
Abstract
Introduction
T
The energy-transducing capacity of mitochondria meets the cellular energy demands, thus supporting metabolic, osmotic, and mechanical functions. Mitochondria are sources of H2O2, and play a pivotal role as mediators of the intrinsic apoptotic pathway. Thus, they play significant roles in the function and plasticity of neurons and are implicated in the pathogenesis of a variety of neurological disorders (155). The most prominent metabolic process carried out by mitochondria is oxidative phosphorylation (OXPHOS) that generates ATP, the universal energy currency. On the other hand, high levels of H2O2 have been associated with mitochondrial redox changes and macromolecule oxidation during aging and are believed to mediate the detrimental effects associated with mitochondrial dysfunction in brain aging (14) and neurodegenerative disorders (18, 20, 214). The cellular composition of the brain consists mainly of terminally differentiated neurons, its regenerative capacity is relatively reduced as compared with other organs, and a dramatic decline in neurogenesis with age and in neurodegenerative diseases may contribute to the impairment of learning and memory (140). Thus, the brain is highly susceptible to neuronal loss due to hypometabolic states and the impairment of redox homeostasis. Age-related changes in energy production and redox status cannot be viewed as independent variables, but rather as an interdependent relationship reflected in the mitochondria energy-redox axis that represents a dual pronged approach to assess the changes in mitochondrial function as a function of age and disease (250).
The generation of H2O2 by mitochondria depends on the respiratory state (faster rates of H2O2 release in state 4 respiration and slower rates in state 3 respiration) and shows an exponential dependence on the mitochondrial membrane potential (32, 132). Mitochondrial H2O2 is implicated in the regulation of the cellular redox status, thus transducing redox signals into a wide variety of responses, such as proliferation, adaptation, differentiation, and cellular death pathways (199). Low to intermediate levels of H2O2 are involved in the regulation of redox-sensitive signaling and transcription, whereas high levels are involved in oxidative damage to cell constituents. The release of oxidants from mitochondria as a function of the mitochondrial metabolic and redox states serves as a coordinated response between these seemingly autonomous organelles and the rest of the cell through the modulation of redox-sensitive signaling and transcription pathways. Conversely, mitochondria are the recipients of cytosolic signaling, such as mitogen-activated protein kinases (MAPKs) and the phosphatidylinositol 3-kinase/protein kinase B (PI3K/Akt) pathway of insulin signaling, which elicit profound changes in the mitochondrial energy-transducing capacity.
This review focuses on the role of a coordinated metabolic triad (Fig. 1) entailing a regulatory devise encompassed by mitochondrial function (maintenance of the energy-redox axis), cytosolic kinase signaling, and transcriptional pathways.

The Mitochondrial Energy-Redox Axis
Mitochondria provide most of the energy needed for cellular functions by the metabolism of fuel molecules into ATP through OXPHOS. The generation of ATP entails the oxidation of acetyl-CoA in the tricarboxylic acid (TCA) cycle with the concomitant generation of reducing equivalents (NADH, FADH2) that flow through the respiratory chain, generating a proton motive force (154); electron leakage leads to the generation of O2
Mitochondrial energy metabolism
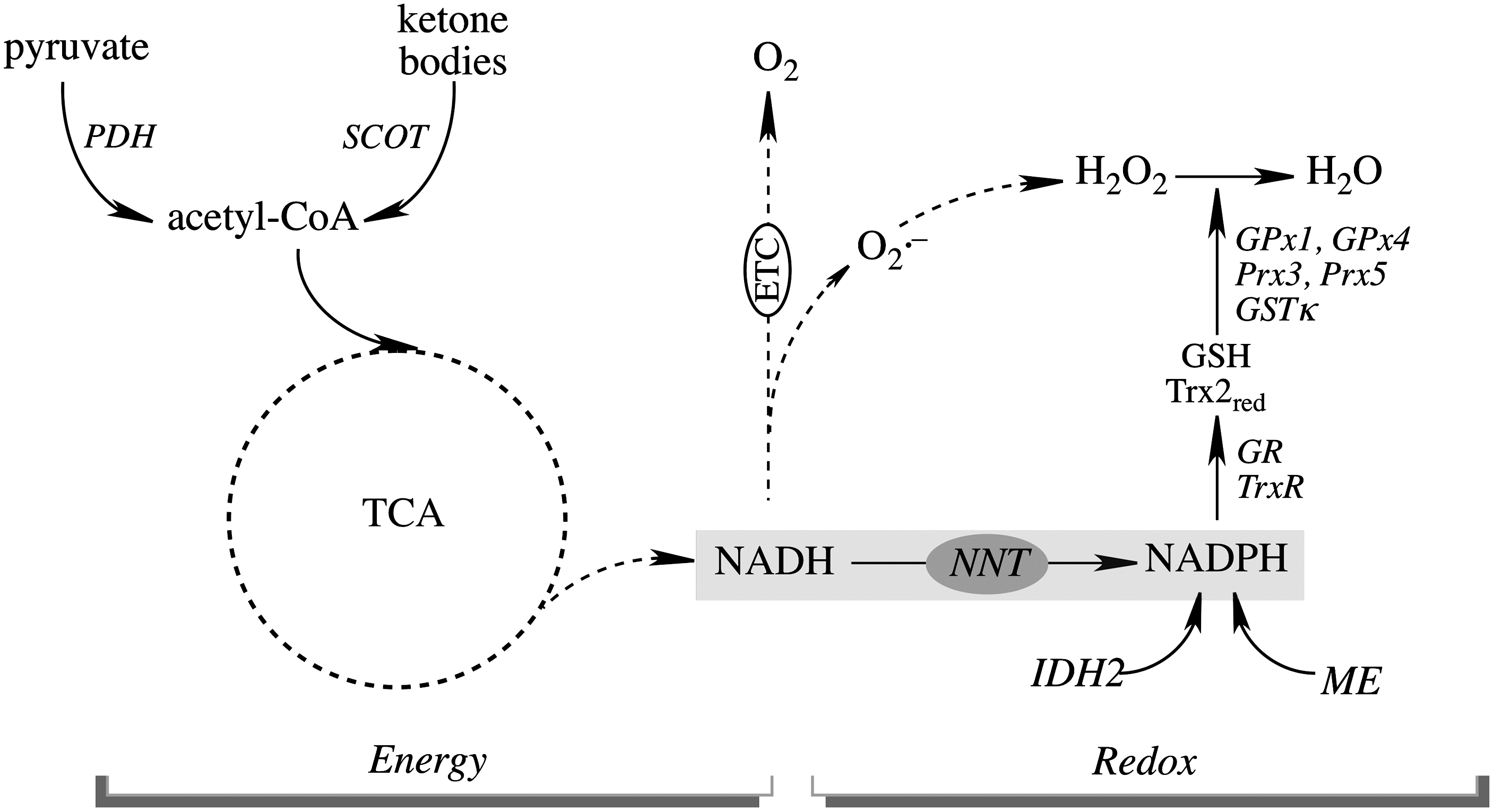
The effects of aging on mitochondrial energy metabolism are tissue specific and are more prominent in tissues whose parenchyma contains mostly postmitotic cells such as brain, heart, and skeletal muscle. Partial loss of the energy-transducing capacity has been documented in mitochondria isolated from aged animals and attributed to changes in protein expression and activities. Glucose is the primary fuel for brain, whereas metabolism of ketone bodies represents an alternative fuel source during glucose deprivation (89) (Fig. 2). Pyruvate, generated from glycolysis, undergoes oxidative decarboxylation by the pyruvate dehydrogenase (PDH) complex to acetyl-CoA that feeds into the TCA cycle. PDH activity in the brain was found to decrease with age (263, 264). In addition, there is an age-dependent decrease in succinyl-CoA:3-oxoacid Co-A transferase (SCOT) activity (137), a key mitochondrial matrix enzyme that metabolizes ketone bodies to acetyl-CoA (Fig. 2); the decreased SCOT activity as a function of age was due to irreversible protein post-translational modifications (137). In a triple transgenic mouse model of Alzheimer's disease, ketone body metabolism is a temporary mechanism that prevents the further decline of brain mitochondrial bioenergetic capacity (248) which is associated with decreased activities of PDH and cytochrome oxidase. 2-Deoxy-D-glucose treatment induced ketogenesis in the same mouse model, and this resulted in increased ketone body metabolism in the brain and a significant reduction of both amyloid precursor protein and amyloid-β (248). The activities of TCA enzymes, such as aconitase and α-ketogluterate dehydrogenase (α-KGDH), also decline as a function of age (251) and the activities of PDH, α-KGDH, and isocitrate dehydrogenase (IDH) are also lower in Alzheimer's disease (37, 249). It may be surmised that alterations in the activities of TCA cycle enzymes and of enzymes controlling the entry of acetyl-CoA into the TCA cycle, such as PDH and SCOT, affect NADH levels and contribute significantly to the decline in mitochondrial bioenergetics during aging and neurodegeneration.

Mitochondrial OXPHOS is a process that encompasses electron transfer through the complexes I, II, III, and IV of the respiratory chain; this exergonic electron transfer is the driving force for the vectorial H+ release into the inter-membrane space and for the H+ re-entry to the matrix through F0 of complex V with ATP synthesis by F1-ATP synthase. Electron transfer in mitochondria decreases in the aged brain (21, 170), with more marked changes in complexes I, III, and IV (135, 168, 169, 173). The inhibition of complex I activity on aging occurs with a decrease in NAD+ levels (26) that leads to the impairment of the turnover efficiency of the TCA cycle, irrespective of the presence of acetyl-CoA. Moreover, reduced electron transfer can also lead to decreased mitochondrial inner membrane potential, which is observed in the aged rat brain (136, 205). The F1-ATPase activity of complex V also decreases with age due to the nitration of Tyr269 close to the Mg++ binding site of the F1β subunit (137).
The operational concepts of mitochondrial metabolic states and respiratory control are defined as state 4 (resting or proton motive force controlled-respiration), with the availability of respiratory substrates but not ADP, and state 3 (active respiration) with ample respiratory substrate and ADP availability (45). Mitochondrial active respiration decreases in aging in terms of a marked decline in state 3 respiration and the related respiratory control ratio and membrane potential, as well as an increase in state 4 respiration (29, 137, 173), all of which indicate a lower energy-transducing efficiency.
Components of the electron transport chain (ETC) exist as large macromolecular assemblies or so-called supercomplexes (208), the ultra-structure of which determines the activity of mitochondrial OXPHOS and, therefore, plays a vital role in mitochondrial phenotype in aging and neurodegeneration (84, 211). The supramolecular architecture of OXPHOS complexes in rat brain cortex is affected by aging: The largest decreases were observed with supercomplexes IlIII2, I1III2IV2, and I1III2IV1 (80).
Mitochondrial function is also regulated by·NO, largely on reversible binding to cytochrome oxidase (36), and at higher concentrations, it inhibits electron transfer at the bc 1 segment of the respiratory chain (190 –192). Although not free of discrepancy (236), multiple studies have shown the occurrence of a mitochondrial nitric oxide synthase (mtNOS) in several tissues [including brain (233) and with a function in rat brain development (200)] by its biochemical activity and by the inhibition of mitochondrial respiration elicited by NOS substrates and inhibitors (167, 171, 172). Mitochondrial metabolic states regulate the diffusion of both·NO and H2O2 from mitochondria to cytosol (32, 200). Interestingly, the tissue levels of mtNOS have been reported to decrease with age, particularly in the brain, and it has been suggested to be a biomarker of brain aging.·NO can signal through the cGMP-PKG pathway to activate Sirt1 and PGC1α (33, 171, 175).
The levels of neuronal NOS (nNOS) in the rat brain increase with age (137), and this is associated with the S-nitrosylation of cytosolic proteins and the nitration of a discreet set of mitochondrial proteins (40, 137). Excessive production of·NO in the brain has been implicated in a number of neurodegenerative diseases, including Alzheimer's and Parkinson's disease (156, 247). Thus, during brain aging and neurodegeneration, the physiological regulation of mitochondrial function by lower concentrations of·NO appears to decrease due to declined mtNOS and, in a separate phenomenon, increased pathophysiological levels of·NO (generated by nNOS and inducible NOS [iNOS]) lead to the replacement of specific·NO signaling by random·NO-mediated modifications to proteins.
Mitochondrial redox homeostasis
After the initial reports on intact heart and liver mitochondria as an active source of H2O2 by Chance and Boveris (28, 31), further work established that superoxide anion (O2
In brain mitochondria, H2O2 is eliminated mainly by glutathione (GSH)- or thioredoxin (Trx)-driven catalysts that depend on NADPH as ultimate electron donors (Fig. 3).
GSH-based systems
GSH, synthesized in the cytosol from glycine, glutamate, and cysteine in a two-step process by the enzymes γ-glutamylcysteine synthetase and GSH synthase (87), is imported into the mitochondria through the dicarboxylate- and oxoglutarate carriers on the inner mitochondrial membrane (88, 262). The role of mitochondrial thiols in redox signaling (165) and cell death pathways (252) has been recently reviewed. The redox potential—which is calculated of the mitochondrial GSH/glutathione disulfide (GSSG) or Trx2red/Trx2ox couples—is ∼–300 and –340 mV (116, 124), respectively. The mitochondrial GSH pool can apparently function autonomously from the cytosolic GSH pool in response to local changes in the production of mitochondrial oxidants (107).
Mitochondrial GSH protects against oxidative stress largely as a cofactor for glutathione peroxidases (GPxs), glutathione-S-transferases, sulfiredoxins, and glutaredoxins (Grxs) (152, 197). GPx1 localizes mainly in the mitochondrial matrix, whereas GPx 4 (also referred to as phospholipid hydroperoxide GPx) (210, 232) occurs in the inner mitochondrial membrane; the latter detoxifies mainly phospholipid hydroperoxides, and its significance is underscored by the embryonic lethality that follows systemic ablation of GPx4, which is explained in part by studies conducted on the expression of GPx4 in the embryonic brain and its role in organogenesis (24). The mouse brain showed a decreased GSH/GSSG ratio and a slight shift toward a more pro-oxidizing redox potential with regard to age (195, 196). Among the glutathione S-transferases (GST), the GST class κ is mitochondrion specific and also exhibits some selenium-independent peroxidase activity; in addition to the GST class κ, the α, π, and μ classes have also been reported in brain mitochondria (85, 194); however, the specific function of these GST classes in brain mitochondria is not clear, except as a response to xenobiotic inducers. It is worth noting that in a cross-species comparison study on the conservation of longevity assurance mechanisms, GST was the common denominator in Caenorhabditis elegans, Drosophila, and mice (158), thus supporting the hypothesis that detoxification reactions, such as those catalyzed by GSTs, are an important part of longevity assurance mechanisms (265).
Protein thiols are sensitive to changes in the redox environment (57): the GSH/GSSG redox couple regulates protein function through the reversible formation of mixed disulfides between protein cysteine sulfhydryls and GSH in a process termed S-glutathionylation. Mixed-disulfide formation affects the activity of enzymes, transcription factors, and transporters, thus enabling them to respond to the redox environment by reversible activation/inactivation (78, 227, 261). Thus, S-glutathionylation reflects the redox status of the mitochondria (207) and is viewed as a regulatory device for the proteins involved in energy metabolism, redox signaling, and cell function (61, 62, 99, 130, 162). A number of proteins have been identified to be S-glutathionylated during oxidative conditions, including components of the energy metabolism: (a) SCOT and the E2 subunit of PDH (82, 107); (b)TCA cycle enzyme such as aconitase (92), α-KGDH (177), and IDH (126), and (c) complexes I (226), II (50), and V (82, 240). S-glutathionylation of SCOT and ATP synthase (F1 complex, α-subunit) in brain mitochondria resulted in a decrease of activity and a reduction potential of −171 mV; supplementation of mitochondria with respiratory substrates to complex I or complex II increased NADH and NADPH levels, restored GSH levels through a reduction of GSSG, elicited deglutathionylation of mitochondrial proteins, and resulted in a more reducing mitochondrial environment (−291 mV) (82). Complex I is persistently glutathionylated under conditions of oxidative stress, and this resulted in increased generation of O2 .−and decreased mitochondrial function (226). Conversely, S-glutathionylation of adenine nucleotide translocase (ANT) protects against mitochondrial membrane permeabilization and apoptosis (193). These data provide evidence of mitochondrial redox changes that modulate energy metabolism through protein thiol modifications.
The reversible formation of protein-GSH mixed disulfides has been suggested as a protective mechanism that masks critical sulfhydryls from irreversible oxidation; the reversibility of this process acquires further significance because of its involvement in the redox regulation of signal transduction (98, 130). Protein-mixed disulfides are specifically reduced by Grxs (Grx2 is the mitochondrial isoform) through a monothiol mechanism (102). Oxidized Grx2 is reduced by GSH, which is regenerated from GSSG by NADPH-supported glutathione reductase (GR). Grx2 is constitutively expressed in neurons and glia in mouse and human brain (122), and the knockdown of cytosolic Grx1 is associated with a loss of mitochondrial membrane potential (203) and is essential for maintaining the functional integrity of brain mitochondrial complex I (125). Grx2 protects cells against oxidative damage (72) involving Akt signaling and the redox-sensitive transcription factor NF-κB and anti-apoptotic Bcl-2 (166). In addition, Grx2 has been characterized as being a part of an iron-sulfur cluster that senses redox changes and controls the activation of Grx2 during conditions of oxidative stress (103), thus expanding the interaction between oxidants, mitochondrial redox status, and protein glutathionylation.
Trx-based systems
The reducing power for peroxiredoxins (Prx) is transmitted through thiols of the Trx system: NADPH→Thioredoxin reductase (TrxR)→Trx→Prx (257). A comprehensive study on immunohistochemical mapping of all six Prx subtypes in the mouse brain revealed that astrocytes and microglia were reactive to Prx6 and Prx1, respectively; immunoreactivity for Prx1 and Prx4 in the nuclei of oligodendrocytes; in neurons, Prx3 and Prx5 were found in the stratum lucidum of the hippocampus and Prx2 in the habenular nuclei (115). Of these Prxs, Prx2 was critical for the maintenance of hippocampal synaptic plasticity against age-associated oxidative damage (129) by a mechanism entailing the oxidant- and age-dependent mitochondrial decay of hippocampal neurons; in addition, the expression of Prx2 in hippocampal neurons increased as a function of age.
Mitochondrial Prx3 and Prx5 (Fig. 3) are involved in the enzymatic degradation of H2O2; Prx5 can also reduce ONOO− (69, 183). Trx2 is highly efficient at reducing disulfides in proteins (163), thus impacting cellular functions such as antioxidant defenses and the redox control of transcription and signal transduction (8, 101). Using a polarographic method for real-time detection of H2O2, it was concluded that the removal of H2O2 by energized brain mitochondria was largely dependent on the Trx/Prx system with a modest contribution by the GSH/GPx system (66). Prx3 and Prx5 are constitutively expressed in human neuroblastoma Sh-SY5Y cells, and their silencing by small hairpin RNAs renders the cells more susceptible to oxidative damage and apoptosis (63). Prx3 protects hippocampal neurons against excitotoxicity, and its up-regulation prevented or reduced gliosis, that is, proliferation of astrocytes in certain areas of the brain (96). The overexpression of Prx-3 reduces H2O2 production and lipid peroxidation and protects cells from hypoxia-, TNFα-, cadmium-, and oxidant-induced cell death (46, 49, 176, 243), and a neuroprotective effect was observed when administered into ischemic brains (108). The increased Prx3 (along with TrxR2 and Trx2) immunoreactivity in the hippocampus of aged dogs as compared with adult dogs was interpreted as a reduction of neuronal damage (against oxidative stress) during aging (3). Prx3 levels are found to be significantly lower in the brains of Alzheimer's disease patients (128), and a deficiency in Prx3 is also associated with multiple neurodegenerative disorders such as amyotrophic lateral sclerosis, Parkinson's disease, and Down syndrome (133, 244). Trx2+/− mice show reduced ATP production and electron-transport chain rates (184); this notion is further supported by the increased apoptosis in early embryos of Trx2−/− mice (embryonic lethal) with mitochondria maturation. The systemic ablation of mitochondrial TrxR-2 also yielded embryonic lethal phenotypes (54). The levels of TrxR2 decrease with age in different tissues and is accompanied by enhanced susceptibility to apoptosis (201), also suggesting that NADPH supply and TrxR activity rather than activities of Trx2 may be rate limiting for the protection against oxidants and be of importance in dysregulated redox status during aging and neurodegeneration (182). The significance of the redox couples and redox catalysts for brain aging and age-related neurodegeneration just mentioned is underscored by the highly oxidized mitochondrial and cellular redox environment that is involved in these processes (146, 168, 252).
Interdependence of energy- and redox components—role of nicotinamide nucleotide transhydrogenase
The ultimate reductant of mitochondrial redox systems is NADPH (supporting the activities of GR and of TrxR) (Fig. 3). The age-dependent decrease in the ratio of NADPH/NADP+in kidney mitochondria (251) indicates that NADPH deficiencies are another factor in mediating mitochondrion-dependent aging. Mitochondrial NADPH is mainly formed through three pathways: NADP+-dependent IDH2, malic enzyme, and nicotinamide nucleotide transhydrogenase (NNT) (Fig. 3). Of these pathways, 50% of the mitochondrial NADPH pool is uncoupler sensitive, thus suggesting that the NNT-catalyzed reduction of NADP+ accounts for more than 50% of the mitochondrial NADPH pool (202). NNT, a nuclear encoded mitochondrial 110 kDa protein located on the inner mitochondrial membrane (100), catalyzes the reversible reduction of NADP+ to NADPH coupled to the oxidation of NADH to NAD+. The proton gradient across the mitochondrial inner membrane strongly stimulates the forward reaction, that is, the generation of NADPH and the subsequent capacity for H2O2 reduction (255). NNT plays an important role in regulating cellular redox homeostasis, energy metabolism, and apoptotic pathways (150, 253). Knockdown of NNT in PC12 cells results in an altered redox status encompassed by decreased cellular NADPH levels and GSH/GSSG ratios and increased H2O2 levels, as well as an impaired mitochondrial energy-transducing capacity. The activation of redox-sensitive signaling (c-Jun N-terminal kinase, JNK) by H2O2 after NNT suppression induces mitochondrion-dependent intrinsic apoptosis and results in decreased cell viability (253). NNT activity provides a link between the mitochondrial metabolic function (energy-transducing activity) and redox homeostasis by coupling NADPH generation to the TCA cycle and active respiration (Fig. 3). This supports the notion that changes in cellular bioenergetics and changes in the redox status of the cell cannot be viewed as independent events, but rather as an interdependent relationship established by the mitochondrial energy-redox axis (250). Disruption of electron flux from fuel substrates to redox components due to NNT suppression not only compromises mitochondrial dysfunction, including energy-transducing capacity and redox homeostasis, but also affects cellular functions through interactive communication between mitochondrion-generated second messengers and cytosolic redox-sensitive signaling (see section III). The modulation of NNT function could be considered important in the collective impairments of the interdependent mitochondrial energy-redox axis and the regulation of cytosolic redox-sensitive signaling that is inherent in several pathophysiological situations. This study (253) potentially explains the underlying mechanisms of the poor response of NNT−/− C57Bl/6J mice to glucose (79). It also explains the shortened life span of mice when the SOD2 deficiency (lethal with ∼3 weeks life span) is combined with NNT deletion (∼1 week life span) (106, 141, 143). NNT activity decreases during brain aging in rats and mice and is up-regulated on caloric restriction. Investigations of the physiological and pathological roles of NNT expands the understanding of the mechanisms that support energy- and/or redox deficits in the early or late stages of neurodegenerative diseases (146, 249, 264), diabetes (119, 149), cardiovascular disease (213), and aging (156, 169, 235).
Mitochondrial dynamic remodeling
Mitochondria are highly dynamic organelles and undergo continuous fusion and fission throughout their life cycle (44). These processes regulate not only mitochondrial morphology, but also their biogenesis, transportation, cellular localization, quality control, and degradation (mitophagy) (44, 231). Mitochondrial fusion is regulated by GTPases optic atrophy-1 (OPA1) and mitofusin-1/2 (Mfn1/Mfn2), which are responsible for the fusion of outer- and inner mitochondrial membranes, respectively (216). Mitochondrial fission is controlled by dynamin-related protein 1 (Drp1) and fission protein 1 homolog (Fis1), with the former as scissors of the mitochondrial membrane and the latter probably as a recruiter of Drp1 to mitochondria (110, 121, 256).
Mitochondrial dynamics are closely related to the energy-redox axis. A coordinated balance between fission and fusion is essential for cell function, and its perturbation is associated with pathologies (43, 47, 48, 144). OPA1 deficiency has been associated with decreased mitochondrial ATP production, increased oxidant production, mitochondrial fragmentation, decreased life span, and neurodegeneration (47, 53, 225, 258). The suppression of Mfn2 results in a loss of mitochondrial membrane potential, reduced OXPHOS complexes expression, and decreased glucose oxidation and respiration (13, 187). Similar effects on mitochondrial metabolism are shown when mitochondrial fission is inhibited: The down-regulation of Drp1 leads to a loss of mitochondrial membrane potential and DNA content, a decrease of mitochondrial respiration and cellular ATP levels, as well as an oxidized cellular redox status and cytochrome c release (77, 181). Repression of Fis1 decreases mitochondrial respiration with the accumulation of oxidized mitochondrial proteins (230). Conversely, the mitochondrial dynamics machinery could also be regulated by the energy and redox change: The energy-consuming OPA1 cleavage reflects the mitochondrial energy-transducing capacity and the inner membrane potential (70). It is also recognized that nitrosative stress activates mitochondrial fission through the S-nitrosylation of Drp1 (SNO-Drp1) by·NO and further induce synaptic loss and neuronal damage (52), and levels of active forms (S-nitrosylated and phosphorylated forms) of Drp1 are higher in brains of Alzheimer's patients than in control subjects (238). It has also been shown that increased generation of mitochondrial O2
The Energy-Redox Axis with Cytosolic Signaling
The energy-transducing and redox-regulation capacity of mitochondria are highly affected in aging and age-related neurodegeneration. Mitochondria generate second messengers (redox: H2O2 and·NO; energy: ATP) that are involved in the regulation of redox/energy sensitive cell signaling pathways, thus coordinating functional responses between mitochondria and other cellular processes. Conversely, mitochondria are the recipients of cytosolic signaling molecules, which translocate to mitochondria under specific conditions and elicit profound metabolic or redox effects in the organelles. The communication between mitochondria and other components of the cell establishes a regulatory device that controls cellular energy levels and the redox environment (Fig. 1); impairment of this regulatory device may serve as the basis for the mechanisms which are inherent in aging and age-related degenerative disorders.
Mitochondrial regulation of cytosolic signaling
Mitochondrial-generated oxidants regulate important signaling pathways such as the insulin and insulin-like growth factor (IGF-1) signaling (IIS) and the MAPK (e.g., JNK) pathways. The PI3K/Akt route of IIS in the brain (Fig. 4) is implicated in neuronal survival and synaptic plasticity via, among other effects, maintenance of the metabolic function of mitochondria. Aging is associated with decreases in the levels of both insulin/IGF-1 and their receptor (81). Mitochondrial H2O2 is involved in the regulation of insulin signaling, which is not surprising given the large quantity of redox-sensitive cysteine residues on the insulin and IGF1 receptors and insulin receptor substrates (IRS): Oxidation of specific cysteine residues promote tyrosine autophosphorylation of the insulin receptor (219) and inhibition of phosphatases (PTEN and PTP1B) involved in the IRS node of insulin signaling on the oxidation of critical cysteines to disulfides (151). Aged cells are more vulnerable to H2O2-induced apoptosis, which is accompanied by reduced activation of Akt, and caloric restriction can prevent this loss of Akt activation (109). In addition, Akt activation is also inhibited by nitrosative stress through tyrosine nitration (55). Akt inhibits GSK-3β on phosphorylation at Ser,9 thereby protecting cell against apoptosis, because activated GSK3β stimulates phosphorylation of the anti-apoptotic member of the Bcl-2 family, Mcl-1, thus leading to its degradation and the ensuing cytochrome c release and apoptosis (157, 180).

The MAPKs are also sensitive to oxidative or nitrosative stress conditions, entailing an enhanced generation of H2O2 or peroxynitrite, which results in their activation (174, 188, 253, 263) (Fig. 5); H2O2 may act at multiple levels to activate JNK (and p38): dissociation of thioredoxin from the ASK-1 complex (204), disruption of the glutathione transferase (GT)-JNK complex (1), or inhibition of MAPK phosphatase activity (76). Basal JNK activity, but not its protein levels, is increased in mouse brain and liver, rat kidney and splenic lymphocytes, and human skeletal muscle during aging (105, 127, 142, 221, 242). Basal activities of ERK and p38 kinase, but not their protein levels, are reported to decrease in the brain cortex during aging, a phenomenon that was prevented by caloric restriction (259); conversely, basal p38 and ERK activities were increased in mouse liver, rat kidney, and human skeletal muscle (104, 127, 242). It is unclear whether or not these discrepancies are due to tissue specificity of p38 and ERK responses to the aging process. Nevertheless, the activation of ERK in response to epithelial growth factor is decreased in cortical brain slices and hepatocytes from old rats (148, 259), indicating reduced sensitivity to stimuli in these tissues during aging.

In addition, JNK also plays a central role in the progression of insulin resistance; a likely mechanism that entails the phosphorylation of the IRS-1 at Ser307, leading to inhibition of the insulin-promoted tyrosine phosphorylation of IRS (2). Conversely, the MLK3-mediated JNK activation is inhibited by Akt on the phosphorylation of MLK3 both in vitro and in vivo (19). Due to the distinct downstream signaling between PI3K/Akt and JNK (survival vs. apoptosis; growth vs. differentiation), the counterbalance of the IIS and JNK pathways induced by different concentrations of H2O2 is expected to determine the coordinated response of the cell. These disparate effects of mitochondrial H2O2 are important, as they refer to a healthy aging or accelerated aging, and they need to be assessed in terms of the cellular peroxide tone, that is, a quantitative assessment of a mitochondrial steady-state concentration of H2O2 in connection with domain-specific signaling. Hence, the mitochondrial energy-redox axis is one of the factors that regulate the peroxide tone of the cell in a domain-specific signaling fashion.
Despite its role in regulating the activities of kinases, phosphatases, and other redox-sensitive enzymes on the time scale of minutes to hours, dynamic H2O2 signaling is also involved in sub-second signaling via ion channel activation in neuronal cells (198). In dopamine neurons of the substantia nigra, mitochondrial H2O2 inhibits neuron firing by activating ATP-sensitive K (KATP) channels, thus linking metabolic state to cell excitability (10, 15). The mitochondrial energy and redox status are also connected with suppressed dopamine release in the striatum through the activation of KATP channels by glutamatergic AMPA receptor-induced H2O2 generation (11, 12). False regulation of H2O2 signaling due to mitochondrial dysfunction could compromise striatal dopamine release and, thus, be implicated in nigrostriatal degeneration and Parkinson's disease (16). H2O2 can also activate the transient receptor potential (TRP) channels that increase the excitability of striatal GABAergic medium spiny neurons (16). The expression of the redox-sensitive ion channels, either inhibitory (KATP) or excitatory (TRP), in coordination with cell-specific mitochondrial metabolic and redox regulation, defines the specificity of dynamic neurotransmission (198).
Cytosolic regulation of mitochondrial function
Mitochondria are also important targets of cytosolic signaling molecules. It was shown that the expression and activation of JNK1 increases in the brain as a function of age and that active (bisphosphorylated) JNK translocates to the mitochondrion, where it triggers a phosphorylation cascade which results in phosphorylation (inhibition) of PDH, a key mitochondrial enzyme complex that bridges anaerobic and aerobic brain energy metabolism. PDK2 is an essential intermediate in this phosphorylation cascade. The outcome is a bioenergetic crisis translated into decreased cellular ATP and an increase in lactate levels (anaerobic glycolysis as a compensatory mechanism) (263, 264) (Fig. 5).
The PI3K/Akt pathway promotes neuronal survival and synaptic plasticity (234) by mechanisms entailing the phosphorylation of proapoptotic Bcl-2 family members (Bad) of GSK3β (thus inhibiting tau hyperphosphorylation) and of FoxO factors (that drives nuclear FoxO factors to the cytosol, thus inhibiting the transcription of some apoptotic genes and those involved in heme degradation) (51). In NIH/3T3 cell lines, mitochondrial H2O2 modulates the entrance of cytosolic Akt (phosphorylated at Ser473) to the mitochondria and induces the further phosphorylation at Thr308 that is required for nuclear translocation (7). Akt translocates to the mitochondrion in human neuroblastoma cells, and its phosphorylation targets are a constitutive form of GSK3β in the mitochondrion and the β-subunit of ATPase (23) (Fig. 4).
ATP is the universal energy currency in the cell, and mitochondrial produced by ATP is transported to the cytosol by ANT in exchange with ADP. The cytosolic ADP/ATP ratio is an important parameter of not only the cellular consumption of ATP but also ATP synthesis as a reflection of mitochondria bioenergetic profile. The ubiquitously expressed adenylate kinases in all cell types, which catalyzed the inter-conversion of adenine nucleotides (2ADP↔ATP+AMP), make AMP/ATP another important indicator of energy status. 5′ adenosine monophosphate-activated protein kinase (AMPK) is a kinase with its activity controlled by intracellular AMP/ATP ratio and, therefore, rendered a sensor of cellular energy status. In fact, recent studies have suggested that AMPK activity can also be regulated by ADP (178, 246). AMPK is activated on various stress conditions, including glucose deprivation, ischemia, and hypoxia, and is also a key player involved in the cellular response to exercise (218). Moreover, AMPK is redox sensitive (237) and interacts with mitochondrial redox status through the mitochondrial aldehyde dehydrogenase (ALDH-2) (60). On activation, AMPK induces multiple responses, including enhanced glucose metabolism and fatty acid oxidation, to switch cellular metabolism from anabolism to catabolism by (a) increasing glucose uptake by stimulating glucose transporters expression (260) and its translocation to the plasma membrane (17, 134); (b) enhancing glycolysis by activating 6-phosphofructo-2-kinase (PFK2) (153); and (c) simultaneously inhibiting fatty acid synthesis and activating mitochondrial β-oxidation by blocking acetyl-CoA carboxylase (ACC1/2) activity (95). AMPK is regarded a central regulator of the pathways that are implicated in aging and a potential longevity regulator in worms (86), and it is also involved in the beneficial effects of caloric restrication (118). Mixed results were reported regarding the change of AMPK activity during aging in different tissues, but growing evidence suggests that decreased AMPK activity and/or decreased responsiveness to AMPK activity is associated with declined mitochondrial function as a function of age (75), thus indicating an inter-relationship between mitochondrial energy status and AMPK activity.
It may be surmised that the cross-talk between IIS, JNK, and AMPK signaling in the brain, their modulation by mitochondrial signaling molecules, and how these signaling impinge on mitochondrial function is of importance in understanding the process of aging and their relevance for some neurodegenerative disorders. Since active JNK can also initiate mitochondrion-dependent apoptosis, impairment of the communication between mitochondrion-supported redox signaling and cytosolic signaling pathways may serve as the basis for the mechanisms inherent in cell death pathways and the loss-of-cell function that is associated with aging and age-related degenerative disorders.
The Energy-Redox Axis and Nuclear Transcriptional Pathways
Transcriptional control of mitochondrial biogenesis
The majority of the mitochondrial proteins are nuclear encoded and cytosol synthesized before being transported to mitochondria (186), and mitochondrial DNA encodes 13 subunits of the complexes in the electron transport chain. The transcription of mtDNA is primarily controlled by mitochondrial transcription factor A (TFAM), while the coordinated expression of nuclear-encoded mitochondrial proteins involves multiple transcription factors such as Sp-1, YY-1, CREB, MEF-2, and nuclear respiration factors (NRFs) such as NRF-1, NRF-2, ERRα, and REBOX/OXBOX, among others (75, 206). The coordination of these transcriptional pathways is integrated by the peroxisome-proliferator-activated receptor γ coactivator-1 (PGC-1) family of transcriptional coactivators, and PGC-1α is currently the best-characterized member (93). The ability of PGC-1α in regulating the transcription of mtDNA by coactivating NRF-1 on the promoter of TFAM and mitochondrial transcription specificity factors TFB1M and TFB2M, as well as its role as a co-activator of major transcription factors involved in nuclear-encoded mitochondrial gene expression, renders PGC-1α the master regulator of mitochondrial biogenesis (Fig. 6).·NO can signal through cGMP-PKG pathway to activate Sirt1 and PGC1α (33, 171, 175).

The decline of mitochondrial function with age and in neurodegeneration is associated with reduced mitochondrial biogenesis and decreased activity of its major regulator, PGC-1α. PGC-1α levels were found to be decreased with age, and the decline was rescued by caloric restriction in skeletal muscle (97); PGC-1α levels and mitochondrial function were positively linked to lifespan extension in several rodent genetic models (4, 123). Overexpression of PGC-1α in a model of mitochondrial myopathy significantly prolonged lifespan (239). PGC-1α knockout mice are more sensitive to the neurodegenerative effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and kainic acid; conversely, increased PGC-1α levels protect neurons against oxidative stress-mediated death by inducing expression of antioxidant genes (217). Mitochondria from a Huntington's disease transgenic mouse brain show reduced PGC-1α levels and OXPHOS gene expression (241), and PGC-1α null mice also show increased degeneration of striatal neurons and lesions in the striatum, which is the primary brain region affected by Huntington's disease (145). Conversely, the overexpression of PGC-1α in the striatum provides neuroprotection by reversing the toxic effects of mutant huntingtin (56). Notably, despite its major role in modulating mitochondrial function, the impact of PGC-1α activity modulation is stimulus- and tissue dependent (5).
As a key regulator of mitochondrial biogenesis and function, PGC-1α is regulated at multiple levels, including its transcription, post-translational modification, localization, and degradation. The regulation of PGC-1α expression involves CREB (245) and mTOR-YY1 (58) pathways, making them important modulators of mitochondrial metabolic function in aging (209). Post-translationally, PGC-1α is activated by Sirt1 by deacetylation after the translocation of the former into the nucleus during stress conditions (6). AMPK is another regulator of PGC-1α activity either by direct phosphorylation of PGC-1α (111) or by indirectly enhancing Sirt1 activity (42) (Fig. 6). The regulation of PGC-1α turnover by GSK3β phosphorylation and subsequent degradation (6) provides an additional layer of control on PGC-1α function. Taken together, the spatiotemporal regulation of PGC-1α at multiple levels and time points enables the fine tuning of mitochondria activity and energy homeostasis by integrating transcriptional pathways driven by Sirt1, AMPK, and mTOR in response to extracellular signals and specific cell demands.
Mitochondrial regulation of transcriptional pathways
The signal communications between the nucleus and mitochondria are not unidirectional. Perturbations of mitochondrial energy and redox status can also be transmitted to the nucleus to induce cellular adaptive or compensatory responses. This process involves several mitochondrion-generated second messengers, such as ATP, H2O2,·NO, and processes involved in the dysregulation of Ca2+ homeostasis and the maintenance of cytosolic NAD+/NADH ratios. As the primary indicator of mitochondrial metabolic status, altered ATP levels in the cells affect AMPK activity and positively modulate energy-consuming anabolic processes through mTOR either directly (64) or indirectly (90, 212) involving a variety of transcription factors, such as p300, HNF4α, MEF-2C, and p53 (94, 159). ATP signaling is also involved in the regulation of mitochondrial biogenesis through the AMPK-PGC1α pathway (41, 111) (Fig. 6). Hence, the mitochondrial bioenergetic state is an important modulator of cell growth and proliferation. It is still controversial whether ATP levels or ATP production rate declines with age (67, 135, 138); thus, future studies should focus more on the sensitivity of these adenine nucleotides signaling pathways to altered mitochondrial energy status during aging.
As important intermediate metabolites, both NAD+ and NADH, and their ratio, affect mitochondrial function by modulating mitochondrial permeability transition pore and Ca2+ homeostasis (254). Cytosolic/nuclear NAD+ can serve as a substrate of Sirt1 to regulate mitochondria biogenesis by the deacetylation of PGC1α (42) and as a substrate of poly(ADP-ribose) polymerase, which is an important DNA nick sensor. The age-dependent decline in intracellular NAD+levels and NAD+/NADH ratio were observed in parallel with decreased Sirt1 activity (34). Although the inner mitochondrial membrane is impermeable to NAD+ and NADH, it was found that the malate-aspartate NADH shuttle could play a critical role in the activation of the downstream targets of caloric restriction, such as sir2 (Sirt1 homolog), and is required for caloric restriction-mediated lifespan extension in yeast (71). This indicates that alterations in the mitochondrial energy status are capable of influencing the cytosolic and nuclear NAD+/NADH pool and further affecting the NAD+-dependent sirtuin pathways (Fig. 6).
A variety of transcriptional pathways are redox sensitive and can be activated on extracellular stimuli or intracellular redox changes (35). Transcription factors, such as HSF-1, p53, and NF-κB, can be directly activated on oxidative stress (74), while some other factors, including NRF-1, NRF-2, and FoxO, can be activated/inhibited indirectly via redox-sensitive kinase signaling pathways such as JNK, ERK, p38, and PI3K/Akt (35) (see Section III). The role of H2O2 in NF-κB activation has been critically reviewed (179), and it was concluded that H2O2 does not function as an inducer of NF-κB but it is largely involved in the regulation of NF-κB-related pathways. Another redox-sensitive transcription factor is nuclear factor erythroid 2-related factor (Nrf2), which on its dissociation from Keap1, activates antioxidant responses. Altered redox status leads to the oxidation of Keap1 (via thiol/disulfide exchange) and the release of Nrf2; the latter translocates to the nucleus and activates the antioxidant response element-mediated Phase II enzyme expression (65). Multiple key endogenous antioxidant enzymes that are involved in GSH synthesis and reduction and O2 •−/H2O2 homeostasis are regulated by Nrf2 (117); the levels of Nrf2 and the expression of its downstream genes decrease with age (220), suggesting the potential role of the Nrf2 pathway in the age-related decline of redox capacity.
Mitochondria are also important regulators of Ca2+ storage and homeostasis. Ca2+ signaling is involved in cell- and stimuli-specific responses through various transcriptional networks such as NFAT, MEF2, TORC, CREB, and NF-κB (160). A comprehensive study on the role of mitochondrial Ca2+ in the regulation of cytosolic and nuclear redox signaling is previously reviewed (224).
Conclusions and Perspectives
Aging and neurodegeneration are associated with declines in energy production in the brain as well as parallel changes in redox status with a pro-oxidant shift that may be due in part through the mitochondrial generation of O2
•− and H2O2. Mitochondria regulate distinct cytosolic signaling pathways and have vital roles: (a) They are the cellular organelles that generate ATP, which supports the cellular energy demands; (b) generate second messengers, such as H2O2 and
Footnotes
Acknowledgments
This review was supported by NIH grants RO1AG016718 to E.C. and PO1AG026572 (to Roberta Díaz Brinton; E.C. as co-investigator in Project 1).