
Abstract
Introduction
Overview of bacterial responses to nitrosative stress: Detoxification, gene regulation, and chemistry
Bacteria have evolved a multitude of detoxification systems to counter RNS, including reductases and microbial globins whose expression is tightly regulated by external stimuli. The major diverse mechanisms identified were reviewed previously (85). This is a rapidly developing field: regulation of gene expression by NO and RNS and the functional identification of resistance mechanisms are being studied in numerous bacteria, as well as fungi and protozoa. Many mechanisms are known to confer NO resistance in bacteria, such as flavohemoglobin-catalyzed NO detoxification by Escherichia coli and Salmonella enterica serovar Typhimurium (hereafter S. Typhimurium), protecting the bacterium from NO-mediated killing in human macrophages (108). Flavohemoglobin (Hmp) catalyzes the reaction of NO with oxygen to give innocuous nitrate via a dioxygenase (36 –38) or denitrosylase (45, 46) mechanism, and hmp gene transcription is activated on exposure of bacteria to NO or nitrosating agents (39, 86, 87). Other globins intensively studied (119) include the two globins in each of Mycobacterium tuberculosis (20, 79) and Campylobacter jejuni (68, 69). However, it is important to recognise that all these systems, as far we can judge from current experiments, are first and foremost concerned with NO detoxification, not with SNO detoxification.
Although the earliest publications on flavohemoglobin regulation (86) and enzymic function (38) were conducted with 'real' NO, many later studies have used substitutes for these agents. Reference 46 provides a clear distinction between NO metabolism and SNO metabolism: it was known that SNO decomposition by E. coli generates nitrite and nitrate, and that NO could arise from the former by autoxidation in solution. A quest for SNO decomposition showed that, although the NO metabolizing activity of E. coli was stimulated by treatment of the culture with S-nitrosocysteine (CysNO), the SNO lyase activity was not.
Confusing the literature, however, is that many experimental studies to unravel the details of NO microbiology are performed with proxies for NO, often SNOs and especially S-nitrosoglutathione (GSNO). The chemical consequences of using NO gas, NO donors, or nitrosothiols appear to be insufficiently understood or considered in much published work, but are considered in detail by Bowman (14). Although NO can be used directly (either by administering the gas or as a solution) (1), it is often more convenient to use NO donors, but appropriate precautions and controls are necessary. It must be determined that the biological outcomes are due to NO and not the donor molecule or donor co-products, the consequences of impurities in the reagents must be considered and, if biological activation is required, this may affect the outcomes (14). Most pertinent to this review is the direct use of SNOs, although S-nitrosation of biological targets can of course also result from exposure to NO. SNOs, typified by GSNO, can do much more than release NO, which is not spontaneous but requires light or a reducing metal (e.g., Cu1+). In addition, SNOs may transfer the equivalent of NO+ (the nitrosonium ion) to another thiol (87) in a transnitrosation reaction, rendering interpretation of data difficult. A later section describes the effects of administering SNOs to microbial cultures.
Another consequence of using GSNO may be modification of a Cys thiol by incorporation of a glutathione (GSH) moiety (S-glutathionylation); even though some species of bacteria contain very high levels of GSH (3.5–6.6 mM in E. coli) (24), the literature on this bacterial process is extremely sparse. The process has been described in some photosynthetic eukaryotes (121) and protozoa (60) and in many higher eukaryotes where it serves critical signaling roles (70). Other donors including sodium nitroprusside, HNO donors, and reagents for applying peroxynitrite to microbes are covered in Ref. 14.
A major research area is the mechanism of gene regulation in response to NO and GSNO. The ability of bacterial pathogens to respond to these species, which are generated in vivo by the NOS enzymes and especially the iNOS of macrophages, and can also arise from dietary or other external sources, appears to be crucial for virulence in many cases (for a review see Ref. 107). The small levels of NO produced may, however, activate SoxR (28), Fnr (21), NorR (23), Fur (22), and NsrR (11). Spiro usefully distinguishes between those that are secondary sensors and those that are 'dedicated' in that their physiological function appears to be detecting primarily NO and then regulating expression of genes that encode enzymes with NO as a substrate (107). The archetype is NsrR, which appears in enterobacteria to be the major regulator for NO-detoxifying proteins like flavohemoglobin. In some pathogens, sensors can detect a range of ligands, such as the redox-sensitive transcriptional regulator, OxyR (62) in E. coli and the DosRST system of M. tuberculosis, which mediates the response to both NO and hypoxia (101).
A word on nomenclature
Before discussing S-nitrosothiols, a note on nomenclature is required. The incorporation of NO to a thiol results in the formation of an S-nitrosothiol; where this thiol group belongs to a cysteine residue of a protein, the incorporation of NO results in the formation of an S-nitrosoprotein. Note that, in the chemical literature, the term 'nitrosation' describes the formation of a nitrosospecies with a 'nitroso' group (-NO) covalently incorporated. S-nitrosation refers to this incorporation via a sulfur atom (usually in a thiol) irrespective of the mechanism. In contrast, nitrosylation describes the formation of a coordination bond between a metal center (for example, the iron of heme) and a nitrosyl (NO) ligand. However, in the biological literature these terms are used interchangeably, particularly where there is mechanistic ambiguity. In this review, whenever S-nitrosothiol formation is shown to occur as a result of exposure to a transnitrosating agent, then the term ‘S-nitrosation’ is used. Wherever there is insufficient data or ambiguity as to the mechanism of S-nitrosoprotein formation, the term ‘S-nitrosylation’ may be employed.
The Bacterial Response to, Uptake, and Endogenous Formation of S-Nitrosothiols
Bacterial uptake of S-nitrosothiols
Small S-nitrosothiols such as GSNO are extensively used in studies in vitro as a source of nitrosative stress, and are widely believed to be of physiological relevance in vivo given the abundance of GSH in cells. The partially overlapping responses to GSNO, NO, and peroxynitrite observed in E. coli as a paradigm are summarized in Figure 1. In general, SNOs elicit a nitrosative stress response at the transcriptional level, resulting in expression of protective functions, notably NO-detoxifying proteins such as hemoglobins and NO reductases. It is important to note that these proteins are not thought to detoxify SNOs like GSNO directly; it is generally assumed that NO is generated in vivo from these SNOs (54) to activate transcription factors, while other cellular consequences of SNO exposure, such as thiol modification, are caused by the SNO itself.
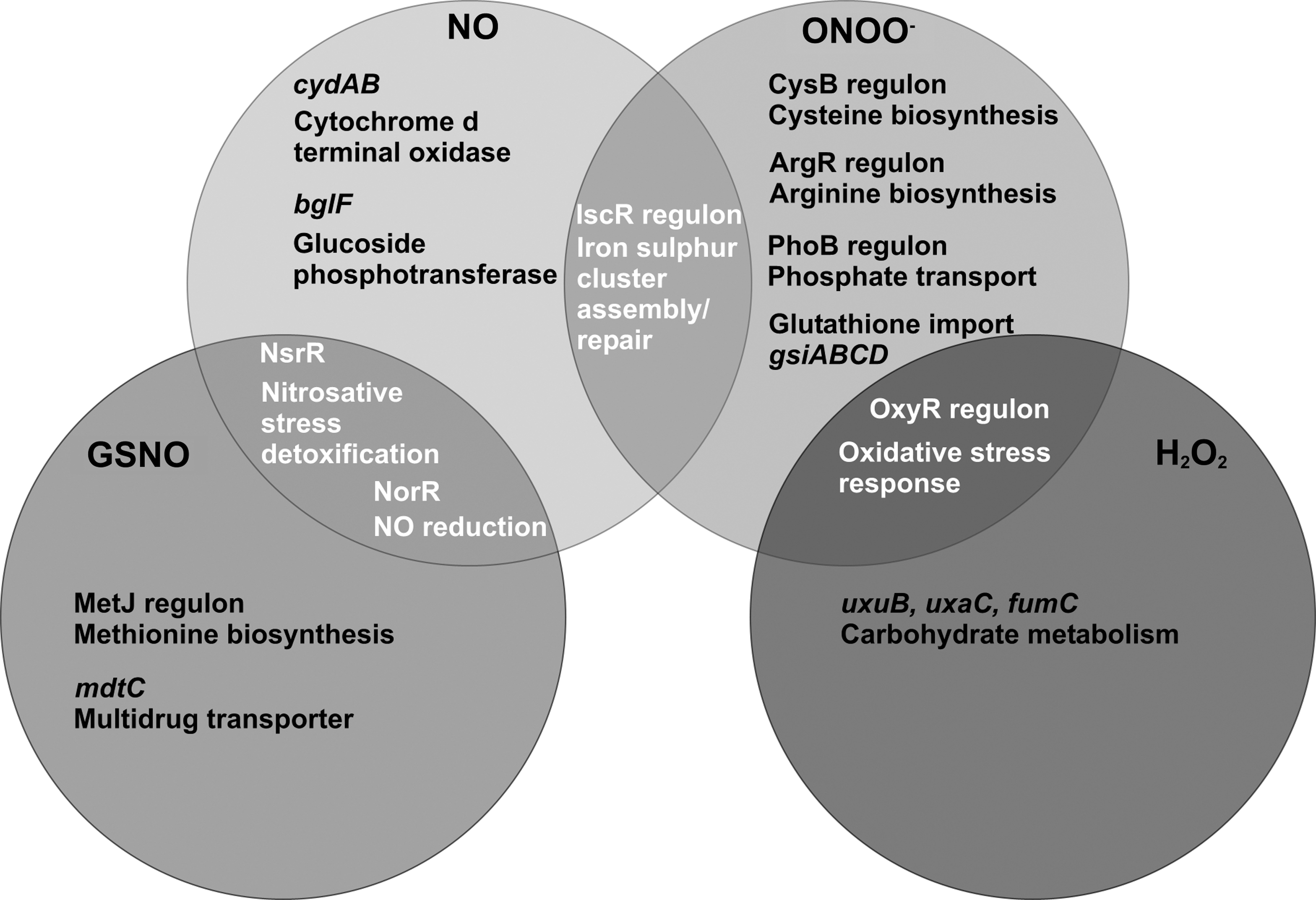
The mechanisms of communication between extracellular and intracellular pools of SNOs are still poorly understood. Transport mechanisms involving cell-surface protein disulfide isomerases (94, 122), γ-glutamyl transpeptidase (26, 48), or anion exchangers (80) have all been proposed. GSNO is reported to transfer NO+ to outer membrane thiols in Bacillus (76), but other studies suggest that active transport of the compound is required for toxicity. Figure 2 summarizes the major mechanisms recognized in bacteria and mammalian cells. Broadly, these are: direct transport of the whole nitrosothiol compound, transport only of the nitrosothiol moiety (by transnitrosation), and mixed transport, as in the breakage of GSNO and transport of CysNO.
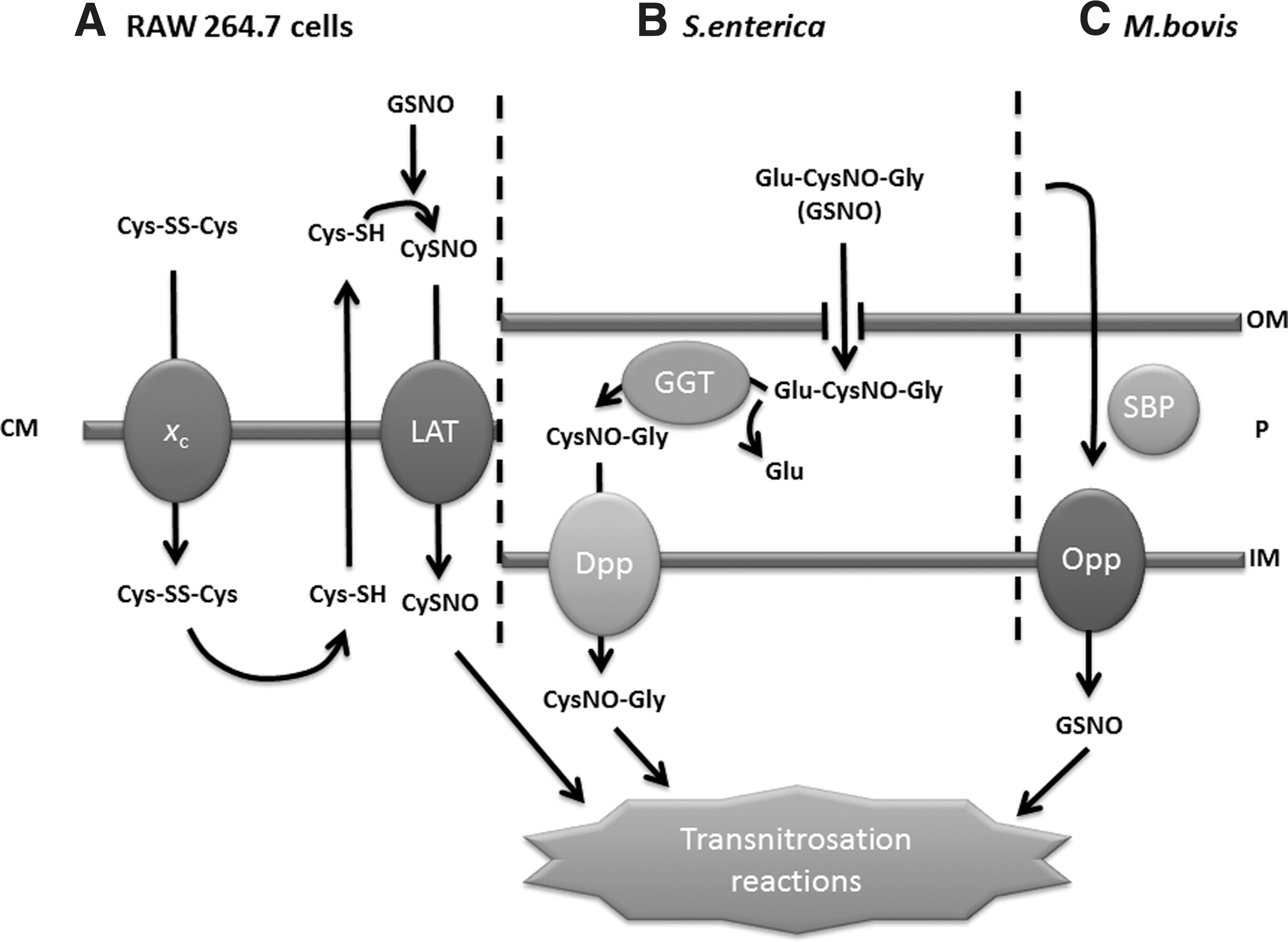
First, in mammalian systems (Fig. 2A), the cellular impact of GSNO or SNAP is influenced by the availability of cysteine or cystine. Indeed, the degradation of GSNO is absolutely dependent on extracellular cystine, which is reduced to cysteine, and in turn reacts with GSNO to form CysNO in a transnitrosation reaction. CysNO is then imported by the amino acid transport system L-AT (123).
Second, in S. enterica (Fig. 2B), although GSNO is bacteriostatic, it is not itself transported into the cell. Highly GSNO-resistant mutants were isolated from a MudJ transposon library and the insertions shown to be in dppA and dppD (26). These genes are part of an operon encoding dipeptide permease, an ABC-family transporter responsible for L-dipeptide import. It is therefore suggested that a periplasmic transpeptidase encoded by the ggt gene removes the γ-glutamyl moiety. The residual dipeptide, S-nitroso-L-cysteinylglycine, is then transported inwards, using the Dpp dipeptide permease. A similar mechanism appears to operate in E. coli since regulatory perturbations inducible by GSNO are dependent on the presence of the Dpp system (54), and a ggt mutant of E. coli is also GSNO-resistant (26). GSNO is also toxic to mycobacteria but, again, the tripeptide is not directly transported to the cytoplasm. The processing of glutathione and GSNO in Mycobacterium tuberculosis is brought about by a γ-glutamyl transpeptidase that cleaves glutathione and S-nitrosoglutathione to the dipeptide (CysNO-Gly), which is then transported into the bacterium by the multicomponent ABC transporter dipeptide permease (25).
Third, in Mycobacterium bovis, the oligopeptide permease operon (oppBCDA) is implicated in GSNO transport (41). Mutation of the oppD gene encoding the ATPase component of this binding protein-dependent transport system elicits resistance to 4 mM GSH in the external medium, a concentration that inhibits the wild-type stain. Importantly, similar results were found with 0.5 mM GSNO, which is bactericidal for the wild-type strain but not the oppD mutant. The resistance of the mutant to GSH is due to diminished import of the thiol, as shown by transport studies using [3H]GSH. In view of the finding that, in S. enterica, it is transport of the dipeptide that carries the NO+ group (26), Green et al. (41) tested whether the opp system in M. bovis transports dipeptides, but none of the tested components of GSH (L-Cys-Gly, L-Cys, Gly) showed toxicity towards this bacterium.
Bacterial formation of S-nitrosothiols
A recent publication by Seth and colleagues demonstrated the endogenous S-nitrosylation of proteins in E. coli anaerobically respiring on nitrate in minimal medium (104). The generation of endogenous S-nitrosothiol was shown to be largely, but not exclusively, dependent on the expression of the NarG nitrate reductase and was inhibited by a chelator of divalent metal cations (104). This is consistent with reports that nitrosation of 2,3 diaminonapthalene (DAN) to produce a fluorescent triazole is predominantly dependent on the expression of NarG (73, 93), which was shown to produce NO from nitrite in the absence of nitrate (56). The DAN assay of nitrosation was shown to proceed in two stages, the biologically significant process being the initial generation of NO from nitrite, followed by NO autooxidation and the chemical nitrosation of DAN (55). However, the molecular mechanisms of S-nitrosylation in the model of endogenous SNO formation have yet to be determined (104). Emerging from this study is a key role of the redox active transcriptional regulator, OxyR, in responding to nitrosative stress. The article identifies a novel regulon that is controlled by S-nitrosylated OxyR (SNO-OxyR) (104).
Bacterial networks of response to S-nitrosothiols
It is not straightforward to prepare aqueous solutions of NO for experimental use and many investigators have turned instead to off-the-shelf reagents to release NO or mimic some of the properties of NO in biological systems. Various S-nitrosothiols (RSNO) are used in this way (1). They decompose in solution to give NO and RS radicals, the latter dimerizing to RSSR. GSNO is the most commonly used and may be purchased or synthesized relatively easily. An important warning, however, has been clearly stated (123) “…the biochemical changes that occur after exposure of cells to S-nitrosothiol, with respect to thiol chemistry, are distinctly different from those observed with NO and do not depend on the release of NO from S-nitrosothiol…the cellular events that are triggered or modulated by S-nitrosothiol exposure should not be automatically attributed to NO formation. S-nitrosothiols may mimic many of the downstream effects of NO but through potentially disparate intracellular mechanisms.” For example, SNOs may act as transnitrosating agents rather than NO donors, provided that appropriate nucleophilic groups are accessible to act as receptors for NO+ (1). Although GSNO can release NO in biological systems, the levels are low. In one study, 500 μM GSNO released much less NO than did 5 μM diethylamine nitric oxide (DEANO) (54). Furthermore, higher concentrations of GSNO are routinely employed as growth inhibitory treatments than are used for NO or NO-releasing compounds. For GSNO, concentrations in the range 0.1–1.0 mM are often used (33, 52, 77), whereas for NO up to 10 μM (52, 74, 90) is generally adequate. These NO levels are close to those reported to accumulate in RAW macrophages (57).
With these caveats in mind, it is useful to consider the effects of GSNO observed in selected microbial examples; for a recent review see (14). S-nitrosoacetylpenicillamine (SNAP) (100) and CysNO (46) are also used. Numerous studies have described the growth inhibitory properties of SNOs. Both NO and GSNO transiently inhibit growth of E. coli but careful growth and transcriptomic studies show that, as described above, the effects are quite distinct. In one study, adaptation to GSNO or SNP increased resistance to subsequent insults by NO (54), but the converse was not true. Thus, the GSNO stress response includes upregulation of mechanisms that protect from NO (such as the flavohemoglobin Hmp), but the NO response network does not include GSNO-defensive elements (54). The tolerance of bacteria to GSNO during growth is markedly influenced by the efficacy of bacterial protective mechanisms (see section on bacterial mechanisms). For example, a strain of S. enterica that overexpresses Hmp as a result of mutation of the negatively acting transcriptional regulator NsrR is hyper-resistant to GSNO. Introduction of a second mutation in the hmp gene abrogates GSNO resistance, showing that Hmp is the major protective mechanism (39).
Flatley and colleagues (33) conducted transcriptome profiling experiments comparing E. coli MG1655 grown under control conditions with cells grown with toxic, sublethal GSNO concentrations (0.2 mM). Cultures were grown in a defined medium lacking amino acids in glycerol-limited chemostats at constant growth rate and pH to obviate other environmental effects. GSNO exposure for 5 min was sufficient to elicit tolerance as judged by the development of NO-insensitive cell respiration. Genes upregulated included those known to be involved in NO tolerance, particularly hmp (encoding the NO-consuming flavohemoglobin Hmp) and norV (encoding flavorubredoxin). Significant omissions from the list were those regulated by the iron-responsive regulator Fur, in contrast to an earlier report (77), and adhC encoding a GSNO reductase (see section on bacterial mechanisms). Several met genes involved in methionine biosynthesis or regulation were significantly upregulated; of these, metN, metI, and metR were shown to be important for GSNO tolerance since mutants in these genes exhibited GSNO growth sensitivity. Exogenous methionine abrogated the toxicity of GSNO. This supported the earlier hypothesis that GSNO nitrosates homocysteine (Hcy) to give unstable S-nitrosoHcy, which decomposes to give NO and the radical Hcy(S.) as proposed by Membrillo-Hernández et al. (72) and also demonstrated in vitro therein. The consequence is that homocysteine is withdrawn as an intermediate from the methionine biosynthetic pathway. In an anaerobic chemostat, seventeen genes showed significant upregulation, of which norV, hcp, metR, and metB were also upregulated aerobically. The results revealed new genes important for nitrosative stress tolerance and demonstrated that methionine biosynthesis is a casualty of nitrosative stress.
Jarboe and others (54) extended these observations by applying network component analysis to transcriptomic data sets and showed that GSNO targets homocysteine and cysteine with disruption of the methionine biosynthesis pathway. Reaction of GSNO with Hcy and Cys results in altered regulatory activity of MetJ, MetR, and CysB, activation of the stringent response and growth inhibition (54). Supplementation with methionine abrogates the GSNO effects (54, 90) but is without effect on NO sensitivity (90). Hcy was earlier reported to be an effective endogenous antagonist of GSNO-mediated cytotoxicity (27). Further distinction between the effects of NO and GSNO is provided by the observation that the upregulation of Hmp via the transcriptional regulator NsrR may be demonstrated to arise from the submicromolar levels of NO released from GSNO, but GSNO internalization is not required for this (54). Figure 1 summarizes the transcriptional networks activated by NO and GSNO in E. coli based on experiments published by our laboratory (33, 71, 90). Note that all data presented were the results of carefully controlled experiments in continuous culture and minimal growth media using identical growth rates (imposed by the dilution rate of the chemostat).
Bang et al. (9) observed an increase in SNO formation upon exposure of Salmonella enterica to GSNO, which is approximately doubled in a hmp mutant. Furthermore, data in Figure 3 show that mutation of the transcription factor nsrR, which negatively regulates hmp results in significant reduction in the formation of SNOs upon exposure to GSNO. Here, S. Typhimurium cultures were grown to mid-log phase in defined medium (71) before injection of 2 mM GSNO and further incubation for 1 h. Cells were washed, lysed, and clarified for subsequent total intracellular SNO quantification by ozone-based chemiluminescence (65). Deletion of both hmp and nsrR results in near-wild-type levels of S-nitrosylation. One possible explanation is that as well as hmp, nsrR also negatively regulates other gene(s) responsible for maintaining low SNO levels within the cell.
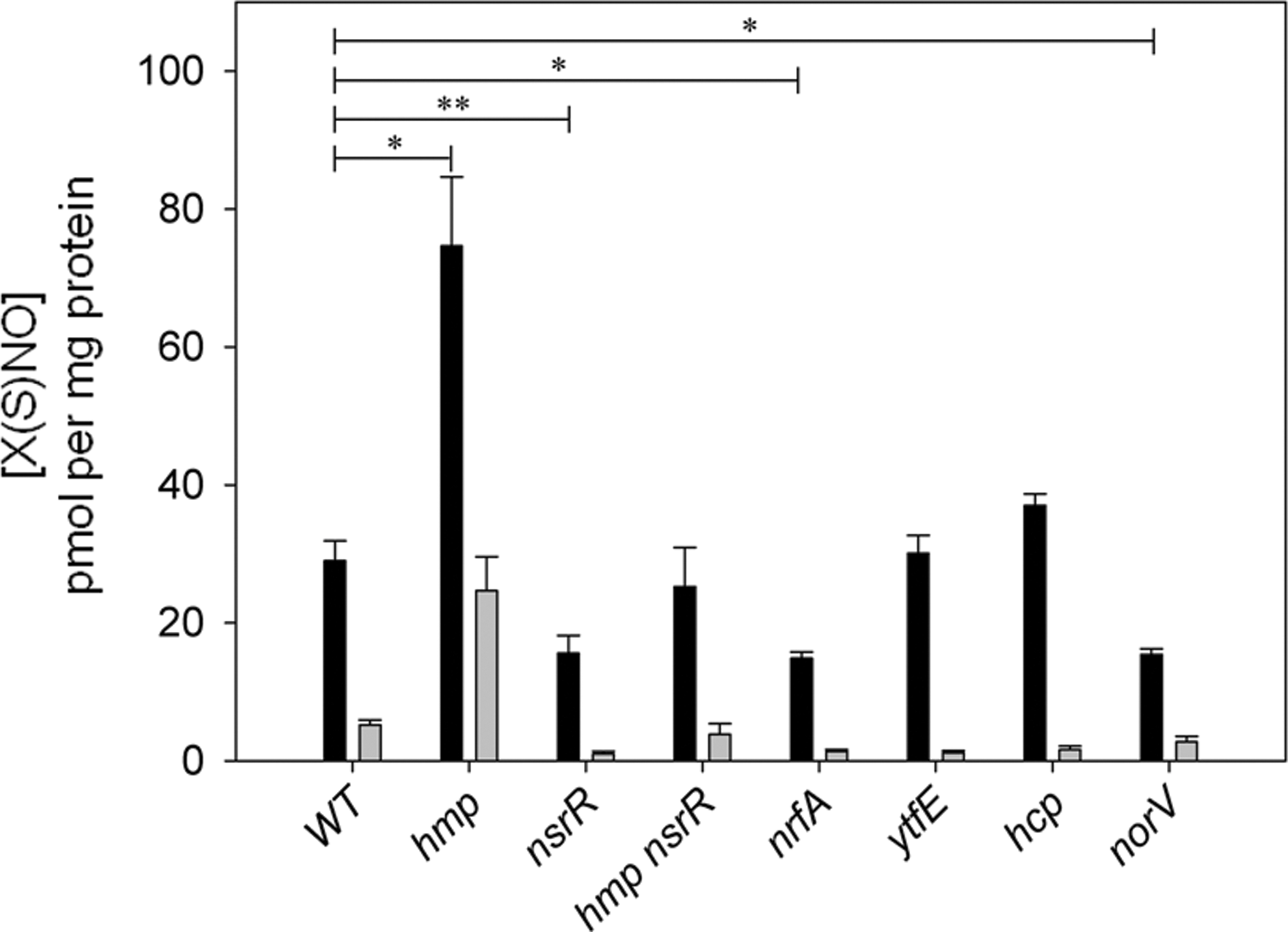
These results, however, appear contrary to data from Moraxella catarrhalis, where an nsrR mutant produced elevated SNO levels in response to nitrite addition (115). It was suggested that elevated expression of the nitrite reductase, aniA, whose expression is repressed by NsrR, accounts for the increase in S-nitrosothiols via an increased capacity of the cells to reduce nitrite to NO. It is also noteworthy that the M. catarrhalis genome does not contain a gene encoding an Hmp-like protein (115).
In Salmonella, deletion of the nitrite reductase gene, nrfA, causes a significant decrease in SNO levels (Fig. 3). This is consistent with findings by Poock et al. (84) that the nrfA mutant shows wild-type sensitivity to GSNO but is more sensitive to another nitrosating agent, SNAP. The authors suggest this could be due to the bacterial detoxification of GSNO, which may not occur for SNAP. The contribution of NrfA to increased SNO levels in GSNO-treated cells remains unclear; Gilberthorpe et al. (40) found that NrfA was not responsible for production of NO from nitrite in Salmonella, although activity under conditions of GSNO stress were not tested. There is, however, evidence that NrfA from E. coli produces NO in the presence of nitrite (19) and makes a relatively modest contribution toward the endogenous formation of SNO in E. coli anaerobically respiring on nitrate (104).
Upon exposure to GSNO, a Salmonella mutant defective in norV, which encodes a flavorubredoxin NO reductase, also forms significantly fewer S-nitrosothiol species than wild type (Fig. 3), whereas deletion of the ytfE or hcp genes, both members of the NsrR regulon, causes no significant change in total SNO levels. However, the expression of both these genes is significantly upregulated under conditions of endogenous nitrosative stress in E. coli (104). The significant attenuation, but not abrogation, of this induction by deleting oxyR suggests that yftE and hcp expression is modulated by SNO-OxyR, though whether this is a direct or indirect effect has not yet been determined. This discovery suggests the gene products of yftE and hcp may play some yet unappreciated role in modulating the bacterial S-nitrosoproteome.
The food-borne pathogen C. jejuni responds to nitrosative stress by activating gene expression from a small regulon under the control of the NO-sensitive regulator, NssR. The extent of the bacterial response to GSNO (0.25 mM for 10 min) has been unraveled in transcriptomic and proteomic analyses of batch- and chemostat-cultured C. jejuni (74). The upregulated genes included many assigned to the NssR regulon, which is considered to be responsive to NO per se. These include two hemoglobins (Cgb and Ctb), but also more than 90 other upregulated genes, notably those encoding heat shock proteins and proteins involved in oxidative stress tolerance and iron metabolism/transport. Upregulation of most of these genes, including cgb and other genes in the NssR regulon, is also elicited by a mixture of the NO-releasing compounds NOC-5 and NOC-7 (each at 10 μM for 10 min) (7a). Mutation of the iron-responsive regulator Fur resulted in insensitivity of growth to NO, suggesting that derepression of iron-regulated genes and augmentation of iron acquisition is a physiological response to nitrosative damage. Interestingly, cells cultured at higher rates of oxygen diffusion were more GSNO-tolerant and had elevated levels of hemoglobins (74) and cells cultured under extreme limitation failed to upregulate the NssR regulon and the globin genes (7a). It will therefore be of interest to assay SNO levels in such cells apparently lacking known defensive mechanisms. NssR mediates the oxygen response, but its molecular basis is obscure. Thus, in addition to NO detoxification catalyzed by the hemoglobins Cgb and possibly Ctb, C. jejuni mounts a more extensive stress response. We have suggested that inhibition of respiration by NO may increase availability of oxygen for Cgb synthesis and function (74).
In a recent study, it was shown that the GSNO produced in vivo by enteric glial cells (components of the enteric nervous system located along the gut) is a key mediator of intestinal epithelial barrier function (102) but also protects against invasion by Shigella flexneri. It is speculated that GSNO nitrosates key proteins involved in S. flexneri invasion, perhaps actin and GTPase. For example, GSNO-induced actin nitrosation might interfere with the process of bacterial actin recruitment, an essential step in bacterial pathogenesis (32). Similarly, glial cell-derived GSNO may inhibit the glucosylating toxins, TsbA and TsbB, of Clostridium difficile by S-nitrosylation of the toxin cysteine protease domain, which displaces the host-derived allosteric cofactor inositol hexakisphosphate and prevents toxin autocleavage and cell entry (103).
Another recent study (31) used GSNO and DETA NONOate to test the hypothesis that xenB expression in Pseudomonas aeruginosa, which encodes a flavoprotein oxidoreductase able to remove a nitro group from nitroglycerin, is inducible by nitrogen compounds. Both GSNO and DETA NONOate were described as 'NO-generating compounds' and each used at 5 mM, in considerable excess over most other studies. Only the NONOate can truly be considered an NO donor (see above). Peroxynitrite, or one of its breakdown products, is also able to form S-nitrosospecies in E. coli, specifically CysNO (71). A transcriptional upregulation of the glutathione import system (gsiABCD) is seen in response to peroxynitrite stress, consistent with data showing that a decrease in glutathione levels can precede the S-nitrosylation of enzymes (10). However peroxynitrite per se is unlikely to be the major cause of S-nitrosylation; the formation of SNOs is more likely due to indirect mechanisms such as the production of nitrite during the breakdown of peroxynitrite and its subsequent reactions with nitrite reductases or other enzymes capable of reducing nitrite to NO. In conclusion, despite numerous published studies, the responses of bacterial cells to SNOs are incompletely understood. A major factor must be the uncritical procedures and principles adopted in some of this work. However, where carefully controlled growth conditions have been adopted and due care taken to compare the effects of a SNO such as GSNO with NO, it is clear that both classes of reagent elicit transcriptional responses to 'nitrosative stress'—typically the expression of flavohemoglobin and other NO defence systems regulated by a small number of dedicated transcription factors, which include in enterobacteria NsrR and NorR. GSNO, but generally not NO, targets thiols and affects methionine biosynthesis. NO, but not GSNO, impacts on FeS clusters as does peroxynitrite (Fig. 1).
S-Nitrosothiols in Bacteria
Lipoamide-dependent metabolism—target of S-nitrosylation
The first comprehensive study to identify a bacterial ‘S-nitrosoproteome’ was conducted by Rhee and colleagues, who identified 29 proteins in M. tuberculosis exposed to 30 mM sodium nitrite (pH 5.5), a 'bactericidal concentration of reactive nitrogen intermediates', for 12 h (97). Though the authors comment that this list is by no means exhaustive, the S-nitrosoproteome contains a disproportionate number of proteins involved in intermediary and lipid metabolism, with approximately half the proteins being essential for bacterial viability (97). Utilizing the biotin switch technique (BST) (53), Rhee and colleagues identified lipoamide dehydrogenase (Lpd) as an S-nitrosoprotein and showed a dose-dependent decrease in its NADH consumption upon exposure to SNAP (97). Lpd, a flavoprotein, contains a pair of redox active thiols and oxidizes dihydrolipoamide to lipoamide during catalytic cycling of the α-keto dehydrogenase complexes, where it constitutes the E3 component (82). Lipoic acid, in the form of lipoamide, is noncovalently associated with the E2 components of these multi-enzyme complexes: dihydrolipoyl transacetylase (pyruvate dehydrogenase) and dihydrolipoyl transsuccinylase (α-ketoglutarate dehydrogenase) (82). In the oxidized, catabolically active form of the enzyme, the vicinal dithiol group of lipoamide exists as a disulfide, but is reduced to two thiol groups during the oxidative decarboxylation of either pyruvate or α-ketoglutarate (81). It is therefore plausible that the reduced form of lipoamide can be S-nitrosylated. Indeed, in a study of proteins with NO-responsive thiols in E. coli, dihydroplipoyl transacetylase (AceF) was identified in a differential thiol trapping technique after exposure to the NO donor compound, DEANO (15). Nitric oxide per se (released from the NO donor compound, ProliNO) was recently shown to inhibit the activity of both the pyruvate and α-ketoglutarate dehydrogenases in Salmonella enterica and, in the same study, that gene expression from NO-treated Salmonella showed significant overlap with a ΔlpdA mutant (99). The same workers previously showed that Staphylococcus aureus, unlike other members of its genus, is able to continue growth following NO exposure, but without the production of acetate, suggesting inactivation of staphylococcal pyruvate dehydrogenase (98). It is worth noting that, while neither of these studies directly demonstrated protein S-nitrosylation, their findings are consistent with the inferences of the proteomic analyses (15, 97). However, the specific site(s) of S-nitrosylation in Salmonella lipoamide-dependent enzymes have yet to be identified. During anaerobic respiration of E. coli in minimal media on nitrate, there is a threefold induction of lpdA expression compared to bacteria respiring anaerobically on fumarate (104). This induction is not observed in a ΔoxyR mutant, suggesting that under these conditions lpdA is part of the S-nitrosylated-OxyR (SNO-OxyR) regulon (104).
Other bacterial S-nitrosoproteins
Five proteins of Helicobacter pylori were recently identified as SNO-proteins, following exposure of bacterial lysates, produced from microaerobic cultures, to 100 μM GSNO and through use of the BST (91). These include alkyl hydroperoxide reductase (TsaA) and the urease alpha subunit (UreA), the latter being reversibly inhibited by GSNO in a concentration-dependent manner (91). S-nitrosylation of a number of proteins from Borrelia burgdorferi has been implicated in the susceptibility of this bacterium to killing by NO (13). Bacteria grown under microaerobic conditions and exposed to DEANO accumulated S-nitrosoproteins in a concentration-dependent manner. The study utilized the BST to visualize S-nitrosoproteins, but did not identify any individual targets (13). Although not identified in the Rhee study (97), the activity of mycobacterial tyrosine phosphatase A (PtpA) has also recently been demonstrated as susceptible to modulation by S-nitrosation (30). A secreted protein, PtpA inhibits phagosome-lysosome fusion by dephosphorylation of vacuolar protein sorting 33B (VPS33B), to which it binds and co-localizes in infected human macrophages (8). Artificial S-nitrosation of PtpA with GSNO occurs on solvent-exposed Cys53, which reduces its activity by ∼60% (30). Similarly, rhodanese (RhdA) of Azotobacter vinelandii undergoes S-nitrosation on Cys230 of its catalytic site (106). In the presence of GSNO, the sulfurtransferase activity of RhdA was reduced to only 7% of controls; likewise, exposure of the protein to the NO donor, NOR-3 resulted in a greater than 95% reduction in activity (106). The crystal structure of RhdA shows that the area surrounding the catalytic residue, Cys230, is only slightly concave (12). The solvent-exposed nature of these residues is in keeping with the general characteristics of S-nitrosocysteine residues, whose distribution was recently shown to be skewed towards surface-accessible areas of proteins (29). The two large exotoxins of Clostridium difficile, TsbA and TsbB, have recently been shown to be S-nitrosylated in vivo and have been identified in extracts of SNO-protein recovered from infected mouse ileum and C. difficile-infected human stool samples (103).
Proteomic approaches to identifying bacterial S-nitrosoproteins
To date, there have been few comprehensive proteomic studies conducted to identify protein S-nitrosylation in bacteria (15, 35, 97, 117). Similar to concerns expressed for transcriptomic analysis, the studies that have been performed have utilized different methodologies and a variety of compounds to induce nitrosative stress. While a review of the methodologies used to detect protein S-nitrosylation is outside the scope of this review and has been attempted elsewhere (92), it is important to recognize that the S-nitrosoprotein profiles generated will be influenced by a number of factors, including growth conditions of the bacteria, the agent(s) used to induce nitrosative stress (if necessary), and the techniques used to isolate and identify the S-nitrosoproteins. Whether certain of these approaches are specific for S-nitrosothiol is still contested (15, 51, 59). Given the recent discovery of endogenous SNO formation in E. coli anaerobically respiring on nitrate and the identification of a novel regulon controlled by SNO-OxyR (104), it is plausible that the studies of Brandes et al. and Wiktorowicz et al., which used aerobically-cultured bacteria as starting material, underestimate the number of E. coli proteins susceptible to S-nitrosylation (15, 117). It makes biological sense that at least a proportion of proteins under transcriptional control of an S-nitrosylated transcription factor would be susceptible to similar post-translational modification, if only as a means to control the SNO flux through the cell. Although the resin-assisted capture technique (SNO-RAC) of Forrester et al. generated a larger list of 44 peptides from E. coli treated with 500 μM CysNO (albeit a nonphysiological concentration), it surprisingly does not include a peptide derived from OxyR ((35), SI). This is most likely attributable to a low abundance of the species in wild-type bacteria, but the absence of detailed culture conditions in the Methods section makes it difficult to comment (35). However, other studies have robustly demonstrated the S-nitrosylation of this protein following its overexpression (47, 62, 104). Characterization of the endogenous S-nitrosoproteome of E. coli anaerobically respiring on nitrate represents an interesting challenge for future studies and may reveal hitherto unappreciated activities for proteins of both known and unknown function. It may also reveal novel targets for new therapeutic strategies utilizing NO or SNOs. In ideal circumstances, identification of pathogenic S-nitrosoproteomes would be performed on samples obtained from a physiologically relevant setting, such as mycobacterial SNO-proteins isolated from bacteria grown inside interferon-treated macrophages (97). However, the methodological constraints of requiring large volumes/high concentrations of starting material and the impracticability of obtaining sufficient quantities from physiologically relevant models mean that use of exogenous sources of nitrosative stress will probably continue. In any case, it is prudent to validate individual sites of S-nitrosylation in a protein of interest by other means, such as targeted mutagenesis of potentially S-nitrosylated Cys residues.
Bacterial Mechanisms for the Processing/Detoxification of S-Nitrosothiols
Denitrosylation, the process of reversing S-nitrosylation by removing NO from thiol side groups, was once considered a random and uncontrolled event. However, mounting evidence suggests that bacteria, akin to mammalian systems, have mechanisms in place to process and deplete SNOs, alleviating the detrimental consequences of nitrosative stress.
GSNO reductase
The enzymatic reduction of GSNO to glutathione disulphide and ammonia, via a glutathione N-hydroxysulfenamide intermediate, is a reaction widely distributed in mammals and is thought to be evolutionarily conserved from bacteria to humans as a means of controlling cellular SNO levels and of protection from nitrosative stress. In E. coli, a GSNO reductase (formally glutathione-dependent formaldehyde dehydrogenase) has been identified (67). Other glutathione-dependent class III alcohol dehydrogenases (AdhC) have also been identified in N. meningitidis, Hemophilus influenzae, and Streptococcus pneumoniae (61, 89, 110) that catalyze the reduction of adducts of glutathione including GSNO. Neisseria gonorrhoeae also shows GSNO reductase activity; however, AdhC does not contribute to GSNO reductase activity here due to the presence of a premature stop codon suggesting a different mechanism of GSNO metabolism in this species (89).
Thioredoxin system
The thioredoxin system, comprising thioredoxin (Trx), thioredoxin reductase (TrxR), and NADPH, has emerged alongside GSNO reductase (GSNOR) as a physiologically important denitrosylase. Evidence of SNO catabolism originated from a kinetic study investigating the breakdown of GSNO by Trx/TrxR isolated from E. coli. Researchers demonstrated that GSNO oxidized reduced thioredoxin in the presence of oxygen, resulting in GSH and NO formation as demonstrated by HPLC analysis. E. coli Trx has a dithiol moiety that is transiently S-nitrosated, resulting in the reduction of SNO-bound proteins. This intermediate decomposes to NO with oxidized thioredoxin as the ultimate reaction product. TrxR subsequently re-reduces Trx by employing NADPH as a reducing equivalent (78). This mechanism has been brought into question by other investigators who found that following the addition of GSNO to E. coli Trx/TrxR proteins, HNO, as opposed to NO, was formed (109). GSNO detoxification was also demonstrated in vitro by the thioredoxin system isolated from M. tuberculosis. This was determined by the oxidation of NADPH by spectrophotometric means and by HPLC analysis of the reaction product, GSH (7).
Both thioredoxins from H. pylori (Trx1 and Trx2) on the other hand have been shown to play a pivotal role in resisting nitrosative stress due to an increased susceptibility towards GSNO and sodium nitroprusside (SNP) in strains deficient in these proteins (18). In addition, the reductase component, TrxR, was shown to be essential in protecting N. gonorrhoeae from NO-mediated killing. However, this was not attributed to the thioredoxin system but, at least in part, to the requirement of TrxR for the expression of both nitrite and NO reductase under microaerobic conditions (88). It is also of note that the gene encoding TrxR in Candida albicans is amongst the most highly expressed following treatment with NO, signifying its role in denitrosylation (50).
γ-Glutamyl transpeptidase
Mammalian γ-glutamyl transpeptidase (GGT), like the GGT enzymes in S. enterica (see section on bacterial uptake and Fig. 2) and M. tuberculosis (25), accelerates the breakdown of GSNO by hydrolysis of the γ-glutamyl moiety, resulting in S-nitrosocysteinylglycine (CG-SNO) formation (3, 48). Studies conducted in vitro with GSNO and GGT isolated from mammalian cells have shown that the product, CG-SNO, is markedly more susceptible to transition metal ion catalysis than GSNO, resulting in NO production (3, 48), a finding that has not been documented in the microbial literature to date.
Glutathione peroxidase
Mammalian GSH-Px, a selenium-containing enzyme, also has a demonstrated capacity to catalyze the breakdown of GSNO, possibly by a selenocysteine-based SNO-lyase mechanism (49). The gene encoding GSH-Px is present in several bacterial pathogens, including N. meningitidis (75) and Streptococcus pyogenes (16), but a role in denitrosylation has yet to be elucidated.
Xanthine oxidase
A flavin-containing enzyme isolated from mammalian cells, XO, demonstrates discrete mechanisms of RSNO catabolism under aerobic and anaerobic conditions. In the presence of oxygen, XO plus xanthine indirectly mediates both GSNO and in part CysNO decomposition via superoxide production. It has been proposed that NO is produced following these reactions which reacts rapidly with superoxide, resulting in peroxynitrite formation. In the absence of oxygen, CysNO, but not GSNO, serves as an electron acceptor substrate for XO, resulting in the formation of NO (112). Xanthine oxidase, like GSH-Px, is distributed across a range of bacterial species (118); however, a physiological role in SNO detoxification has not been demonstrated.
Nitroreductase
Microarray studies have revealed that the ntrA gene, which encodes a putative nitroreductase (NtrA), is induced by GSNO in S. aureus (111). Further investigation revealed that NtrA is a bifunctional protein able to reduce both the antimicrobial nitrofuran complexes and GSNO but not NO, CysNO, or S-nitrosohomocysteine. The K m for GSNO of NtrA was approximately 4-fold lower than that of the GSNO reductase from E. coli (67). Sequence searches revealed no ntrA homologue amongst the nitroreductases found in the enterobacterium; indeed NtrA appears to be part of a distinct group of nitroreductases not found in E. coli.
In summary, bacteria display a number of potential mechanisms that might process and/or detoxify SNOs. However, for many, a clear role has not yet been established and further work is required. Now that conditions that induce SNO formation have been established, careful transcriptomic studies followed up by mutagenesis should prove fruitful.
S-Nitrosoprotein Formation in Host Cells During Infection: Bacterial Interference in S-Nitrosothiol Homeostasis
As many bacterial proteins regulated by S-nitrosylation are essential for viability (see section on S-nitrosothiol in bacteria), it is likely that this process will play an important role during the interaction between bacteria and their mammalian hosts. A wide range of targets have been identified in vitro for a number of bacteria, but this is unlikely to accurately reflect the situation that exists in vivo. Although acquiring sufficient quantities of intracellular bacteria for S-nitrosoproteome analysis is technically demanding, it would be extremely interesting to determine how this process affects the proteome of the bacterium and in turn how this impacts on bacterial metabolism/survival. However, S-nitrosylation and denitrosylation are dynamic and fluid processes (2). In the context of infection, they are events taking place among a myriad of confounding factors, making their study in isolation less biologically relevant. One such factor is the detoxification of NO by bacteria, which, in this laboratory, has been shown to impact upon the formation of S-nitrosothiols in the host cell (64). Infection of mammalian cells with bacteria containing NO detoxification mechanisms results in dramatic depletion of host cell SNOs, probably due to restriction of de novo S-nitrosylation. Through the rapid conversion of NO per se into less active nitrogen species, bacterial mechanisms of NO detoxification are metabolically epistatic to the formation of host cell S-nitrosothiols, a model of which is represented in Figure 4. Though these proteins, such as bacterial flavohemoglobin (Hmp) or nitric oxide reductases (NorB), have no direct interaction with S-nitrosoproteins (i.e., they do not catalyze the breakdown of S-nitrosothiols in the same way as GSNO reductases), their activities effectively remove one of the substrates necessary for SNO formation. The result is that the balance between SNO formation and decomposition is tipped in favor of decomposition, as demonstrated by a lower burden of S-nitrosospecies in murine macrophages infected with wild-type bacteria as compared to uninfected controls (Fig. 5).
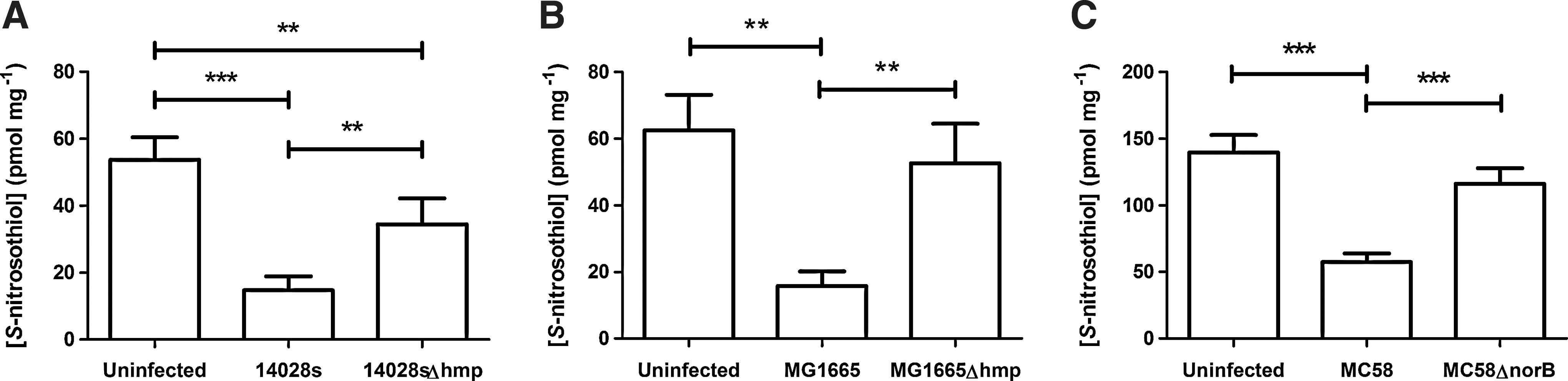
That bacterial infection is potentially associated with impairment of specific S-nitrosylation events is of great interest, as such interaction could lead to physiological dysfunction of the host cell, with profound implications for the outcome of an infection. Eukaryotic targets that are potentially susceptible to changes in SNO abundance, resulting in cell dysfunction, might include proteins involved in innate immunity and intracellular signalling (see two examples below). Currently this has not been investigated, but is an important topic in future research as it may lead to new therapeutic targets for sepsis.
Modulating innate immunity: S-nitrosylation of surfactant protein D
Surfactant protein D (SP-D) functions as a dodecamer, or a tetramer of trimers, and acts as a calcium-dependent pattern recognition receptor (PRR), binding to pathogen-associated molecular patterns (PAMPs) as part of the innate immune system (reviewed in Ref. 63). SP-D has been shown to bind to both Gram-positive (44) and Gram-negative (83) bacteria, though the precise role of SP-D varies and is dependent on the bacterial strain involved (66, 96, 120). SP-D has both pro- and anti-inflammatory properties dependent upon which interaction it makes with host cell receptors. SP-D can be S-nitrosylated, which occurs on Cys15 and Cys20, the N-terminal cysteine residues responsible for the higher order multimerisation of SP-D trimers (42). Addition of Cys NO to bronchoalveolar lavage fluid (BAL, to form ‘SNO-BAL’) resulted in the formation of S-nitrosated SP-D trimers and monomers (SNO-SP-D), which due to the presence of the NO moiety were unable to multimerize into the dodecameric form. These lower order multimers bind to calreticulin/CD91 and stimulate macrophage chemotaxis, a pro-inflammatory process. Reduction of SNO-BAL with ascorbate ablated this effect and permitted the formation of dodecamers, which bind to SIRP-α and block pro-inflammatory signaling (42). These findings have subsequently been corroborated in vivo (5, 6). For a more in-depth review of the S-nitrosylation of SP-D, see Ref. 4).
Dynamin2, eNOS, and the regulation of epithelial cell invasion
The members of the dynamin family are large (∼100 kDa) GTPases involved in the formation of intracellular vesicles (reviewed in Ref. 95). Through a direct protein–protein interaction with endothelial nitric oxide synthase (eNOS), dynamin2 (DynII) selectively manipulates intraflavin electron transfer through eNOS and enhances the synthesis of NO (17). The proximity of DynII to eNOS results in S-nitrosylation of DynII in its GTPase domain (58), which results in increased GTPase activity and an increase in the rate of cell surface receptor internalization (114). Wang and colleagues demonstrated the importance of S-nitroso-DynII in the invasion of bladder epithelial cells (BEC) by uropathogenic E. coli (UPEC). By inhibiting the synthesis of NO or specifically mutating the residues involved in the formation of S-nitroso-DynII (Cys86 and Cys607), there was marked reduction in the number of viable intracellular UPEC recovered from infected BEC (114, 116).
This model is particularly interesting because it identifies the constitutive nitric oxide synthase eNOS as the regulatory source of NO and suggests that lower levels of NO can profoundly influence the early stages of an infective process. In the context of the work of Wang et al., the recent demonstration that mechanisms of bacterial NO detoxification are capable of influencing the concentration of host cell S-nitrosothiols (64) suggests that bacterial NO detoxification may plausibly have important roles to play in processes preceding the expression of iNOS, when overall NO abundance is lower and the amount of NO detoxified by these proteins constitutes a significantly larger proportion of the total.
Conclusion and Perspective
The formation and degradation of S-nitrosothiols has emerged as an important mechanism of post-translational protein modification and is ubiquitous in biology. In bacteria, small nitrosothiols are transported into the cells and subsets of bacterial proteins susceptible to S-nitrosothiol formation have been identified. The consequences of bacterial SNO formation are evident from transcriptomic studies, which also serve to distinguish clearly between the antimicrobial effects of SNOs (such as GSNO), NO, and peroxynitrite, for example. Future studies in this area should seek to distinguish more clearly between these agents; in particular, the widespread use of small molecular weight SNOs as surrogates for NO needs urgent reconsideration. In addition, the demonstration of conditions suitable for endogenous SNO formation in enterobacteria (specifically E. coli) should be exploited and our knowledge of this phenomenon broadened. Methodological difficulties hamper characterization of the S-nitrosoproteome, particularly in the case of pathogens where intrinsic NO detoxification mechanisms influence S-nitrosation events in host cells.
Footnotes
Acknowledgments
JRL, LJH, and RCR are supported by the Meningitis Research Foundation (project 0902.0). SM is supported by the BBSRC through a grant to RKP, and LAHB is supported by the European Commission Framework 7 through a grant to RKP
Author Disclosure Statement
No competing financial interests exist.