
Abstract
Introduction
Redox Homeostasis in Leishmania
Reactive oxygen species (ROS) in Leishmania are generated by means of cellular metabolism, uncoupled electron transfer in mitochondria, drug metabolism, exogenous agents, etc. Right after phagocytosis, as the first line of defense, the macrophage produces ROS, like superoxide anion (O2 •−) and H2O2, with the help of NADPH oxidase, a phenomenon known as oxidative burst. Once the infection is established, the proinflammatory cytokines like tumor necrosis factor-α (TNF-α) and interferon-γ activate the macrophage to produce reactive nitrogen species (RNS) by upregulating inducible nitric oxide synthase expression (25). Also, the production of hypohalide by myeloperoxidase or eosinophil peroxidase and peroxynitrite (RNS) as secondary oxidants is observed as a result of oxidative burst (21). Small (physiological) amounts of ROS are a cellular requirement, because they are involved in signaling pathways, inducing and regulating a variety of cellular activities for example, growth, differentiation, and gene expression (32). However, excessive ROS have the potential to induce significant biological damage and, hence, cells possess many antioxidant systems for maintaining the ROS threshold at physiological concentrations (Fig. 1). Oxidative stress can occur when ROS production is increased or when the mechanisms involved in maintaining the normal reductive cellular milieu are impaired. Redox active thiol groups in proteins, superoxide dismutase (SOD), catalase, and low-molecular mass compounds, such as ascorbic acid, tocopherol, thiols including glutathione, and uric acid play key roles as redox buffers that balance any disturbance of the intracellular redox state (43, 60). Depending on the level and extension of the oxidative stress, the cells may establish short- or long-term adaptive responses (40). The redox homeostasis in Leishmania parasites appears to be efficiently regulated since they can successfully withstand the oxidative burst during host infection and perfectly adapt to the different metabolic and environmental conditions imposed by their digenetic life cycle (40). The Leishmania are equipped with SOD, tryparedoxin (TXN), TXN-dependent peroxidases and ascorbate peroxidase (APX) to maintain cellular redox equilibrium (3, 8, 9, 24, 26, 27, 52, 63). In addition, Leishmania produces significant amounts of low-molecular mass thiols such as trypanothione, glutathione, and ovothiol A (2, 16, 24). Of these, only glutathione is present in cells of the host (30, 49). All the three thiols are directly or indirectly maintained in a reduced state by trypanothione reductase (TR) (64). Most of the glutathione content in Leishmania is found in the form of trypanothione (N1,N8-bis(glutathionyl) spermidine), a unique thiol consisting of two glutathione molecules joined by a spermidine linker that is produced by trypanothione synthetase (51). As in other organisms, in Leishmania also, the enzymes γ-glutamylcysteine synthetase and glutathione synthetase successively act for the synthesis of glutathione (49). The second order rate constants for reactions with H2O2 have proved ovothiol A to be functionally less efficient than trypanothione; however, in promastigote stages of L. major, L. Mexicana, and L. aethiopica it can be considered as the principal thiol, particularly in the late logarithmic and stationary phases of growth (2). Ovothiol A and trypanothione can reduce H2O2 nonenzymatically. The anti-oxidative armamentaria of Leishmania also include TXN and tryparedoxin peroxidases. TXNs belong to the thioredoxin-fold family but trypanosomatids lack thioredoxin reductase (58). Unlike typical thioredoxins, which are directly reduced by NADPH-dependent flavoreductases, Leishmania TXNs are oxidoreductases requiring trypanothione as the mediator to take up electrons from the flavoprotein TR. Two classes of peroxidases occur in trypanosomatids: the non-selenium peroxiredoxin and the cysteine GPX members, which reduce H2O2 using electrons from TXN (9). Leishmania can synthesize ascorbic acid, which is a powerful antioxidant (4, 68). Very recently, in L. major an ascorbate-dependent peroxidase has been detected as a part of the organism's antioxidant defense system. As biochemical and functional characterization of LmAPX is the subject matter of this review, a brief introduction about peroxidases is given below.

Peroxidases: An Overview
Peroxidases hold a venerable position in enzymology. This is one of the most extensively studied group of enzymes and literature has been enriched with the reviews and a large number of basic research articles dating back to the early part of the 19th century. Peroxidases are mainly classified into heme- and non-heme-containing peroxidases. The native heme peroxidases contain a heme prosthetic group, usually ferriprotoporphyrin IX, with four pyrrole nitrogens bound to the Fe (III). The fifth coordination position on the proximal side of the heme is usually the imidazole side chain of a histidine residue. The sixth coordination position is vacant in the native enzyme on the distal side of the heme. The distal cavity is the region in which the peroxidase reactions occur. The peroxidative reaction cycle follows the initial formation of compound I followed by compound II formation and regeneration of native enzyme. Compound I and compound II are the two and one electron-oxidizing equivalents higher than native ferric state of peroxidase respectively (21). Hugo Theorell was awarded Nobel Prize in 1955 for the discovery of horseradish peroxidase (HRP) compound I. A major breakthrough was the determination of the X-ray crystal structure of yeast cytochrome c peroxidase (CCP) in the year 1980. These discoveries established both HRP and CCP as model peroxidases, based on which structure-function studies of other peroxidases are carried out. Heme peroxidases are distributed throughout plant and animal kingdom. All currently known heme-containing peroxidases can be divided in two main superfamilies (peroxidase–cyclooxygenase and peroxidase–catalase superfamily) and three families (di-heme peroxidase, dyp-type heme peroxidase, and haloperoxidase) (73). The peroxidase–catalase superfamily is further divided into three distinct classes. Class I peroxidases include intracellular peroxidases of prokaryotic origin-like plant chloroplast and cytosolic APX, yeast CCP, and gene duplicated catalase-peroxidase. Analyses of the amino acid sequences of catalase-peroxidase indicate that the double length of the bacterial peroxidases can be ascribed to gene duplication (66). Class II peroxidases comprise fungal secretory peroxidases like lignin and manganese peroxidases from Phanerochaete chrysosporium and the ink cap mushroom peroxidase from Coprinus cinereus (21). Examples of Class III peroxidases are endoplasmic reticulum mediated classical secretory plant peroxidases like HRP isoenzyme C. During 1940–1960s a large number of animal peroxidases were characterized and their physiological functions revealed. Plant ascorbate dependent peroxidase was discovered in 1979 (29). APX from pea is well characterized biochemically and the crystal structure has been solved in great detail in the year 1995 (55). In plants, different isoforms of APX are shown to localize in chloroplast, cytosol, and microbody where they eliminate photosynthetically generated H2O2 by glutathione/ascorbate cycle and control H2O2-mediated redox signaling (47).
GPXs and vanadium haloperoxidases lack heme in their active sites. GPXs share a common catalytic core formed by selenocysteine (Sec), tryptophan, and a glutamine residue. However, in some GPXs, a Cys replaces the Sec residue. Such substitution, characteristic of the so-called nonselenium GPX-like enzymes, confers decreased peroxidase activity to these proteins (33). Vanadium haloperoxidase contains a vanadate prosthetic group and utilize H2O2 to oxidize a halide ion into a reactive electrophilic intermediate (69).
Ascorbate and APX in Leishmania
Physical and Spectral Characteristics of LmAPX
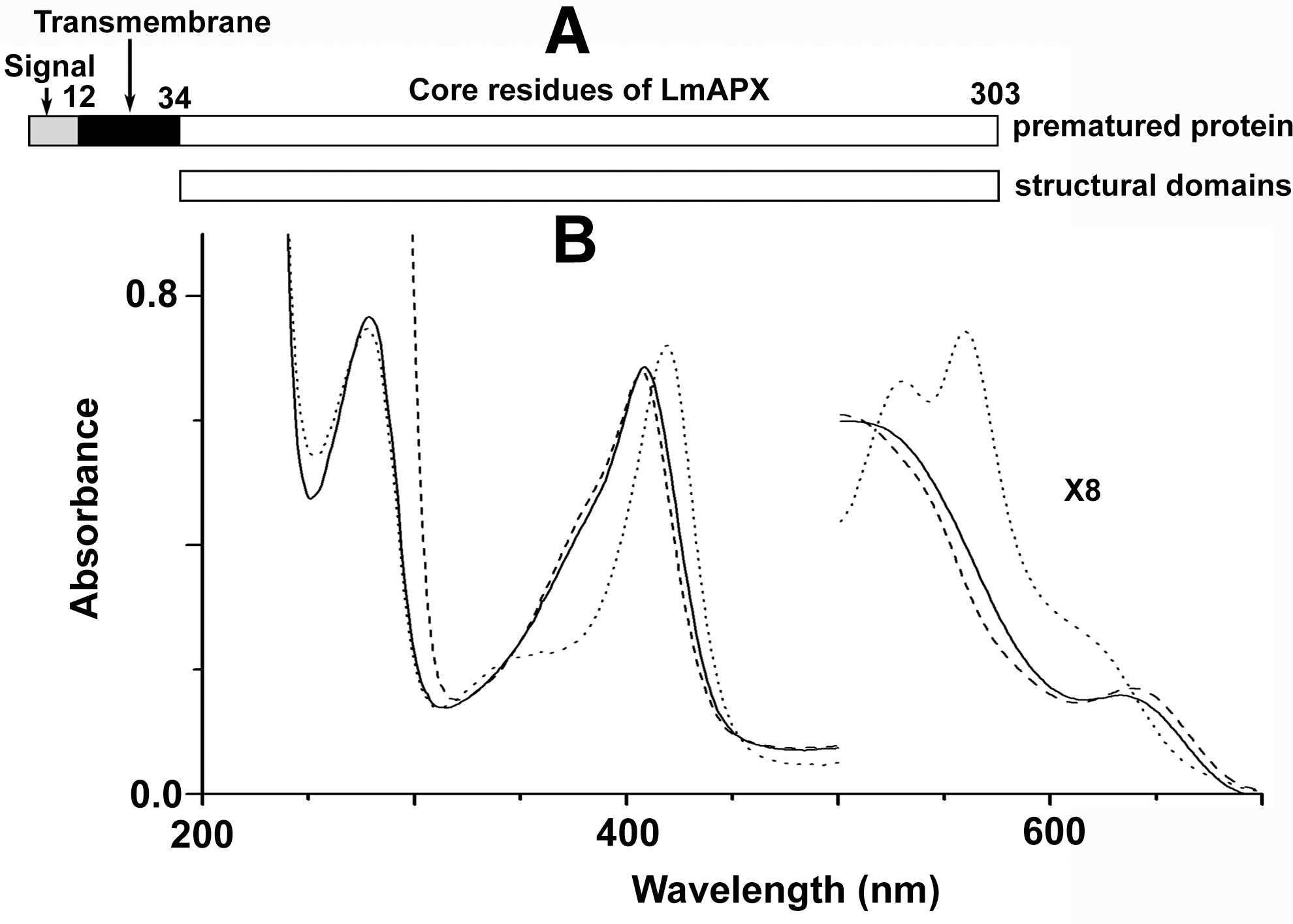
LmAPX primary sequence has the high level of sequence identity with both yeast CCP (∼35%) and the pea APX (∼36%) (70). In silico analysis of LmAPX sequence (TargetP V1.0) predicts (22) that the N-terminal 12 amino acids constitute a signal sequence, which is followed by a stretch of 22 amino acids containing a hydrophobic region resembling a transmembrane domain (Fig. 2A). All the key residues on both distal and proximal sites of the heme are conserved among LmAPX, yeast CCP, and plant APX. However, it lacks both the cytochrome c-binding residues (Asp34/Glu35, Tyr39, and Glu290 in yeast CCP) and critical ascorbate-binding residue (Arg 172 in pea APX) (7, 56, 59). The proximal cation (K+) binding loop of the LmAPX is more similar to pea APX, whereas the c-terminal insertion and proximal Trp radical stabilizing Met residues of LmAPX are more similar to CCP (35, 70). The gel filtration results indicate that Δ34 LmAPX (34 amino acids deleted from the N-terminus sequence of LmAPX) is a monomeric enzyme with a molecular weight of ∼33 kDa (1). The UV-visible spectra of ascorbate-free Δ34 LmAPX shows the Soret peak at 408 nm with charge transfer peaks at around 500 and 640 nm (Fig. 2B). The calculated purity number, Rz (A408/A280), for Δ34 LmAPX is 0.98 (Fig. 2B). Addition of an equimolar H2O2 to resting state of Δ34 LmAPX produces compound I* within 0.75 ms absorbing at 420 nm at Soret region with visible peaks at 532 and 560 nm. Extensive spectroscopic studies have demonstrated that the Soret band of compound I* is similar to an oxyferryl tryptophan cation radical of yeast CCP compound I species (23).
Catalytic Mechanism of LmAPX
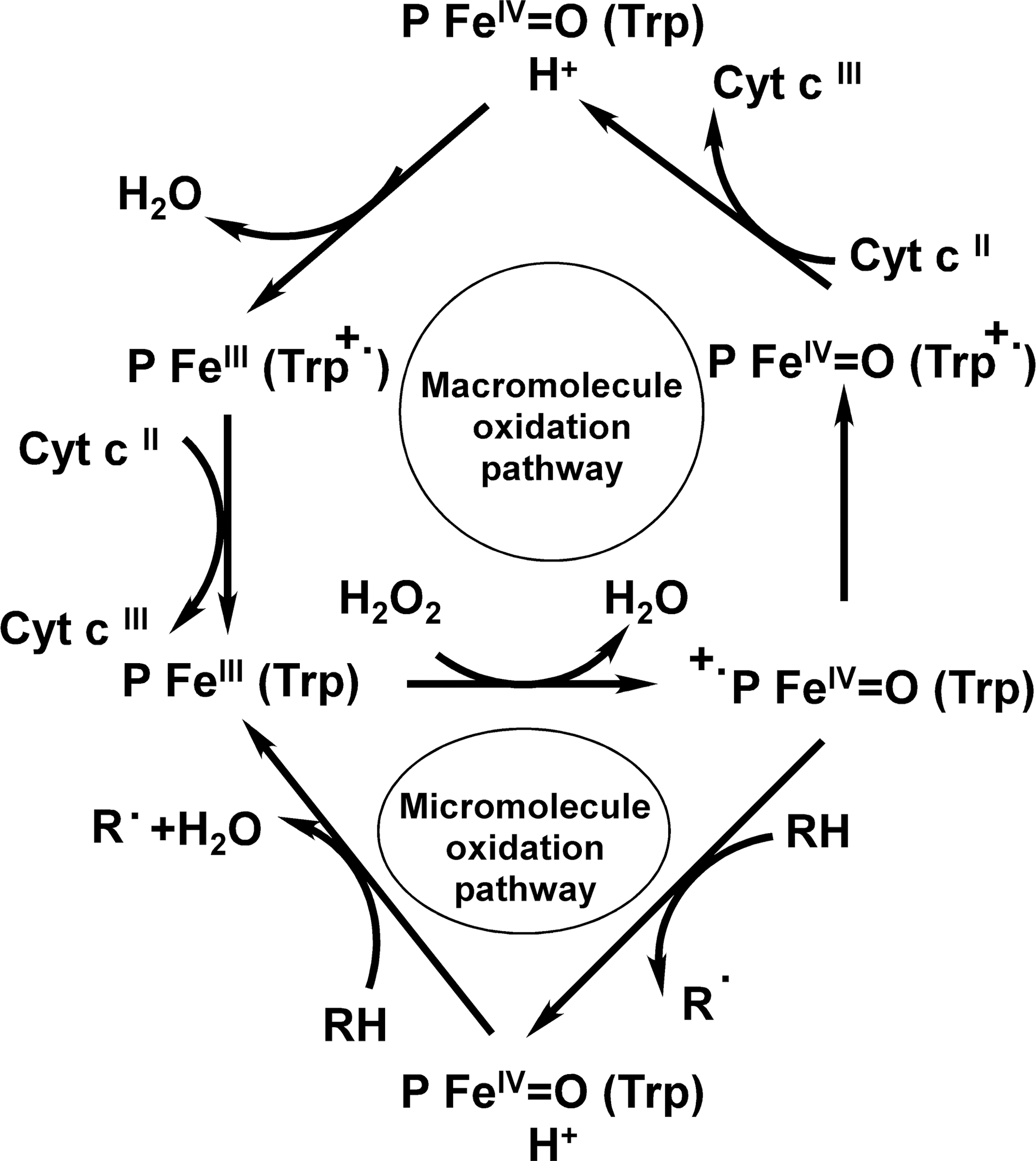
Based on previous yeast CCP, plant APX, and current LmAPX results (35, 54, 70, 72), a plausible mechanism for the redox reaction catalyzed by LmAPX may be suggested as shown in Figure 3. In the presence of low-molecular mass (micromolecule) electron donors like ascorbate or guaiacol (2-methoxyphenol), the reaction of H2O2 with the resting form of LmAPX (FeIII,P,Trp) leads to the formation of CMPI (FeIV=O,P•+,Trp), which contains an oxyferryl heme FeIV=O and a porphyrin π-cation radical. Low-molecular mass electron donor initially reduces π-cation radical of CMPI (FeIV=O,P•+,Trp) to produce compound II (FeIV=O,P,Trp), which contains an oxyferryl heme FeIV=O. A second molecule of the electron donor then reduces the oxyferryl heme in CMPII (FeIV=O,P,Trp) to form the resting enzyme (FeIII,P,Trp). However, in the presence of cytochrome c, the situation is altered. Indeed, one of the curious features of LmAPX is that, in contrast to plant APX, its CMPI (FeIV=O,P•+,Trp) is most probably comparatively unstable and, it leads to the generation of another species CMPI* (FeIV=O,P,Trp•+), which contains an oxyferryl heme FeIV=O and a Trp•+ radical cation located on the indole ring of Trp-208. Ferrocytochrome c initially reduces the Trp-208 radical cation in CMPI* (FeIV=O,P,Trp•+) to produce compound II (FeIV=O,P,Trp). By internal electron transfer in CMPII (FeIV=O,P,Trp) the Trp radical cation in CMPII* (FeIII,P,Trp•+) is regenerated, which is then reduced by cytochrome c to form the native enzyme (48).
Pseudocatalase Activity of LmAPX
In addition to peroxidase activity of lactoperoxidase and thyroid peroxidase, several reports have shown that in case of iodide as electron donor, the intermediate enzyme with bound hypoiodous acid reacts with a second molecule of H2O2 in a catalase-like reaction, with liberation of O2 (34, 44, 45). The pseudohalide, SCN−, cannot replace I− as a promoter of pseudocatalase activity, even though SCN− is readily oxidized by thyroid peroxidase or lactoperoxidase in presence of H2O2 (34, 44, 45). In this case, H2O2 is utilized solely for the oxidation of SCN−, in contrast to the catalytic cleavage of H2O2, which occurs in the presence of low I− concentration (34, 44, 45). Like well known peroxidases, LmAPX catalyzes the degradation of H2O2 with concomitant formation of O2 under low ascorbate concentration at physiological pH (18). The reaction is initiated by its peroxidase activity with the generation of ascorbate-free radicals via one-electron-transfer mechanism and then regeneration of ascorbate by reduction of ascorbate radical with H2O2. The requirement of enzyme, H2O2 and ascorbate indicates that monodehydroascorbate plays a vital role in free radical mediated H2O2 consumption, rather than the disproportionation reaction of the free radicals.
Structural Investigation of the Catalytic Domain of LmAPX
The structure of the distal pocket, proximal pocket, and the cation-binding loop in LmAPX, yeast CCP, and plant pea APX is shown in Figure 4. The 1.76 Å resolution of LmAPX structure demonstrates 10 alpha helical bundle folds, characteristic of peroxidases (35). The distal Arg-Trp-His and proximal His-Asp-Trp arrangements in LmAPX are typical of Class I peroxidases, which include CCP and APX. The electron density of LmAPX shows two cations, a proximal K+ that is visible up to 20 σ in the Fo-Fc difference map and a distal Ca2+ visible up to 26 σ in the same map (35). As expected from sequence alignments, LmAPX can be considered as a hybrid peroxidase that shares structural features with various classes of heme peroxidases. Like class III peroxidases such as HRP, LmAPX contains two cation-binding sites. The following structural features are similar between yeast CCP and LmAPX (35): (i) presence of the triple beta strand, (ii) the proximal Met residues (Met248 and Met249) that are responsible for formation of the stable proximal Trp208 radical (35, 72), and (iii) a stretch of acidic residues (D225, E226, and D227) before a c-terminal sequence insertion (228–240 aa), one of them (E226) is responsible for proper maintenance of active site conformation (71). Again, some plant APX-like structural elements, present in LmAPX are: (i) the insert region between A and B helices, which provides contacts with cytochrome c in the CCP–cytochrome c complex (56), is much shorter in LmAPX and forms part of the ascorbate binding pocket and (ii) the monovalent nature of the proximal cation (35). Analysis of the proximal K+ cation-binding loop in LmAPX indicates that three main-chain ligands, three side-chain ligands, and one water ligand are bonded to K+ (35). Those seven coordinations inside the loop are formed by residues 193 (164 in pea APX and 176 in yeast CCP) to 218 (189 in pea APX and 201 in yeast CCP) in LmAPX. In LmAPX, T193 (side chain OH, back bone CO), T209 (side chain OH), D211 (back bone CO), G214 (back bone CO), and S218 (side chain OH) amino acid residues, and H2O ligand contribute to form coordinate bonds with K+. LmAPX and pea APX differ in coordination bond formation: (i) H2O molecule and D211 residue in LmAPX form one coordinate bond each with K+, whereas the corresponding residue N182 in pea APX forms two coordinate bonds. (ii) One coordinate bond is formed by S218 in LmAPX but the corresponding residue S189 in pea APX does not form coordinate bond. Instead, D187 residue in pea APX takes part in coordinate bonding. However, LmAPX has Phe201 residue in the ascorbate-binding site instead of Arg residue (Arg172 in plant pea APX) (56, 59), which may account for the lower APX activity of LmAPX, 32 min−1 compared with ∼250 s−1 for plant APX (42). The main structural dissimilarity between LmAPX and CCP involves the presence of Cys197 in LmAPX, whereas the analogous residue in CCP is Thr180. Mutational and electron paramagnetic resonance studies suggest that this major structural difference plays a key role in LmAPX for stabilization of proximal Trp radical in the presence of K+ ion (35). Other reports reveal that in plant APX and LmAPX, the K+-binding residues function in maintaining the protein structure in the heme vicinity to favor the enzymatic activity by controlling the specific structural conformation of the proximal and distal histidines (14, 53). Recently, a model of the L. major APX–cytochrome c complex, using the yeast system as a guide, suggests that its complex is very close to the yeast CCP–cytochrome c crystal structure (36). KM and the efficiency (kcat /KM) of LmAPX for L. major cytochrome c are 6 μM and 1.6×108 M −1 s−1, respectively, similar to those exhibited by CCP in the yeast system (36, 38). It has been suggested that the biological function of LmAPX is to act as a CCP (36).

Localization and Regulation of LmAPX
Localization and regulation of a protein provides vital information for a group of many co-operating proteins, which are responsible for an integrated physiological function. Thus, exact information regarding the localization of LmAPX will be supportive for predicting the possible physiological function of this protein. TargetP 1.1 Server predicts that the N-terminal signal sequence of LmAPX is a mitochondrial targeting signal. Subcellular fractionation, Western-blot analysis, and confocal microscopy suggest that the enzyme is localized in the mitochondrion of Leishmania (19). Mitoplast (mitochondria without outer membrane) preparation by treatment of digitonin, sub-mitochondrial fractionation, and activity measurement of kynurenine hydroxylase (outer membrane marker), succinate dehydrogenase (inner membrane marker), and malate dehydrogenase (matrix marker) enzymes have revealed that LmAPX is localized in the inner membrane of the mitochondria with its catalytic domain in the intermembrane space (19), which is similar to the location of CCP in yeast (13, 28, 37). Therefore, LmAPX is more likely to be accessible to scavenge endogenous ROS produced in the mitochondrial inter membrane space. Although, chloroplasts and peroxisomes in plants are the main organelles in terms of both ROS formation and breakdown (50), yet intermembrane space of mitochondria is clearly the major site of ROS generation in yeast and animal systems. The expression of yeast CCP gene and plant APX gene was also increased on treatment with various oxidative stressors (41, 61). Similarly, upregulation of LmAPX gene expression by H2O2 suggests that the parasite sequentially uses this enzyme to overcome oxidative stress (19).
Physiological Function of LmAPX
In a wide variety of species, ranging from bacteria to higher mammals, the mechanisms of H2O2-detoxification are grossly distributed into variations of two mechanisms: mediated either by the heme proteins and/or by non-heme proteins like selenium-containing GPX. Detailed cell biological approaches have successfully established the biochemistry and physiological significance of LmAPX enzyme in Leishmania promastigotes (see working model on Fig. 5) (19, 20, 52). Overexpression of LmAPX in L. major protects promastigote cells against oxidative stress-induced apoptosis (19, 20). Clearly, knocking the gene out of the cell has thrown the parasite under a constant level of oxidative stress as evidenced by the higher intracellular H2O2 content (52). This basal level of oxidative stress acts as the driving force for the early onset of metacyclogenesis producing infective metacyclic promastigotes from non-infective procyclic ones. Unless favorable condition (high temperature and low pH, characteristic of the condition prevalent in the mammalian host) is reached, the metacyclics proceed toward apoptosis; in this respect, metacyclogenesis is comparable to terminal differentiation. The infectious parasite population in the midgut of infected sandfly or in vitro stationary phase cultures contain both metacyclic and apoptotic parasites and a combination of these two types of cells are needed for infectivity of the parasite within host macrophages (65). In accordance with this, deprived of LmAPX, the stationary phase culture contains a higher number of metacyclic and apoptotic parasites compared with both wild-type and LmAPXverexpressing cells (52). The apoptotic cells suppress macrophage activation by downregulating the inflammatory cytokines like TNF-α and upregulating the anti-inflammatory cytokines like transforming growth factor-β, while the virulent metacyclics proceed to invade macrophages and cause disease. These data establish vital contributions of LmAPX: (i) the enzyme has a direct role in effective detoxification of ROS, which is significant enough considering the absence of any other potent ROS scavenging heme enzyme in Leishmania. (ii) The indirect effect of the enzyme was revealed on creation of the knock out mutant, where its absence enhances cellular differentiation and virulence mediated by oxidative stress. Generally, attention is given to the genes whose loss leads to decreased virulence, pathogenicity, or infectivity. LmAPX represents a new class of genes whose deletion helps in the infectivity of the parasite. Thus, the enzyme possesses great evolutionary significance, being effectively utilized by the parasite at different conditions, which is not surprising, considering the high degree of adaptability of the phylum, protozoa. Hence, our research presents an ideal model system to study parasite virulence.
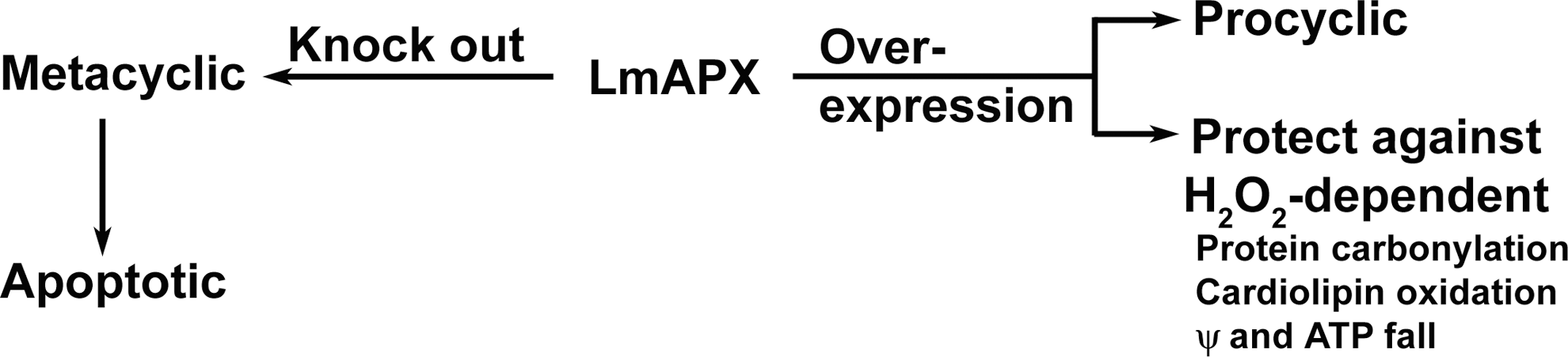
Potential of the Defense System Involving LmAPX in Oxidative Stress
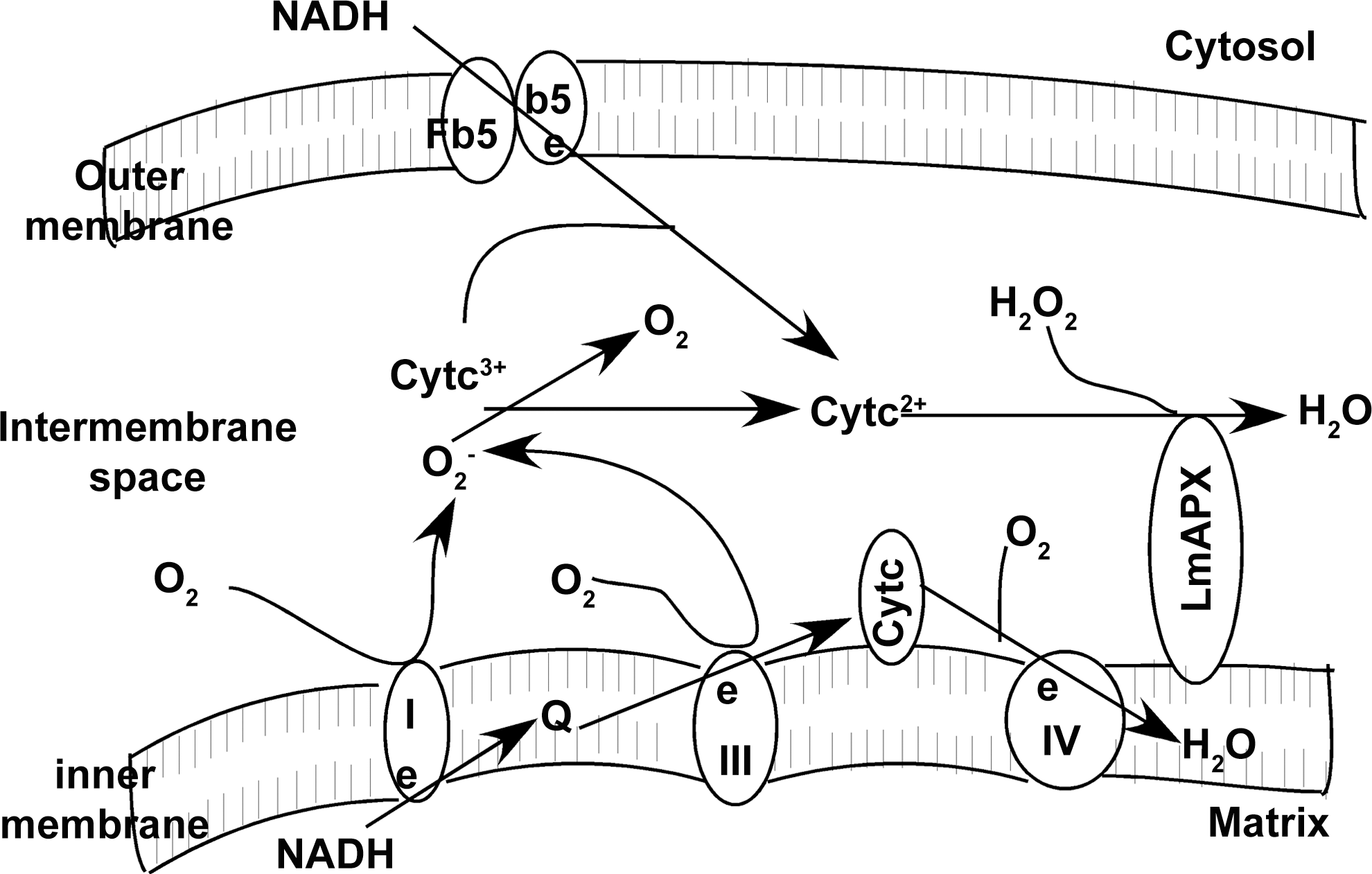
Like yeast CCP, one of the electron donors of LmAPX was found to be ferrocytochrome c (18), which is co-localized at intermembrane space side of the inner membrane. The fact that cytochrome c is a much better substrate than ascorbate, as observed from the kcat values for the reaction of these molecules with LmAPX, suggests that the biological function of the enzyme is similar to yeast CCP (35, 36, 72). Since LmAPX can oxidize ferrocytochrome c in presence of endogenous H2O2, it can be considered as an excellent candidate for playing key functions in mitochondria-related redox control, such as elimination of H2O2 and O2 −•, and resistance to mitochondrial ROS. Although the relative contribution of mitochondria to ROS production in green plants is very low (57) yet mitochondria is the major source of ROS in non-photosynthetic organisms (31). The plausible antioxidant role of mitochondrial LmAPX in Leishmania (catalase absent) is illustrated in Figure 6. The processes possibly involved in the LmAPX-linked defense mechanism are indicated in this model. LmAPX could detoxify H2O2 in the presence of reduced cytochrome c. The reduction of cytochrome c could occur either by NADH-cytochrome b5 reductase-cytochrome b5 pathway or O2 −•, which was generated by complexes I and III of the respiratory chain. Thus, the cytochrome c-LmAPX system appears to play an important antioxidant role in L. major cells by scavenging both H2O2 and O2 −•.
Conclusion
Living cells require ROS for their normal growth and proliferation and at the same time must possess appropriate scavenging system to get rid of the excess ROS, which, otherwise, might be detrimental. So, there is a constant struggle for maintaining the balance, leading to a variety of adaptations, honed through evolution, which is evident from the redundancy of antioxidant systems as well as intelligent use of a single protein in diverse physiological processes. Single-celled trypanosomatids are adroit adaptors, which is not unexpected considering their digenetic life cycle, exposing them to very different environments. Structural and functional analysis of LmAPX has pointed to its novel evolutionary consequence as a hybrid of APX and CCP (35, 70, 72). LmAPX is located in the intermembrane space side of inner mitochondrial membrane, where cytochrome c is present (19). The high in vitro oxidation rate for cytochrome c (35, 36, 72) suggests that this molecule likely is the probable physiological substrate of the enzyme. Also, the enzyme has been shown to have pseudocatalase activity (18) and its role in protection of the cell against oxidative stress mediated cardiolipin oxidation, extensive protein damage, and apoptosis (19, 20, 52) has also been established. It is also possible that the loss of LmAPX activity has secondary effects on leishmanial gene expression caused by an alteration in the redox balance in the protozoa. As a consequence, modulation of surface lipophosphoglycan (LPG) of L. major (an approximate doubling in length of LPG and capping of majority of the galactose sugars with arabinose residues) and simultaneous differentiation of the promastigotes into infective metacyclic form may occur (46, 52). Thus, in L. major APX acts to protect the cells from ROS-mediated apoptosis and may have a major role in cellular differentiation. Future works will more precisely determine the signaling pathways involved in the processes.
Footnotes
Acknowledgments
This work was supported by the Council of Scientific and Industrial Research (CSIR) Network Project NWP 0038. Dr. Swati Pal is supported by an individual fellowship from the CSIR, Government of India.