria to transmit mitochondrial membrane potentials to regions with low oxygen tension in muscle cells (476). Mitochondrial fission and fusion depends on guanosine triphosphate GTPase dynamins, such as dynamin-related protein1 (Drp1), mitofusins (Mfn1 and Mfn2), presents in the mitochondrial outter membrane and optic arthropy protein 1 (OPA1), present in the inner membrane (553). OPA1 dysfuction impairs mitochondrial fusion and is the cause of most cases of dominant progressive external ophtalmoplegia. (240). On the other hand, Mfn2 mutations have been reported in patients with Charcot-Marie-Tooth neuropathy type 2A, a disorder that is characterized by the gradual degeneration of peripheral neurons (102). OPA1 isoforms are generated in mammalian cells by alternative-splicing and proteolytic processing by AAA-proteases, such as AFG3L2 and SPG7, also known as the paraplegin group. Nevertheless, all OPA1 isoforms are able to interact with Mfn1 and Mfn2 (209). Pathogenic mutations in paraplegin genes were identified in patients with progressive weakness and spasticity of the lower limbs due to the degeneration of corticospinal axons. More recently, pathogenic mutations in the metaloprotease AFG3L2 were described in patients with dominant cerebellar ataxia, a remarkable feature considering that the autosomal dominant pattern of inheritance is more frequently associated with progressive external ophtalmoplegia (132).
In yeast, mtDNA also becomes unstable when the synthesis of cardiolipin is altered (97). Cardiolipin is usually associated with the respiratory complexes and other mitochondrial proteins involved in mitochondrial biogenesis and apoptosis, being particularly susceptible to ROS-induced lipid peroxidation, a typical feature of neurodegenerative diseases. Cardiolipin biosynthesis occurs inside mitochondria, and depends on acyl groups remodeled by tafazzin (TAZ1). Impairment in this step generates aberrant species of cardiolipin, and patients are commonly diagnosed with Barth syndrome (459).
Deficiency in the synthesis of enzymatic oxidative phosphorylation prosthetic groups associated with metals can also affect mtDNA stability through increments in ROS release (182). For instance, in Friedreich ataxia, patients have a specific deficiency in frataxin, a mitochondrial matrix enzyme that is an iron chaperone functioning in the biosynthesis of iron-sulfur clusters (429). Patient cells depleted of frataxin function show multiple Fe-S-dependent respiratory chain deficiencies, with complexes I, II, and III compromised. This disease may manifest in adolescence with progressive cerebellar, limb, and gait ataxia, and cardiac hypertrophy. Antioxidant treatment of patients with idebenone has been used in many trials, and positive effects were observed in the cardiopathic component of the disease (446).
Pathogenic mutations in complex I nuclearly encoded subunits and assembly factors are the most frequent group of mitochondrial diseases, responding for 1 out of 3 of all known oxidative phosphorylation deficiencies (513). Mutations in genes required for the biogenesis and assembly of complexes III, IV and V, as well tricarboxilic acid cycle enzymes, are less frequent.
Complex I is the largest respiratory complex with 45 structural subunits. Approximately 150 patients were already shown to have pathogenic mutations in 22 genes involved in complex I structure and assembly (513). Patients with mutations in one of these subunits regularly manifest symptoms of neuromuscular deficiencies. Encephalomyopathy, cardiomiopathy, and Leigh syndrome are more frequently identified during childhood. Interestingly, some complex I deficiencies and associated pathologies, such as tumorigenesis, can be partially overcome through the heterologous expression of yeast NADH dehydrogenase (NDI1) (21). The benefits achieved with yeast NDI1 launched it as a putative gene therapy protocol (558). In fact, Ndi1p delivered by protein transduction results in cardioprotection in models of ischemia/reperfusion (414).
Complex I is an important ROS production site (as discussed earlier), and studies using cell lines from patients containing complex I deficiency revealed a correlation between ROS production and mitochondrial morphology (275, 554). Severe complex I impairment results in higher ROS production and mitochondrial fragmentation, while mild impairment leads to discrete ROS elevation and normal elongated mitochondrial morphology (143). One attractive explanation for this would be that mitochondrial fragmentation restricts the local effects of ROS, limiting damage extension (143). The correlation between oxidative imbalance and mitochondrial dynamics has been corroborated recently by studies on Drp1 (553). The GTPase activity of Drp1 is sensitive to NO• (543), while the S-nitrosylation of Drp1 (SNO-Drp1) promotes mitochondrial fragmentation (367). The presence of SNO-Drp1 is increased in brains of human Alzheimer's disease patients, and, consequently, may be an important player of the pathologycal development of neurodegeneration (101, 367).
Pathogenic mutations in complex II subunits and fumarate hydrase have been described as causes of rare late-onset Leigh syndrome and of tumor formation (68). The mechanisms by which depressed complex II activity favors tumor formation are still poorly understood, but one likely hypothesis is that succinate accumulation induces the hypoxia-response pathway, which activates the transcription of genes related to tumorigenesis and angiogenesis (466).
Nuclearly encoded mutations for complexes III, IV, and V are rare—only four gene mutations for structural proteins of these three complexes have been described—but mutations in another handful of auxiliary proteins involved in the assembly of these complexes have been identified. For instance, BCS1L plays a role in the synthesis and insertion of the active-site iron-sulfur cluster of the Rieske center in complex III (388). Patients harboring mutations in BCS1L have been identified with variable phenotype conditions and with different levels of neurological degeneration, including GRACILE and Björnstad syndromes (37).
Together with NADH dehydrogenases in complex I, the protonmotive Q-cycle in complex III is the major site for electron leakage and consequent formation of ROS (281). Therefore, it is somewhat expected that complex-III-deficient patients have an elevated level of ROS (37). In the case of BCS1L mutated patients, oxidative imbalance is worse due to iron overload (534). Nevertheless, in cases of complex III and IV dysfunction, assembly of complex I is diminished, indicating a possible regulatory attempt to diminish electron leakage and consequent ROS generation (1, 137).
Primary complex IV deficiency is more commonly identified in patients with mutations in assembly factors of the holoenzyme (28). Mutations in COX10, COX15, SCO1, SCO2, and SURF1 promote cardioencephalopathy, hypotonia, lactic acidosis, hepatic failure, and Leigh syndrome (136). COX10 and COX15 mutants are defective in the synthesis of heme a (28). SCO1 and SCO2 function in the copper delivery route toward the CuA site of COX2 subunit and in the regulation of copper efflux under excess conditions (304). The SURF1 gene product is associated with an early step of cytochrome c oxidase assembly (177). In model organisms, the disruption of complex IV assembly factors already associated with human diseases such as COX10, COX15, and SCO1 results in an increment in ROS production (30, 265). Conversely, to our knowledge, there is no specific indication of ROS increments in complex IV patient cell lines, perhaps because of compensatory mechanisms such as complex I depletion (137).
Curiously, cytochrome c oxidase is secondarily affected in patients containing mutations in ETHE1, a sulfur dioxygenase localized in the matrix and involved in catabolism of sulfide (134). Deficiency in this enzyme elicits ethylmalonic encephalopathy, an early-onset encephalopathy, normally fatal, characterized by sulfite accumulation, which is a potent inhibitor of cytochrome c oxidase (134).
Disorders in F1-FO ATP synthase assembly due to nuclear mutations are very rare (288). Recently, a number of cases of primary ATP synthase deficiency were elucidated with the description of mutations in TMEM70, an auxiliary factor of ATP synthase assembly (485).
Combined deficiencies in the oxidative phosphorylation system can occur due to defects in iron metabolism, lipid organelle protein import, or mitochondrial translation. Mitochondrial translation defects arise from mtDNA mutations in tRNA and rRNA genes or nuclear gene mutations. Patients with mendelian traits that are defective in this process have been recently identified (173, 552). Considering that yeast has a considerable number of translational activators for mitochondrially encoded polypeptides and only TACO1, a COX1 activator, has been identified in humans (511, 552), it is probable that many cases still remain to be identified.
Finally, the respiratory chain also depends on electron carriers such as coenzyme Q and cytochrome c. Coenzyme Q (CoQ, Fig. 9) is responsible for transferring electrons from NADH and FADH2 dehydrogenases to complex III. Although lower CoQ levels are identified in many mitochondrial disorders, just recently, primary deficient cases of CoQ have been described (511). ROS overproduction was detected in cell lines of patients with primary CoQ deficiencies, and the level of ROS production correlated with the severity of the CoQ defect, a result also observed in yeast mutants with CoQ dysfunction (69, 294, 425).
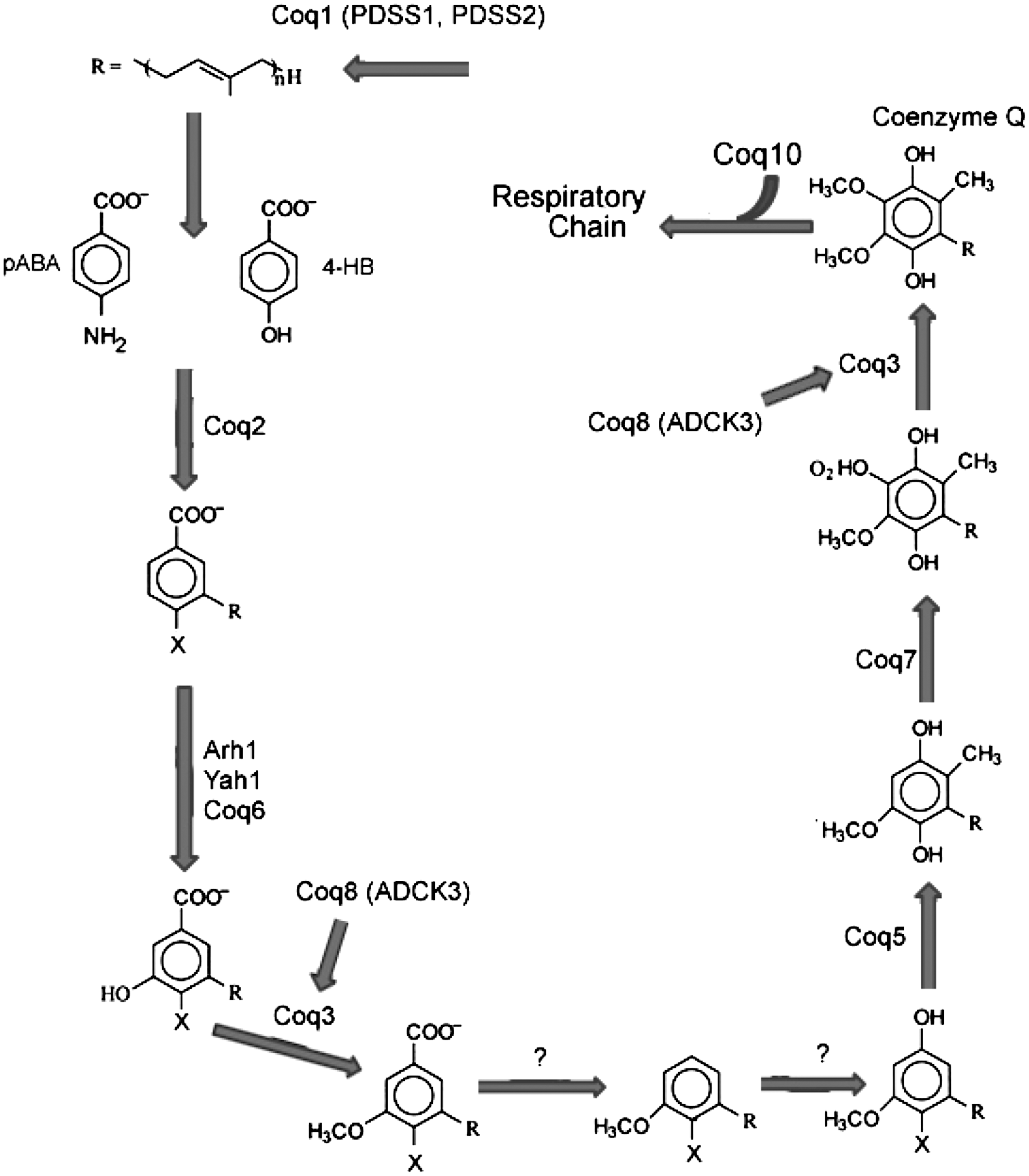
The heterogeneity of mitochondrial dysfunction generates an enormous difficulty to establish therapeutic approaches. Nevertheless, one therapy regularly applied is the administration of CoQ and its synthetic analogue idebenone. As discussed earlier, idebenone treatment as an antioxidant has also been effectively employed in Friedreich ataxia (446). CoQ administration is considered harmless even in treatments using 2000 mg daily (141), and has improved the clinical symptoms of many patients, probably by lowering the levels of ROS being produced in dysfunctional mitochondria as well shuttling electrons directly from cytossol to complex III, elevating the synthesis of ATP in patients with defective complex I (161). On the other hand, in patients with primary CoQ deficiency, it is expected that the rapid identification and early treatment can prevent the irreversible damage to the nervous system (511).
SOD2 allelic variations, which are obviously directly involved with the cellular antioxidant system, were recently investigated as a susceptibility risk factor in diseases where oxidative imbalance is considered an important factor (6). Impairments in MnSOD were detected in C47T polymorphism, the resultant amino acid change (Ala16Val) affects MnSOD transport into the organelle (495). C47T polymorphism has also been associated with overall coronary artery disease risk (504), and nonalcoholic fatty liver fibrosis (6). However, because of the heterogeneity of population studies, more studies are necessary in order to understand the susceptibility and risk factors in SOD2 polymorphisms in the cellular antioxidant machinery.
B. mtDNA alterations in mitochondrial disorders
The human mitochondrial genome is extremely streamlined, encoding for only 13 polypeptides, 22 tRNAs, and 2 rRNAs. While the vast majority of genes from the original bacterial genome were transferred to the nucleus throughout evolution, these 13 polypeptides were retained in the mtDNA, likely due to their high hydrophobicity and central location in the assembled respiratory complexes. The 13 mitochondrially encoded proteins are essential for proper function and assembly of oxidative phosphorylation complexes I, III, and IV and of the ATPsynthase (Fig. 8). For example, in the assembly of Complex I (seven mtDNA-encoded), mutations in three mtDNA-encoded subunits ND1, ND4, and ND6 result in a marked decrease in Complex I activity and protein levels (106, 234, 337), while mutations in ND2 cause impaired assembly and lead to accumulation of assembly intermediates (170).
The hypothesis that alterations in mtDNA could associate with human diseases was initially put forth in the late 1970s, from matrilineal inheritance patterns in some forms of ataxia, such as Frederich's ataxia and blood diseases (175). However, it was only after the sequencing of human mtDNA, in 1981 (8), that a comprehensive search for mtDNA mutations and deletions in human syndromes with clinical features of mitochondrial dysfunction started. In 1988, Holt et al. (235) found two distinct deletions (4.9 and 5.9 kb) in muscle cells from two patients with mitochondrial myopathies. However, seven other patients investigated in this study showed similar, but not identical, deletions. Other deletions of varying sizes were identified soon after in patients with different mitochondrial diseases, from a 0.4 kb deletion in a patient with encephalomyopathy (315) to deletions as large as 7.4 kb, in a patient with a form of progressive external ophthalmoplegia (499). It is not unusual for mtDNA molecules with different deletions to coexist in the same patient, for reasons which will be discussed later. Moreover, deletions are also detected in some tissues of “healthy” subjects, and, thus, the diagnostic value of one particular deletion, taken out of the context of other markers of mitochondrial dysfunction, is somewhat limited (350, 503).
A unique aspect of mitochondrial genetics is the fact that, since each mitochondrion can contain more than one copy of the mitochondrial genome (451), and each cell can contain from a few dozens to several hundreds of mitochondria, the number of copies of mtDNA in one single cell can be significant variable. However, more importantly, these copies can vary slightly from one to another, both in size (as a consequence of insertions/deletions) as well as in sequence (as a result of base substitutions). Both processes likely result from mtDNA damage accumulation and incomplete processing of these lesions by DNA repair pathways (124, 285). The degree of sequence variation among mtDNA molecules within a single cell is termed heteroplasmy (49, 481). If these different copies have different mutations, and individually would result in impaired gene expression, a corollary of this feature is that different copies of the mtDNA can complement each other within an individual organelle (197), as well as within the cell, especially in light of recent results suggesting a role for mitochondrial fusion and fission in mtDNA organization (for a review of this topic, see (461)).
In most cases of patients showing mtDNA deletions, the deleted molecules are highly heteroplasmic, suggesting de novo events, and enriched in the affected tissue, but not in unaffected cell types (538). They often do not follow a matrilineal inheritance patterns as well (473). It later became clear that while mitochondrial dysfunction, and therefore the clinical outcome of the disease, is directly related to the relative amount of deleted mtDNA molecules (243), these events are secondary to mutations in nuclear genes whose protein products are involved in mtDNA maintenance, such as DNA pol γ and the twinkle helicase, as discussed earlier.
However, a specific mtDNA deletion syndrome has been identified in which family history indicated the presence of germ-line alterations. Kearns–Sayre syndrome is a distinctive type of progressive external ophthalmoplegia that is characterized by pigmentary degeneration of the retina, heart block, elevated concentration of cerebrospinal fluid protein, and abnormal muscle mitochondria. While several sporadic cases are attributed to defective mtDNA metabolism, as in patients with defects in ribonucleotide reductase (419), the identification of familial cases (459) led to the identification of a large 4.9 kb germline mtDNA deletion, later known as the common deletion (360, 563). This deletion has been shown to accumulate with age as well (115), although to which extent this accumulation is functionally relevant still remains unclear (114, 237, 411).
Human mtDNA displays an extremely high gene density (
From an evolutionary point of view, mutations can be deleterious, neutral, or advantageous. Since mtDNA encodes 13 essential polypeptides, it is likely that mutations in its coding sequence will have deleterious consequences. Moreover, in animals, mtDNA mutates at a faster rate than does nuclear DNA (61, 542). This elevated mutation rate was initially attributed to inefficient DNA repair in mitochondria, although it is now clear that mitochondria from both lower and higher eukaryotes are proficient in repairing several types of lesions, including oxidative DNA modifications, which are thought to be the most prevalent lesions in mtDNA (347). Interestingly, a work by Kienhofer et al. (266) has demostrated that mtDNA colocalizes with antioxidant enzymes, thus indicating the occurrence of a close and integrated mechanism of protection against mtDNA oxidatiion. Due to the impact of heteroplasmy, pathological mutations in mtDNA only cause disease if they accumulate above a tissue-specific threshold. Thus, the epidemiology of diseases caused by mutations in mtDNA is quite distinct from that of mitochondrial diseases caused by mutations in nuclear DNA. In a study with neonatal cord blood samples, looking for de novo mtDNA mutations, Elliott et al. found that at least 1 in 200 individuals harbor a pathogenic mtDNA mutation (156). Clinically, mitochondrial diseases present in 1 in 10,000 individuals, making them a major group of inherited human diseases (100), although it should be pointed out that the clinical classifications do not distinguish diseases caused by mutations in mtDNA from those caused by mutations in nuclear genes. Nevertheless, the extremely high frequency of mtDNA mutations in the general population also emphasizes the importance of the nuclear background in determining the clinical outcome of carrying a mtDNA mutation.
Mutations in mtDNA can occur at all regions, but disease-causing mutations are most often found in tRNA genes and in the protein coding genes. The first mutation in mtDNA causing a human syndrome was identified in 1988, in a patient suffering from Leber's hereditary optic neuropathy (541). This mutation was a base substitution in a highly conserved arginine in the ND4 subunit of Complex I. Since then, more than 300 pathogenic mutations have been identified in human mtDNA (207). However, it is noteworthy that most mtDNA mutations cause variant forms of a few complex syndromes, with overlapping phenotypes. For an updated and comprehensive review of clinical symptoms of mitochondrial diseases, please refer Greaves et al. (207).
As is the case for mitochondrial diseases caused by mutations in nuclear DNA, therapeutic approaches to diseases of mtDNA are scarce and often unsuccessful (447, 460, 461). Pharmacological or nutritional interventions similar to those discussed earlier have been tried. However, some effort has also been put into gene replacement therapy. The allotropic expression of a mammalianized copies (since mitochondria use a different genetic code from the nucleus) of Leber's hereditary optic neuropathy-mutated ND4 gene has been successful in restoring respiration in a cellular model of the disease (212). An efficient allotropic expression of the protein was also observed in the eyes of mice infected with viral constructs (211). Other approaches being tested include the manipulation of heteroplasmic levels of mutated mtDNA by expressing mitochondrially targeted endonucleases which recognize only the mutated molecules (18), and manipulation of signaling pathways that control mitochondrial function, such as the AMPK/PGC1-α axis (535). Another recent approach was the long-term expression of parkin, an E3-ubiquitin ligase that targets mitochondria to degradation (15) in cells with heteroplasmic COXI mutations (490). The authors observed a shift in mtDNA heteroplasmy toward the wild-type genomes, suggesting that the molecular mechanisms which maintain mitochondrial quality control may also be therapeutic targets in diseases with high levels of mtDNA mutations in heteroplasmy. These results are somewhat encouraging, but the feasibility of such approaches as effective human therapies remains to be determined. Nonetheless, given the high prevalence of mitochondrial diseases, and implication of mitochondrial dysfunction in more common pathologies, efforts to devise effective therapies should be a priority.
VI. Redox Imbalance and Cancer
Progressive changes in ROS and RNS production are observed during malignant transformation (498, 507), and experimental, clinical, and epidemiological evidence indicates that these reactive species are causally involved in malignant transformation and progression. Indeed, large association studies report a strong linkage between polymorphisms in antioxidant enzymes (e.g., GSTs and SOD2) and genetic predisposition to cancer (19, 153). Moreover, increased H2O2 and O2 •;− levels have been associated with tumor aggressiveness and poor disease outcome (289, 409).
The precise pathways leading to oxidative and nitrosative imbalance in tumor cells remain to be uncovered. Mitochondrial dysfunction and oncogenic stimulation by key oncogenes and tumor suppressor genes are intrinsic factors known to cause redox imbalance in tumor cells. In addition, extrinsic factors such as inflammatory cytokines, nutrient deprivation, and the hypoxic tumor environment have also recently been associated with oxidative imbalance in tumor cells (17, 169, 421).
The intrinsic mechanisms causing increased redox imbalance in tumor cells have been more comprehensively studied when compared with extrinsic mechanisms. In particular, the association between oncogenic stimulation and increased mitochondrially derived ROS has been well investigated. Activation of key oncogenes and inactivation of tumor suppressor genes (e.g., K-Ras, C-Myc, p53) are known to raise H2O2 and O2 •;− levels in tumor cells (245, 448, 518). In a recent study, Hu et al. (238) showed that K-Ras activation led to suppressed respiratory chain complex I activity and a decrease in the mitochondrial transmembrane potential, by affecting the cyclosporin A-sensitive MPT, leading to increased ROS production, decreased respiration, and elevated glycolysis (238). Moreover, gene expression studies on RAS-transformed fibroblasts revealed significant changes in the expression of nuclearly encoded genes involved in mitochondrial biogenesis and function (99). Interestingly, as we will see next, oncogenic activation also induces a feedback antioxidant response that enables tumor cells to escape severe oxidative damage (129).
Mitochondrially generated ROS and RNS have been historically associated with nuclear DNA damage and genome instability during malignant transformation (90). DNA mutation is a critical step in malignant transformation, and endogenous ROS and RNS account for a significant fraction of nuclear DNA damage in tumor cells. Elevated levels of oxidative DNA lesions have been noted in various tumors, and more than 100 oxidative DNA adducts have been identified to date (40, 340, 342). The most extensively studied oxidative DNA lesion is the formation of 8-OHdG (430), which has been considered a potential biomarker of carcinogenesis (519).
Although nuclear DNA is susceptible to ROS-mediated damage, mtDNA presents a more vulnerable target due to the lack of protective histones and its close proximity to the electron transport chain. As mentioned earlier, mitochondria from both lower and higher eukaryotes are proficient in repairing different oxidative DNA lesions and, recently, Kienhofer et al. (266) demonstrated the co-localization of mtDNA with both mitochondrial glutathione peroxidase (GPx1) and manganese superoxide dismutase (SOD2). Based on these observations, they proposed the existence of an integrated antioxidant system in the nucleoid structure of the mitochondria to further protect mtDNA from superoxide-induced oxidative damage (266).
Interestingly, the frequency of random mtDNA mutations seems to be ∼70% lower in tumors when compared with patient-matched normal tissues (162). By examining the spectrum of random mtDNA mutations in carcinomas, adenomas, and normal colonic samples, Ericson et al. (162) observed that oxidatively mediated mutations, such as C:G to T:A transitions, occured less frequently in the tumors samples and proposed that the lower frequency of random mtDNA mutations was associated with a decrease in ROS production in the mitochondrial matrix as a result of the metabolic shift from oxidative phosphorylation to glycolysis—a metabolic feature of tumors known as the “Warburg effect.” Alternatively, but not mutually exclusively, tumors might also have a more efficient nucleoid antioxidant system to protect mtDNA from superoxide-induced oxidative damage, although this remains to be addressed.
In contrast to random mutations, clonally expanded mtDNA mutations resulting in frameshifts or nonsynonymous substitions have been observed in most cancers. Of particular interest is the presence of mutations potentially affecting mitochondrial respiratory chain function in cancer patients, which can lead to leakage of electrons and the generation of O2 •;−. Inherited mutations affecting ND5, ND2, and ND1, CYTb and COX1 genes have been reported in esophageal squamous cell carcinomas and in breast cancer patients (330). Recently, Ishikawa et al. demonstrated that mitochondrial mutations compromising respiratory function favor cancer metastasis through the generation of mitochondrial ROS (246). Mutations affecting mitochondrial respiratory chain function, whether in the nuclear DNA or in the mtDNA, may result in increased ROS production, leading to a vicious cycle of increasing DNA damage and mitochondrial dysfunction (Fig. 10). However, the functional relevance of the majority of these alterations to tumorigenesis remains to be determined, as clonally expanded mtDNA mutations have also been frequently detected in adenomas, suggesting that expansion can occur before malignancy (162).

In addition to DNA damage and genomic instability, in the last decade, ROS have been shown to act as second-messenger activating signaling pathways and transcription factors directly involved in tumorigenesis (318, 550). During malignant transformation, tumor cells escape from senescence and apoptosis, survive as immortalized cells, and undergo uncontrolled proliferation to form primary tumors. At a latter step in malignant progression, tumor cells induce the formation of new blood vessels to support tumor growth, invade adjacent tissues, reach the circulation, and proliferate in distant organs, forming metastases. The multistep development of human tumors, thus, requires the acquisition by the tumor cells of biological capabilities known as the “hallmarks of cancer” (223). “Hallmarks of cancer” include replicative immortality, resistance to cell death, sustained proliferation, evasion to growth suppressors, induction of angiogenesis, activation of invasion and metastasis, deregulation of cellular metabolism, and evasion of the immune system (223). There has been an increased focus on the role of redox signaling in malignant transformation and progression, and evidence exists in the literature supporting the participation of redox signaling mediated by H2O2 in the acquisition of most of the hallmarks of cancer [(404, 549), Fig. 10].
Increased levels of ROS have been shown to activate different pathways important for tumor cell proliferation and survival. H2O2 and O2 •;− accumulate in the cell following the activation of tyrosine kinase growth factor receptors (EGF, PDGF, and VEGF) by cognate ligands. Growth-factor-stimulated ROS generation can mediate intracellular signaling pathways by inhibiting protein tyrosine phosphatases (e.g., PTEN), activating several nonreceptor protein tyrosine kinases involved in malignant transformation and progression (e.g., the Src family, Janus kinase, Mitogen-Activated Protein Kinase [MAPK], Extracellular Signal-Regulated Kinase and Protein Kinase B), and regulating redox-sensitive transcription factors (395, 521, 550). However, in many of the reported studies, growth-factor-induced ROS generation is considered to be mainly via NAD(P)H oxidase and a specific role for mitochodrially generated ROS remains to be demonstrated.
Increased levels of ROS have also been shown to activate different pathways, leading to angiogenesis, deregulation of cellular energetics, and metastasis. Central to these pathways are the HIF transcription factors, consisting of HIF1A and HIF1B subunits (544). The latter is stable under normal oxygen levels, while HIF1A is hydroxylated by prolyl hydroxylases under the same conditions, and subsequently ubiquitinated by the HIF-specific Von Hippel-Lindau (VHL) ubiquitin ligase, targeting HIF1A for proteasome-mediated degradation (247). Mitochondrially derived H2O2 is involved in the stabilization of HIF1A and activation of HIF transcriptional activity under low oxygen conditions, through the inactivation of the prolyl hydroxylases, which target HIF1A for proteasome-mediated degradation (269).
Once stabilized, HIF1A dimerizes with HIF1B and regulates the expression of genes mediating angiogenesis, metastasis, and cellular metabolism (467). HIF1A stabilization is a major stimulus for increased vascular endothelial growth factor (VEGF) production and the coordinated expression of other angiogenic factors by tumor cells. Changes in cell polarity, adhesive properties, and motility have also been associated with HIF transcriptional activity via the up-regulation of c-Met/HGF-R and the matrix-modifying enzyme lysyl oxidase. Finally, HIF transcriptional activity is also responsible for increased expression of glucose transporters, glycolytic enzymes (e.g., hexokinase and lactate dehydrogenase), and pyruvate dehydrogenase kinase, promoting a global shift in cellular metabolism from oxidative phosphorylation to glycolysis and providing tumor cells with important metabolic intermediates (nucleic acids, proteins, and lipids) that are necessary for cell proliferation and tumor growth (468).
Since tumor cells actively produce high levels of ROS and are continuously exposed to endogenous oxidants, it is not surprising that they also develop mechanisms to protect themselves from intrinsic oxidative stress. Higher levels of ROS-scavenging enzymes (e.g., SOD, GPx, and Prx) and antioxidant molecules have been observed in tumors compared with normal tissues (239, 250). Moreover, treatment of normal epithelial cells with low levels of exogenous oxidants has been shown to confer cellular resistance to subsequent oxidative challenges (104). These observations support the idea that persistent ROS stress may induce an antioxidant response, enabling cancer cells to escape severe oxidative damage. Recent evidence also suggests that such adaptation contributes to malignant progression and resistance to anticancer therapy (297, 412, 463, 556).
ROS levels are tightly controled by an inducible antioxidant program regulated by the nuclear factor-erythroid 2-related factor 2 (NRF2) and its cytosolic repressor protein Kelch ECH associating protein 1 (KEAP1). NRF2 is a transcription factor that binds to antioxidant response elements and regulates the expression of antioxidant enzymes and detoxifying enzymes such as Prx, thioredoxin, catalase, Cu/Zn SOD, glutathione S-transferase A2, and NADPH quinone oxidoreductase 1 (378). Under normal conditions, NRF2 transcriptional activity is associated with and suppressed by KEAP1, which binds to NRF2 and promotes its proteasome-mediated degradation, keeping antioxidant gene expression tightly regulated. However, on ROS exposure, KEAP1 undergoes a conformational alteration, freeing NRF2 to translocate to the nucleus and driveing the expression of antioxidant and detoxifying enzymes (324). Increased NRF2 activity in tumors can be promoted by somatic mutations that disrupt NRF2-KEAP1 interaction or by oncogenic activation as previously mentioned. In a recent study, DeNicola et al. (129) observed that endogenous expression of the KRAS and BRAF oncogenic alelles increased NRF2 transcription, activating an antioxidant program and lowering intracellular ROS levels.
Since tumor cells have higher levels of ROS when compared with normal cells, they should be, at first sight, more vulnerable to death by ROS-promoting agents. Therefore, increasing ROS levels could represent an effective way to selectively kill cancer cells without causing significant toxicity to normal cells and undesirable side effects. Indeed, many commonly used anti-cancer drugs such as doxorubicin, paclitaxel, and platinum-based drugs promote ROS-mediated cell killing. However, redox adaptation, through the activation of anti-oxidants mechanisms, confers tolerance to exogenous stress and contributes to the drug-resistant phenotype of tumor cells [(297, 412, 463), Fig. 10]. The thioredoxin system, for example, is up-regulated in tumor cells, and elevated levels of this thiol-based antioxidant system correlate with poor prognosis and drug resistance (85). Thus, the use of compounds that abrogate the antioxidant response in tumor cells represents an alternative strategy to selectively kill cancer cells. This concept has recently been proved by Raj et al. (428) in a search for small molecules that selectively kill cancer cells. Using small-molecule screening and quantitative proteomics, they found that Piperlongumine, a natural product isolated from the plant species Piper longum L., increased ROS levels and apoptotic cell death in cancer cells. Piperlongumine also exhibited in vivo antitumor and antimetastatic effects in a mouse model (428).
Indeed, compounds that either enhance ROS generation or abrogate key antioxidant mechanisms in tumor cells have been used in combination with radio- and chemotherapy to improve therapeutic response and to circumvent chemotherapy resistance, with a significant clinical impact [(357, 463), Fig. 10]. Several compounds that promote ROS generation such as mitochondrial electron transport chain modulators (e.g., arsenic trioxide) and redox-cycling compounds (e.g., motexafin gadolinium), or which abrogate antioxidant mechanisms such as GSH-depleting agents (e.g., buthionine sulphoximine) and SOD inhibitors (e.g., 2-methoxyestradiol), are currently being used in clinical trials for different types of cancer (357). Motexafin gadolinium is being used in phase II clinical trials for treating refractory chronic lymphocytic leukemia and brain metastases (316). The combination of arsenic trioxide and ascorbic acid-mediated GSH depletion was also shown to improve treatment efficacy in refractory multiple myeloma (20), and phase I trials combining the use of Imexon—a GSH-depleting compound—and docetaxel are ongoing for lung, breast, and prostate cancer (362). Finally, brusatol, a compound found in plant extracts, increases ubiquitination of NRF2, inhibiting its activity and rendering xenografts more sensitive to the chemotherapeutic agent cisplatin (432).
Improving therapeutic selectivity is a major goal in the development of novel anti-cancer drugs, and exploration of the biochemical differences between normal and tumor cells leads to the emergence of several molecular-targeted drugs (e.g., Herceptin, Avastin, Gleevec), with enhanced therapeutic activity and fewer side effects (214). As we saw in this section, increased levels of ROS, coupled to redox adaptation to protect tumor cells from intrinsic oxidative imbalance, are responsible for the appearance and maintenance of the cancer phenotype. Targeting these biochemical properties of tumor cells with redox-modulating compounds will likely prove in the near future to be an effective way to selectively kill tumor cells and to circumvent drug resistance.
VII. Neuronal Damage and Disorders Associated with Mitochondrially-Generated ROS
Since neurons are longlived and highly dependent on oxidative metabolism, they are uniquely predisposed to accumulate injury by endogenously produced ROS. Even under nonpathological conditions, human brain tissue presents cumulative effects of ROS, described early on in anatomopathological evaluations of elderly brains as accumulation of granules of lipofuscin, a yellowish-brown pigment composed of protein and lipid oxidation products and localized within the lysosomes of cells [for review, see (62)]. An important role for mitochondrially generated ROS under nonpathological conditions in the accumulation of brain oxidative products was recently demonstrated in mice chronically treated with the mitochondrial uncoupler dinitrophenol (72). A decreased accumulation of protein carbonyls and 8-OHdG in brain tissue was observed after 1 or 5 months of continuous treatment of mice with dinitrophenol in the drinking water. Although an increased survival was observed in dinitrophenol-treated mice (72), future studies seek to evaluate whether these mice will perform better in behavioral tests.
An indication for mitochondrial uncoupling as an important regulatory mechanism in the central nervous system was the characterization of three mitochondrial uncoupling proteins (UCP2, UCP4, and UCP5) in brain tissue. Furthermore, rats submitted to a 3 min period of sublethal ischemia, which leads to ischemic preconditioning, showed an increased expression of UCP2 in hippocampal CA1 field (346), indicating a role of UCP2 in neuroprotection. In fact, several studies demonstrated that UCP2 overexpression is associated with neuroprotection in experimental models of ischemia (35, 126, 346), seizures (135, 491), and trauma (346). In addition, mice overexpressing UCP2 present decreased 1-methyl-4-phenyl-1,2,5,6 tetrahydropyridine (MPTP)-induced nigral dopaminergic cell loss (10). UCP4 (320, 549) and UCP5 (267, 290) have also been associated with neuroprotection. In this framework, mitochondrial redox imbalance has been implicated in neural cell death associated with various disorders. Next, we will present evidence for the involvement of mitochondrial redox imbalance in stroke and Parkinson's disease.
Brain lesion after stroke is not an immediate process. Within the penumbra region, cell death becomes prominent between 24 and 72 h after reperfusion after a 30 min period of middle cerebral artery occlusion in mice (142, 160). Redox imbalance may be one of the events that determine neuronal cell death in the penumbra region. In fact, early experimental evidence indicates that some antioxidants, such as lazaroids and N-tert-butyl-alpha-phenylnitrone (220, 416), are potent protective agents in animal models of brain ischemia.
In addition to different systems for oxidant generation, such as NOS, NADPH oxidase, and the cyclooxygenase and lipoxygenase pathways, mitochondria are also a source of ROS in stroke. In fact, hippocampal mitochondria isolated after transient global ischemia show an increased production of ROS at time points that precede neuronal death (185). In addition, some reports have provided in vivo evidence of increased mitochondrial ROS generation in experimental models of brain ischemia using microdialysis and the salicylate trap (417) or hydroethidine oxidation (268) techniques. Mitochondrial aconitase, a citric acid cycle enzyme inactivated by O2 •;− (190), has also been found to be inhibited in cortical neuron cultures subject to oxygen-glucose deprivation (312).
An impairment of the respiratory complexes may be the underlying cause of higher mitochondrial production of ROS in stroke [for a review, see (361); Fig. 11]. There are several studies indicating dysfunction of mitochondrial respiratory chain complexes I-IV and ATP synthase in experimental models of brain ischemia/reperfusion (7, 376, 426, 475). Respiratory chain complexes may be the target of RNS or ROS (356, 361). Activation of nonmitochondrial ROS generation during brain ischemia or reperfusion may lead to the formation of these reactive species.

One of the early triggers of cellular and mitochondrial dysfunction during stroke is excitotoxicity (444) (Fig. 11), a central nervous system process in which an increased glutamate release due to energy deprivation results in neuronal necrosis or apoptosis (176). This glutamate-mediated neuronal cell death is promoted mainly by activation of N-methyl-D-aspartate receptors, resulting in Ca2+ and Na+ influx (103, 444). The involvement of redox imbalance in excitotoxicity was demonstrated by several studies showing the protective effect of antioxidants in in vitro (293, 410) or in vivo experimental models (368, 462). Under excitotoxic conditions, mitochondria are the main organelles responsible for Ca2+ sequestration (as discussed earlier), an event associated with neuronal cell death as long as inner membrane potentials are maintained by respiration or ATP hydrolysis (82, 382).
As previously discussed in this review, increased Ca2+ concentrations in the mitochondrial matrix may induce mitochondrial redox imbalance and MPT (Fig. 4). Schild and Reiser (458) reported that brain mitochondria subjected to hypoxia/reoxygenation in the presence of low micromolar Ca2+ concentrations generated higher levels of H2O2 during reoxygenation. Mitochondrial Ca2+ accumulation may also result in a reversible inhibition of H2O2 elimination (509). In addition, Vaseva et al. (523) have shown that p53 accumulates in mitochondria and trigggers MPT in response to redox imbalance during brain ischemia. In fact, neuronal death after hypoglycemia and brain ischemia is prevented by MPT inhibitors (184, 344). However, the participation of MPT in excitotoxicity is controversial (63, 82, 313, 333). A possible explanation for the lack of or limited participation of MPT in several experimental models of excitotoxicity is the presence of high levels of endogenous inhibitors of this phenomenon, such as ADP and Mg2+ (449).
Finally, mitochondrial ROS production can also comprise a protective signal for brain organelles in stroke. We showed that ROS produced at the respiratory chain activate brain mitochondrial ATP-sensitive K+ channels, a process associated with protection against excitotoxicity (178).
Parkinson's disease is characterized by rigidity, resting tremor, bradykinesia, and postural instability, accompanied by predominant degeneration of dopaminergic neurons in the substantia nigra. Most cases of Parkinson's disease are considered idiopathic and sporadic. In addition to altered protein handling and inflammation, mitochondrial dysfunction and redox imbalance have been indicated as important factors in the pathogenesis of Parkinson's disease [for review, see (74, 453)].
Postmortem studies have provided evidence of a partial inhibition (20%–40%) of mitochondrial respiratory chain complex I (454, 455) and oxidative damage (130, 167) in brains from idiopathic Parkinson's disease patients. Respiratory complex I inhibition was also observed in peripheral tissues (45, 408). Although the origin of respiratory chain complex I deficiency is unknown, it may be related to cumulative mtDNA deletions (36). The importance of complex I inhibition in Parkinson's disease pathology is also supported by the fact that humans exposed to MPTP, whose metabolite 1-methyl-4-phenylpyridine (MPP+) inhibits complex I (383), develop parkinsonism (298). In addition, Greenamyre's group also showed that systemic treatment of rodents with the classical complex I inhibitor rotenone leads to anatomical, neurochemical, behavioral, and neuropathological features of Parkinson's disease (42, 75).
The mechanism involved in complex I inhibition-mediated dopaminergic neurodegeneration probably involves increased mitochondrial ROS generation [(29, 453, 488); Fig. 12]. Complex I inhibition in isolated brain mitochondria results in a marked stimulation of mitochondrial H2O2 production through a mechanism linked to an increased NAD(P)H/NAD(P)+ ratio (488). In addition, a partial inhibition of complex I activity can contribute toward excitotoxicity in a mechanism that involves impaired mitochondrial oxidative phosphorylation (557).
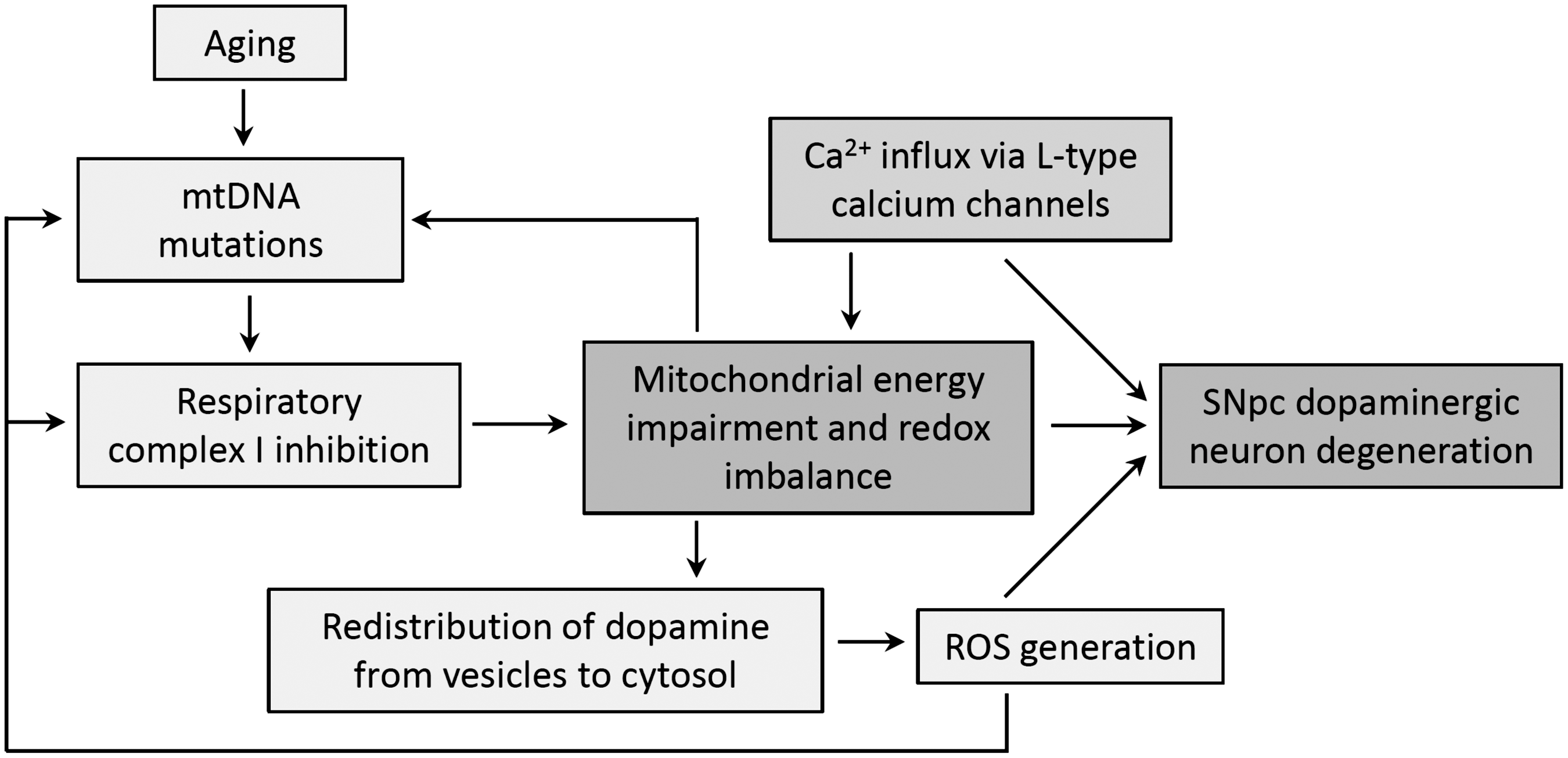
Why dopaminergic neurons are more sensitive to complex I inhibition is still unknown; of note, dopaminergic and nondopaminergic presynaptic synaptosomes present similar bioenergetic capacities (105). One possibility would be that an increased mitochondrial ROS production due to complex I inhibition may lead to dopamine redistribution from vesicles to the cytosol (546). Dopamine is readily oxidized in the cytosol into H2O2, O2 •;− and various dopamine metabolites (327). The higher susceptibility of substantia nigra pars compacta dopaminergic neurons to degeneration may also be related to increased cytosolic Ca2+ concentrations due to Ca2+ influx via L-type calcium channels [(91, 494); Fig. 12]. In this regard, Ca2+ and complex I inhibition synergistically increase brain mitochondrial ROS release and neuronal death (482, 483), in a mechanism associated with MPT induction (537).
Parkinsonism can also be associated with disturbance in mitochondrial quality control (371). Mitochondrial membrane depolarization leads to an increased expression level of PINK1 on the outer mitochondrial membrane (343, 371, 536) and accumulation of PINK1 signals toward the recruitment of parkin from the cytosol to the impaired (depolarized) mitochondria (370). Parkin, an E2 ubiquitin ligase, promotes the ubiquitinization of outer membrane proteins, ultimately leading to degradation of the organelle by autophagy (371). A failure in this process may explain neurodegeneration observed in the familiar form of Parkinson's disease due to loss-of-function mutations within the PARK2 locus, which encodes parkin. In addition, Johnson et al. (257) have shown that parkin has an antiapoptotic role by ubiquitinating and, consequently, inactivating the proapoptotic protein BAX in the cytosol.
VIII. Redox Imbalance in Cardiovascular Diseases
Cardiovascular complications are a major public health problem; acute myocardial infarction and heart failure are leading causes of morbidity and death worldwide. According to the World Health Organization, more than 7 million people die of ischemic heart disease every year. Consequently, fundamental mechanisms responsible for the establishment and progression of cardiovascular diseases, as well as the generation of novel pharmacological and nonpharmacological interventions, should be extensively studied. In this section, we describe the critical role of mitochondrial metabolism and redox signaling in cardiac pathophysiology.
Mitochondria have emerged over the last decades as the critical organelle in maintaining redox balance and in regulating survival and death pathways in cardiomyocytes (236). Considering their high demand for ATP synthesis and elevated oxygen uptake rate, cardiac cells have the highest volume density of mitochondria in the entire body (512)—mitochondria constitute 30% of myocardial mass (98). In cardiovascular diseases, mitochondria in the heart (i.e., cardiomyocytes and endothelial cells) are thought to be the major source of ROS and RNS (351, 391, 399). Specific forms of ROS and RNS generated during acute (ischemia-reperfusion injury) or chronic (hypertension and heart failure) cardiovascular diseases by dysfunctional mitochondria include H2O2, O2 •;−, NO•, and ONOO−. These species can react with membrane phospholipids, DNA, and proteins, which, in turn, exacerbate mitochondrial dysfunction, oxidative stress, and accumulation of lipid reactive species (i.e., 4-HNE) in a feed-forward loop. This cyclic process results in protein aggregation, cellular collapse, and decreased cardiac viability (107, 401).
A. Redox imbalance during ischemia-reperfusion
Acute myocardial infarction is characterized by changes in biochemical properties during ischemia and reperfusion. The heart can survive a short period of ischemia by reducing myocardial contractility, increasing glucose uptake, and switching metabolism to glycolysis. However, considering that the heart is one of the most energy-demanding tissues in the body, sustained oxygen and nutrient deprivation results in irreversible damage. Thus, reperfusion of the ischemic heart is a prerequisite for survival. Paradoxically, reperfusion can further increase the myocardial damage that occurs during ischemia. The severity of reperfusion injury depends on the duration of the preceding ischemia and the effectiveness of blood flow during reperfusion. Several lines of evidence demonstrate that reperfusion injury is directly associated with cardiac mitochondrial dysfunction and increased ROS and RNS generation (95, 96). Hearse et al. (228) noted that reperfusion of isolated hearts after ischemia resulted in abrupt cardiomyocyte death. Following this paper, several studies showed that ischemia reperfusion is associated with a burst of H2O2, O2 •;−, NO•, and ONOO−, but the exact mechanism of their generation is debated. Although some ROS may be generated by NADPH oxidase and xanthine oxidase, it is likely that complexes I and III of the mitochondrial respiratory chain are the main sources of ROS during myocardial ischemia reperfusion (95, 405). In fact, studies using mitochondrial respiratory inhibitors show that the electron leak along the oxidative phosphorylation most likely occurs at the Fe-S centers of complex I and at some components of complex III (70, 323, 389). During the early stages of reperfusion, ROS generation levels increase drastically (477). Interestingly, low amounts of ROS generated by mitochondria during brief and intermittent episodes of ischemia, termed ischemic preconditioning, significantly protect the heart against prolonged ischemia (364).
Ischemic preconditioning-mediated cardioprotection is driven by activation of cytosolic, nuclear, and mitochondrial signaling pathways. Data indicate that many of these intracellular signals converge to the mitochondria, resulting in lower mitochondrial oxidative damage during prolonged ischemia and reperfusion events (364). Interestingly, ischemic preconditioning benefits are lost by a co-intervention with antioxidants, an evidence of its redox signaling nature. Several studies have demonstrated that avoiding exacerbated ROS and RNS generation by preconditioning is a crucial step in protecting mitochondrial key proteins against oxidation during ischemia and reperfusion (73, 364). Although the functional benefits of ischemic preconditioning are well described, the underlying molecular mechanisms are still uncertain. Of interest, activation and further mitochondrial translocation of specific cytosolic kinases such as PKCɛ, ERK, and AKT seems to play a key role in priming organs against sustained ischemic events (66, 354).
Over the last decade, oxidative protein post-translational modifications such as oxidation and nitration have been associated with cardiac ischemia-reperfusion-related damage (514, 565). Conversely, NO•-mediated protein S-nitrosylation plays a pivotal role in preconditioning-mediated cardioprotection (399, 505). During a prolonged ischemia-reperfusion episode, exacerbated ROS generation oxidizes key proteins involved in cellular metabolism, contractility, and protein quality control in the heart, favoring cell dysfunction and death (399). Moreover, O2 •;− reacts with NO•, forming ONOO−, a key effector of cardiomyocyte cell death (328). Peroxynitrite affects cardiomyocyte viability through different mechanisms, including mitochondrial dysfunction, DNA damage, activation of poly(ADP-ribose) polymerase, and protein inactivation (reacting with tyrosine and thiol groups) (328, 398). However, at low concentrations, even ONOO− has been reported to be cardioprotective through the reaction of its subproducts NO2 − and NO3 − with glutathione, forming S-nitrosoglutathione (73).
Excessive ROS and RNS production can induce MPT and cell death (551). ROS-induced MPT collapses the mitochondrial membrane potential, increases ROS and RNS release, triggers mitophagy, and results in cardiomyocyte death, characterizing a positive feedback loop of ROS-induced ROS release [(287); Fig. 13]. In this case, mitophagy functions as an early cardioprotective response, removing damaged mitochondria. Zorov et al. (569) have shown that intracellular photoactivation of tetramethylrhodamine compounds triggers exacerbated ROS release in isolated cardiomyocytes, which results in mitochondrial depolarization, along with prolonged MPT opening and a consequent burst of ROS and RNS from mitochondria.

Despite the increased knowledge regarding the molecular basis of mitochondrial ROS and RNS generation and redox balance in cardiovascular diseases, there is still uncertainty in the literature regarding the cellular node/transducer molecules responsible for communicating mitochondrial and cytosolic signals during pathological conditions such as ischemia-reperfusion injury. The serine threonine protein kinase C family (PKC) is a good candidate for mediating the cross-talk between mitochondria and the cytosol signaling under cardiac redox imbalance conditions, as PKC contains domains that are susceptible to oxidative modification, including zinc-binding and cysteine-rich motifs (206). Among different PKC isozymes expressed in the heart, δPKC seems to play a key role in ischemia-reperfusion injury (Fig. 13).
The first evidence for hydrogen peroxide-induced δPKC translocation from the cytoplasm to the mitochondria was provided by Majumder et al. (336). In fact, mitochondrial translocation of δPKC was associated with the loss of mitochondrial membrane potential and the release of cytochrome c. The biological relevance of this phenomenon was supported by the demonstration that H2O2-induced apoptosis was blocked by the inhibition of δPKC translocation to mitochondria. Since then, several publications have described the role of δPKC in aspects of mitochondrial biology, such as ROS generation, cytochrome c release, and phosphorylation. Given the effects of δPKC on mitochondrial function, it could be targeted in therapeutic interventions against ischemia-reperfusion injury. Interestingly, selective inhibition of δPKC using the peptide inhibitor δV1-1 reduces ischemia-reperfusion injury in cardiac myocytes, ex vivo Langendorff-perfused hearts, and in animal models of acute myocardial infarction (244). Moreover, intracoronary delivery of δV1-1 during cardiac reperfusion may promote myocardial protection in patients with acute ST-elevation myocardial infarction (33).
Reperfusion leads to accumulation of δPKC within the mitochondria (sixfold increase), which results in reduced ATP levels, increased mitochondrial Bad/Bcl-2 ratios, elevated proapoptotic cytochrome c release, PARP cleavage, and DNA fragmentation (107, 366). Inhibition of δPKC translocation during cardiac reperfusion leads to a faster recovery of ATP levels in the heart (244). Nguyen et al. (379) demonstrated that δPKC inhibits F1Fo activity via an interaction with the “d” subunit of F1Fo-ATP synthase in neonatal cardiomyocytes. Treatment of cardiomyocytes with selective peptides that block the interaction between δPKC and F1Fo-ATP synthase abolishes the phorbol ester (a nonselective PKC activator)-induced inhibition of F1Fo-ATP synthase activity (379).
δPKC also regulates mitochondrial morphology through regulation of the Drp1, a large GTPase protein required for mitochondrial fission in mammalian cells. A number of stimuli lead to the translocation of Drp1 from the cytosol to the mitochondria, where it binds to Fis1, a protein located in the mitochondrial outer membrane. Cell culture studies demonstrated that excessive mitochondrial fission is associated with mitochondrial dysfunction and apoptosis in neurons (31). Activation of δPKC induces aberrant mitochondrial fragmentation and mitochondrial dysfunction in neuronal cells and in a rat model of hypertension-induced encephalopathy (424). During oxidative imbalance, δPKC promotes mitochondrial fission by interacting with and phosphorylating Drp1 (424), resulting in further increased Drp1 GTPase activity. Importantly, inhibition of δPKC using either pharmacologic or molecular tools reduced mitochondrial fission and fragmentation and conferred protection in cultured neurons and in the brain (424). During cardiac ischemia–reperfusion, mitochondria also undergo excessive fragmentation, which is associated with decreased mitochondrial membrane potential, opening of MPT pore, and apoptosis (394). However, the role of δPKC in this process remains to be clarified.
Important insights into ROS-mediated δPKC activation have been derived from findings that ROS induces δPKC phosphorylation (Tyr-512 and Tyr-523) in a lipid cofactor-independent manner (274). Moreover, S-cysteinylation or glutathione depletion results in δPKC activation-induced apoptosis in response to H2O2. Recently, Zhao et al. (566) demonstrated that both lipid agonist binding and oxidation release the zinc-finger centers of PKC, which contributes to its activation (566). This process seems to coordinate mitochondrial δPKC activation through a δPKC/p66Shc/vitamin A/cytochrome c complex (2). Therefore, ROS-mediated δPKC activation may be a key factor in reperfusion injury. In addition, selective activation of δPKC stimulates ROS generation. Inhibition of δPKC translocation to mitochondria completely abolishes ROS generation in acute renal failure (271). In the heart, inhibition of δPKC translocation to mitochondria during the first minutes of reperfusion blocks δPKC-mediated impaired mitochondrial function and increased ROS production (458). In δPKC knockout mice, ischemic preconditioning fails to produce ROS and shows exacerbated postischemic damage, which is related to a decreased anti-oxidant capacity in these mice (308, 348). These findings suggest that the redox-sensitive δPKC works in a feed forward loop to regulate mitochondrial ROS production and may provide an important switch, allowing the system to respond to ischemia reperfusion in a graded fashion (Fig. 13).
To date, cardiac mitochondrial ROS-generating targets for δPKC have not been clearly identified. There are several interesting candidates, including the NADPH oxidase-like activity of complex I (567), pyruvate dehydrogenase kinase 2, and cytochrome c (321). Overall, translocation of δPKC to mitochondria is a hallmark of cardiac injury after ischemia reperfusion. At the early stages of reperfusion, ROS levels quickly increase within the cardiomyocyte, which can activate δPKC through oxidative modifications. δPKC translocates to mitochondria, impairs mitochondrial metabolism, and amplifies ROS generation through the following mechanisms: activation of pyruvate dehydrogenase kinase 2, which can block complex I activity (108); activation of the NADPH oxidase-like activity of complex I; and activation of the phospholipid scramblase 3, which increases cardiolipin oxidation and cytochrome c derangement and release. These changes would be expected to increase cellular oxidative imbalance, further activating δPKC and resulting in a positive feedback loop. Thus, δPKC could be defined as a redox node/transducer, modulating a tunable system in which cell damage only occurs if the initial stimulus is large enough to evoke a prolonged response.
Another protein kinase C isozyme (ɛPKC) has been considered a key regulator of cellular signal transduction involved in ischemic preconditioning-mediated cardioprotection. Analysis of ɛPKC subcellular distribution in rodents overexpressing a constitutively active cardiac ɛPKC revealed that ɛPKC forms signaling complexes with a number of mitochondrial proteins (25). Further, in wild-type mice hearts, ɛPKC was colocalized in the mitochondrial vicinity; whereas in constitutively active ɛPKC mice or as a result of ischemic preconditioning, ɛPKC was present in the mitochondrial matrix (64). Several mitochondrial targets of ɛPKC have been described over the last years, including those regulating ion transport, MPT, the electron transport chain, and ROS generation (25, 65, 95, 171). Finally, one of the cardioprotective mechanisms mediated by ɛPKC is an increased aldehyde dehydrogenase 2 (ALDH2) activity during ischemia, which increases the capacity to metabolize the excess of cytotoxic reactive aldehydes (i.e., 4-HNE) derived from mitochondrial dysfunction-mediated ROS generation (95).
B. Redox imbalance in hypertension
Although ROS work as intracelular messengers in the healthy vasculature by promoting short-term signaling events, they are also involved in the establishment of diseases related to vascular dysfunction, such as hypertension and atherosclerosis (26).
Hypertension is a degenerative disease characterized by endothelial dysfunction and neurohumoral disruption of vascular tone. Irrespective of its etiology, hypertension has been associated with increased H2O2, O2,•− and ONOO− generation along with lower NO• bioavailability in the vasculature. At the cellular level, this redox status is closely related to activation of protein kinases (δPKC, MAPK, and tyrosine kinases), transcription factors (AP-1 and HIF1), and proinflammatory genes (358), which further contribute to elevated peripheral resistance and increased blood pressure.
Since recent studies have implicated a role for H2O2 and O2 •;− in the pathogenesis of hypertension (138, 146), an extensive interest in finding the main source of ROS in the cells of vasculature wall has emerged. Among different nonenzymatic and enzymatic systems that generate free radicals, hypertension-related ROS production is mainly associated with NADPH oxidase, xanthine oxidoreductase, mitochondrial enzymes, and uncoupled NO•synthase (138). Haque and Majid (226) have recently shown that mice lacking the gene for gp91PHOX, a subunit of NADPH oxidase, are more resistant to high salt-diet-induced hypertension. Moreover, Doughan et al. (146) and Wosniak et al. (555) demonstrated a cross-talk between NADPH oxidase and mitochondrial O2 •;− production in angiotensin-II-exposed endothelial cells. Elucidating data on the role of mitochondrial oxidative stress in hypertension were provided by Dikalova et al. (140). These authors showed that in vivo treatment with mitoTEMPO (a mitochondrially targeted antioxidant) or overexpression of SOD2 reduced mitochondrial O2 •;− generation and NADPH oxidase activity in hypertensive animals. These changes were followed by increased vascular NO•availability, improved endothelial-dependent vasorelaxation, and promoted a 30 mmHg drop in blood pressure (140). Administration of TEMPOL (a nontargeted antioxidant) did not promote the same effects.
C. Redox imbalance in heart failure
Heart failure is a clinical syndrome characterized by cardiac dysfunction and neurohumoral activation, which further leads to low cardiac output, fluid retention, and decreased survival. A broad variety of defects in respiratory chain machinery and components of oxidative phosphorylation complexes has been related to the progression of heart failure (442). In fact, reduction of mitochondrial oxidative phosphorylation is associated with an increase in electron leakage and O2 •;− generation in complexes I and III, as discussed earlier. Recent clinical and experimental studies have demonstrated that mitochondrial H2O2 and O2 •;− generation directly contributes to the pathophysiology of cardiac remodeling and heart failure (3, 53). Sustained increases in H2O2, O2,• − and ONOO− generation in failing cardiomyocytes are catastrophic to the heart, as they result in mtDNA damage, activation of a broad variety of kinases, and transcriptional factors related to cardiac hypertrophy and disruption of excitation-contraction coupling (401) (Fig. 14). Generation of excessive H2O2 stimulates mast cell degranulation, cardiac inflammatory response, and fibroblast proliferation, along with activation of matrix metalloproteinases (401). These processes are tightly related to the development and progression of pathological cardiac remodeling and failure.

In the setting of haemodynamic overload, Oka et al. (393) recently demonstrated that damaged mitochondria which escape from autophagy are capable of inducing cardiac inflammation and dilated cardiomyopathy, linking mitochondrial damage to other features of heart faillure. The mechanism behind this process is very interesting, as it involves mtDNA (leaked from damage mitochondria) activation of Toll-like receptor 9. Worth noting, due to the endosymbiotic origin of mitochondria, mtDNA is similar to bacterial DNA and can trigger a sterile inflammation. So, disrupted mitochondria could be involved in the genesis of chronic inflammation in failing hearts (393).
Overexpression of antioxidant enzymes such as SOD or GPx confers resistance against myocardial oxidative damage and heart failure in mice (345). These effects were related to the attenuation of cardiomyocyte hypertrophy, inflammation, and interstitial fibrosis, which contributes to improved survival in animals. Similarly, overexpression of either GPx or SOD reduced the severity of diabetic cardiomyopathy in animals (391, 512). Conversely, cardiac-specific SOD-deficient mice developed heart failure associated with defects in mitochondrial respiration and excess O2 •;− formation (391).
Although early efforts focused on the harmful effects of ROS and RNS in the failing myocardium, recent work revealed that accumulation of cytotoxic lipid peroxides derived from ROS-mediated stress can impair cardiac function [(97); Fig. 14]. Excessive peroxidation of phospholipids localized within the inner mitochondrial membrane (i.e., cardiolipin) changes the composition of the cardiac mitochondrial membrane and results in a drastic decline in the rate of oxidative phosphorylation and ATP synthesis in heart failure (442). Heart failure patients present increased circulating lipid peroxide levels, and the concentration of circulating lipid peroxides level such as 4-HNE and malondialdehyde inversely correlates with left ventricular function (338).
Beside its effect on mitochondrial membrane composition, augmented ROS-mediated lipid peroxidation during heart failure results in extensive generation and accumulation of toxic aldehydes, which can form adducts with lipids, DNA, and proteins, and negatively affect the function of these macromolecules (171). Selective activation of the mitochondrial enzyme responsible for removing these aldehydes, ALDH2, protects the heart against pathological remodeling and failure (493).
Recent evidence highlights monoamine oxidases as another important source of mitochondrial H2O2 in cardiovascular diseases, including ischemia-reperfusion injury, cardiac hypertrophy, and heart failure (73, 261). Monoamine oxidases are a class of flavoenzymes located in the outer mitochondrial membrane responsible for oxidative deamination of neurotransmitters such as epineprine, norepinephrine, and dopamine. The products of monoamine oxidases are aldehydes, ammonia, and H2O2 (261). In experimental animals, the pharmacological inhibition of monoamine oxidase or its genetic ablation protects the heart against acute (i.e., ischemia-reperfusion injury) and chronic (i.e., heart failure) insults (44, 262). The likely benefits of monamine oxidase inhibiton under such conditions may also include higher cathecolamine availability in addition to lower H2O2 formation (261).
Altogether, these findings highlight the central role played by mitochondria in regulating different forms of cardiovascular diseases. Mitochondria do not only adapt their metabolism to cope with the demand for energy and substrates, but also influence cellular morphology, function, and survival in an ATP-independent manner, through the excessive generation of oxidants in the damaged heart. Finally, the abnormal generation of ROS and RNS, as well as their byproducts, clearly contributes to the establishment and progression of cardiovascular diseases in both animal models and humans.
IX. Final Remarks
Overall, we discussed here a wealth of mechanisms that promote and regulate mitochondrial oxidant production, as well as the consequences of this production on mitochondrial and cellular signaling events and within diseases. Given the enormous progress in this filed in the last few years, it will certainly be exciting to see what future studies will uncover in this field.
Footnotes
Acknowledgments
Financial support has been rendered by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Instituto Nacional de Ciência e Tecnologia de Processos Redox em Biomedicina (INCT Redoxoma), Núcleo de Apoio à Pesquisa de Processos Redox em Biomedicina (NAP Redoxoma), and The John Simon Guggenheim Memorial Foundation. T.R.F. is currently supported by a postdoctoral FAPESP fellowship.