
Abstract
Introduction
O
Here we discuss potential pathways how NO and ROS, in part, under the control of mitochondrial integrity, affect the HIF-system and how HIF-1/HIF-2 has power over mitochondria themselves. While mitochondrial respiration can adjust HIF, their function and mass are reduced under prolonged hypoxia to adjust metabolic demands of cells under hypoxia, a situation relevant, for example, cancer cells. HIF-1 fulfills a unique role in sensing redox changes, transmitting signals, and adjusting the mitochondria for their demands under prolonged periods of hypoxia, thus working as a mitochondrial hypoxia relay system.
The Mitochondrial Redox State Under Hypoxia
Consequences of a reduced O2 availability at the cellular level are a rapid inhibition of oxygen-dependent enzyme activities. Inhibition of cytochrome oxidase (COX), consuming most of the cellular O2, provokes a rapid decline in electron transport and results in NADH accumulation (62). Although fluorescent detection of NADH in cells or tissues does not discriminate between NADH versus NADPH signals, the fluorescence increase under hypoxia refers to an increase in NADH only. Rather, the capacity to produce NADPH decreases under hypoxia, rendering cells more susceptible to oxidation (91). NADPH forms the basis for mitochondrial redox homeostasis due to its capacity to reduce oxidized glutathione (GSSG). Glutathione (GSH) is produced in the cytosol and imported into mitochondria, where it reaches concentrations up to 10 mM, which are comparable to those found in the cytosol (60). Mitochondrial glutathione reductase reduces GSSG by consuming NADPH, most likely provided by IDH2 (isocitrate dehydrogenase 2). Under hypoxia, the GSH/GSSG ratio decreases in some organs or cell lines (59, 80), with the further notion that especially reduced mitochondrial GSH is linked to cell viability (56). This suggests that hypoxic cells are more sensitive to oxidative stress and/or implies that hypoxia increases the production of reactive nitrogen species and ROS.
Two major sources of hypoxic ROS are discussed; one being the mitochondrial ETC and the others are NADPH oxidases (NOX). Electron leaks at the ETC complexes are believed to contribute to enhanced ROS production under hypoxia and also add to hypoxic signaling (see below). Among the ETC complexes, the Q-cytochrom C oxidoreductase (complex III) and three sites at complex I are well-documented O2 − sources (52). O2 − produced by the ETC is rapidly converted to H2O2 by Mn-superoxide dismutase. Mitochondria do not contain catalase, and therefore GSH-dependent detoxification by glutathione peroxidases (Gpx2 or Gpx4), peroxiredoxins, and thioredoxins represent the major defense systems requiring reduced NAPDH. As the reduction of NADP+ to NADPH might be impaired under hypoxia in mitochondria (because IDH2 uses NADPH for reductive carboxylation of α-ketoglutarate [α-KG]), an increase in oxidized protein thiol groups or fluorescent dyes might be indictors for a reduced antioxidant capacity under hypoxia rather than enhanced ROS production. Along the same line, under hypoxia, attenuated H2O2 production was shown in isolated mitochondria (44) and O2 − flashes observed in single mitochondria were less abundant under hypoxia (94). Alternative sources of mitochondrial ROS under hypoxia are NOX. Five NOX isoforms are characterized in mammalian tissues and hypoxia activates NOX1 or 4 by a mitochondrial-protein kinase C-ɛ (PKCɛ)-dependent mechanism (82). Most NOX isoforms are organized as multienzyme complexes that need activation to assemble to the active state (76). NOX4 is expressed in several tissues and is localized to the endoplasmic reticulum (ER), mitochondria, or the nucleus. Mitochondrial NOX4 is a likely source for hypoxic ROS as treatment with siRNA diminished ROS production in pulmonary arties and adipose-derived stem cells (50, 66). Still mitochondrial localization needs confirmation and may be specific for certain cell types, limiting hypoxic ROS production by NOX only to those tissues. Moreover, assuming that enhanced protein expression accounts for an activity increase of NOX4, likely via HIF-1α, NOX4 does not qualify as a source of enhanced mitochondrial ROS under immediate hypoxia.
Another consequence of hypoxic inhibition of COX is a reduction of the mitochondrial membrane potential (ΔΨm) (42). Mitochondrial membrane potential is tightly linked to autophagy and apoptosis. To prevent induction of apoptosis and for maintaining ΔΨm, mitochondria use succinate to preserve complex II activity or use glycolytic ATP hydrolysis by complex V to maintain the proton gradient (25, 42). Inhibition of complex V, subsequently causes translocation of B-cell CLL/lymphoma 2 (Bcl2)-associated X (Bax) protein from the cytosol to mitochondria, allowing permeability transition opening and initiation of apoptosis (34). To maintain the proton gradient across the mitochondrial inner membrane under normal conditions accounts for 20%–40% of the respiration rate (40). How much this portion increases under hypoxia is not clear, but maintaining ΔΨm is a price hypoxic cells need to pay, to ensure viability even when energy is limited.
Additional hypotheses have been put forward that are suited to understand early perturbation of the mitochondrial redox homeostasis. Although multiple indirect evidence for the existence of a mitochondrial NO synthase (mt NOS) are published, no gene sequence for such an enzyme exists and the protein nature of this enzyme remains obscure (36) and indeed reports arguing against an mt NOS exist (53). More logic, targeting constitutive NOS, preferentially endothelial type NOS, to the outer membrane of mitochondria may allow NO formation in close proximity of the mitochondrial compartment (33). Assuming that mitochondria provide the two reactants NO and O2 −, peroxynitrite generation might be the consequence. Indeed, nitrated proteins have been identified in mitochondria, but as clear evidence for a reversibility of this post-translational modification still is rare (101); its involvement in highly dynamic and fully reversible responses to hypoxia awaits experimental proof. In line, mitochondrial peroxynitrite formation and subsequent oxidative stress has been discussed in the context of apoptosis, a pathway that appears irreversible once mitochondria are heavily damaged (36). An NOS-independent source of NO, ensuring the generation of NO under limited oxygen availability might be nitrite (10). In mammals, the formation of NO from nitrite under hypoxic conditions is carried out by diverse enzymes, among others, metalloproteins, including heme-containing enzymes and molybdenum enzymes, that is, xanthine oxidase (XO). XO is a potential source for both, O2 − and nitrite derived NO. Under hypoxia, the acidosis and the increased concentrations of xanthine and NADH will probably be sufficient to support the XO-mediated nitrite reduction physiologically. Alternatively, nitrite reduction by liver mitochondria at low oxygen concentrations of 20 μM has been ascribed to COX, that is, mitochondrial complex IV (9). Data reinforce the idea that multiple proteins may function as nitrite reductase under low oxygen tension. Unfortunately, precise conditions when nitrite reductase is activated under hypoxia and whether its activity follows hypoxic gradients and adds to modulate the oxygen-sensing machinery are missing. Theoretically, nitrite reduction under hypoxia circumvents the need of oxygen-dependent NOS activity and may contribute to regulating mitochondrial responses and/or impinge on the HIF-system.
Hypoxic HIF Regulation
Under normoxic conditions, HIFα subunits are often kept at undetectable levels due to continuous 26S proteasomal degradation. Lowering oxygen retards degradation of HIFα subunits because impaired hydroxylation at conserved proline residues. These post-translational modifications are mediated by prolyl hydroxylases (PHDs), whose activity is regulated by oxygen availability thus, serving as oxygen sensors linking cellular O2 concentrations to HIF molecular responses [(85) and reviewed in ref. (48)]. HIF proteins are heterodimers of an O2-regulated α- and a constitutively expressed β-subunit, which both are members of the basic helix-loop-helix/

Both, PHDs and FIH hydroxylate targets beyond HIF-α proteins. Well characterized are proteins of the nuclear factor of kappa light polypeptide gene enhancer in B-cells (NF-κB) pathway. FIH hydroxylates p105 and nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor (IκBα) at conserved ankyrin repeat domains (16). Additionally, inhibitor of kappa light polypeptide gene enhancer in B-cells (IKKβ) is hydroxylated by PHDs, which causes its inactivation (17). Thus, inhibition of PHDs activates NF-κB that is low compared to activation during infections, but still detectable in vivo (31). Although, HIF appears a major regulator of hypoxic responses, additionally pathways are affected.
Inflammatory conditions are characterized by increased vascular permeability, resulting in the emigration of fluid, proteins, and immune cells from the vasculature to inflamed areas. Reduced circulation combined with an increasing metabolic demand from immune cells and pathogens provokes local hypoxia. Thus, in inflamed tissue specimens from patients with arthritis, atherosclerosis, and autoimmune diseases or during wound healing, activation of HIF has been observed [(1) and reviewed in ref. (68)]. Moreover, activation of HIF in a variety of tumor cells and cells of the tumor microenvironment is appreciated, with the notion that immune cell attraction and activation favors the production of a variety of cytokines, ROS and NO.
NO and the HIF-1 responses
Besides oxygen, also NO can affect PHD activity. NO fulfills diverse physiological functions, including vasodilatation, neurotransmission, and immune responses, with macrophages and tumor cells being predominant producers of NO via expression regulation of the inducible NOS (iNOS) (90). Chemical diverse NO donating compounds or an active iNOS in macrophages provided evidence that NO stabilizes HIF-1α protein and causes transactivation of HIF-1 under normoxia [for review (5, 8)]. Based on in vitro studies, it is assumed that NO steady state concentrations of 100–300 nM NO are required to stabilize HIF-1α (90). Observations have been recently extended by showing that NO can react with a truncated PHD2 that expresses the catalytic domain (PHD2181–426) both by coordinating the active Fe(II), as shown by electron paramagnetic resonance (EPR) analysis, and/or via S-nitrosylation of cysteine residues as shown by mass spectrometry (MS) studies (13). Although the biological significance of S-nitrosylation is not fully elaborated it is possible that liberation of NO from S-nitrosylated PHD2 can result in inhibition via reaction at the active site Fe(II). This concept may be consistent with proposals based on cellular studies that NO decreases PHD activity, implying that hypoxia and NO at the molecular level use the identical target, that is, interfere with PHD activity, to stabilize HIF-1α. The mechanism might be of importance in certain forms of human cancers, where inhibition of iNOS expression interferes with the accumulation of HIF-1α (79). Of relevance, suppressing HIF-1 activation reduces tumor progression at multiple levels, that is, reducing angiogenesis, invasion, and metastasis (97). Considering the similarity in reactions catalyzed by PHDs and FIH, it is not surprising that FIH activity is also inactivated by NO (75). In addition, stabilization of HIF-1α through S-nitrosylation at Cys533 within the oxygen-dependent degradation domain, with NO being generated from neighboring macrophages, is shown in tumor tissue following ionizing radiation (54). Importantly, this mechanism interferes with the HIF-1α breakdown, occurs independently of the PHD-based destruction pathway and is achieved by impaired binding of pVHL to HIF-1α.
The impact of NO reverses, when HIF-1α expression is analyzed under hypoxic conditions [reviewed in refs. (8, 72)]. As illustrated in Figure 2, this paradox can be explained by the observation that NO competes with O2 for the binding to mitochondrial COX, which consumes most of the oxygen within a cell. With low amounts of oxygen being present, NO efficiently binds to COX, attenuates enzyme activity and thus, residual O2 consumption [reviewed in refs. (15, 90)]. Oxygen spared by mitochondria is now available to nonrespiratory oxygen-dependent enzymes, such as PHDs, enabling the continued hydroxylation and degradation of HIF-1α. This situation is noticed under hypoxia/NO or during inhibition of the mitochondrial respiratory chain and accounts for reduced accumulation of HIF-1α under these conditions (29, 39). As the effect of NO is noticed at low concentrations, that is, linked to the activation of the constitutive NOS, a physiological role appears likely. The in vivo relevance was approached when tumor-bearing mice were treated with nitroglycerin, a NO mimetic, to attenuate tumor growth (3). While this study does not provide a direct cause–effect relation between NO supplementation and HIF-1α expression/activity, the physiological in vivo relevance of this signaling circuit awaits further clarification. However, NO-dependent inhibition of mitochondrial oxygen consumption in the distribution of oxygen in vivo is demonstrated (93). This might be crucial for the adaptation of tissues, including the vessel wall itself to hypoxia and potentially to the accumulation of HIF-1α/HIF-2α.
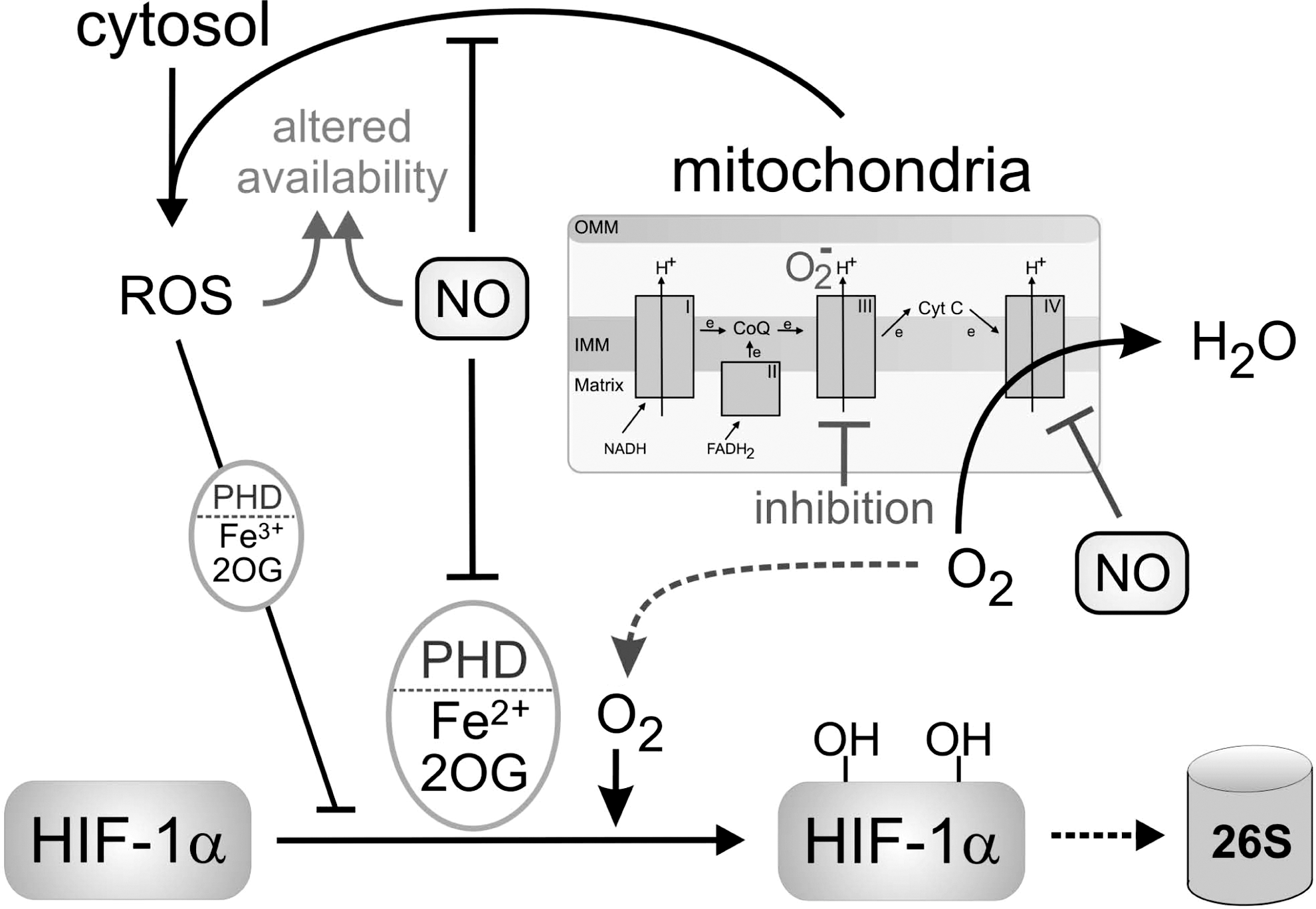
It is of interest that an intimate cross talk between the HIF-1α- and the NO/iNOS-system exists. iNOS has been established as a HIF-1 target, bearing a classical HRE site. However, hypoxia alone is insufficient at expressing iNOS (28, 43) with the further notion that low oxygen (1% O2) also attenuates NO formation from
ROS and the HIF-1 responses
Inflammatory responses often provoke the formation of ROS either by NOX assembly and/or mitochondrial alterations. ROS act as signaling compounds, but may be destructive if their production is uncontrolled or if their removal/detoxification is impaired. Based on the enzyme catalysis proposed for PHDs, ascorbate (vitamin C) is required to keep the iron in PHDs reduced (48, 49, 97). However, vitamin C is not required for oxygen sensing in vivo as GSH fully substituted for its requirement of all three PHD isoforms in vivo, with the further possibility that besides the central iron other redox-active cysteine residues exist (69). At the cellular level there is the risk that antioxidants like ascorbate may become limiting in protecting against oxidative stress. Indeed, ascorbate deficiency limits PHD function in human tumors, and consequently increases HIF-1α protein (48). The transcriptional regulator JunD (jun D proto-oncogene) increases antioxidant levels and its deletion stabilizes HIF-1α and HIF-2α because of increased ROS formation (35). This study offered a potential link between oxidative stress and PHD inhibition via altered Fenton chemistry leading to an accumulation of iron in the oxidized ferric (Fe3+) state, being unable to catalyze the hydroxylation reaction. Recent data indicate that FIH is more sensitive toward peroxide stress than PHDs (61). Cells treated with H2O2 show a marked reduction in asparagyl, but only minor effects on prolyl hydroxylation. FIH inhibition was reversed by iron chelation, pointing to the Fenton chemistry in inactivating dioxygenases.
Already in 1998, a report from the Schumacker Lab proposed that mitochondria, as the primary oxygen-consuming organelle, act as the oxygen sensor in activating HIF-1α in hypoxia by generating ROS (11). The work that heavily relied on pharmacological tools was met with some skepticism, especially as others noticed that cells lacking mitochondria still stabilized HIF under hypoxia [reviewed in ref. (88)]. However, a variety of approaches were used to substantiate the early observations. A fluoresence resonance energy transfer (FRET) probe containing a redox-sensitive dye was used to confirm the still controversially discussed issue that hypoxia increases ROS production. siRNA technology to inactivate the Rieske iron–sulfur protein of complex III or cells with a disrupted cytochrome c locus were used to strengthen the link between inhibition of ROS production and impaired HIF induction by hypoxia [reviewed in refs. (47, 88)]. A detailed insight into compartmentalization of ROS production under hypoxia was achieved by using the redox-sensitive fluorescent protein RoGFP to visualize redox changes (96). Hypoxia causes a decrease in ROS formation in the mitochondrial matrix, but increased oxidation of RoGFP in the mitochondrial inner-membrane space as well as in the cytosol. ROS formation under hypoxia may involve additional sources, as mitochondrial-derived ROS under hypoxia provoke activation of NOX through PKCɛ activation, suggesting that mitochondria, but also other cytosolic and/or membrane ROS-generating systems may communicate to the overall response [reviewed in refs. (18, 95)]. In addition, pathologically recognized compounds, such as oxidized low-density lipoprotein, known to generate ROS, have been linked to increased expression of HIF-1α (78, 86).
Challenging ROS formation under hypoxia, it is argued that genetic or pharmacologic approaches to block the ETC are expected not only to interfere with O2 − production, but also to inhibit mitochondrial oxygen consumption and thus, appear not suited to distinguish between ROS production and a mechanism of simply sparing mitochondrial oxygen consumption for PHD activity. This assumption is based on a study showing that all ETC inhibitors tested, regardless of which complex they inhibit, decrease HIF-1α protein half-life under hypoxia (14). This suggests that any ETC inhibitor has the same effect on HIF-1α degradation under hypoxia. As these inhibitors tend to have varying effects on O2 − production at complex III and had different effects on cellular O2 − concentrations, mitochondrial ROS production was ruled out to account for stabilization of HIF-α by hypoxia. In addition, FIH is more sensitive toward oxidative stress compared to PHDs, while PHDs are more oxygen-sensitive (61). These contrasting sensitivities are an argument for O2 rather than ROS as the primary signaling molecule. Many data would be also consistent with the idea that the mitochondrial oxygen consumption rate is the key determinant of HIF-α stability in hypoxia. Inhibiting cellular oxygen consumption prevents HIF-α accumulation in hypoxia because oxygen is spared and increased cellular oxygen is available for PHD activity that, based on in vitro estimates, show KM values for oxygen in the range of 250 μM (88). The spared oxygen hypothesis is well suited to also explain the impact of NO in lowering HIF-1α under hypoxia, may allow to rationalize how CO interferes with HIF-1α under hypoxia and eventually also account for the HIF-1α lowering capacity of H2S, as all these gas molecules can interfere with oxygen binding at COX. Fully compatible, inhibition of COX by cellular poisons, such as NO, CO, cyanide, or azide, has been known to shift the ETC to a more reduced state, a condition that enhances not only the formation of ROS, but also reduces mitochondrial oxygen consumption, predicting that these molecules contribute to HIF-1α stability regulation under reduced oxygen tension (67). The intracellular oxygen concentration has been reported to be as low as 10 μM and with the notion that oxygen gradients exist may indicate that this value could be even lower. At 10 μM O2, the IC50 of NO for COX is predicted to be around 20 nM, which indicates that NO concentrations are within the range of those (10–450 nM) detected in tissues [reviewed in ref. (67)]. The ability of NO to divert oxygen from mitochondria, making it available for bioluminescence, is used by fireflies, indicating that other systems use NO-dependent variations in O2 consumption for triggering physiological signaling cascades.
The spared oxygen hypothesis is further supported by studies controlling proteins that alter mitochondrial abundance or efficiency of ETC complexes. Overexpression of peroxisome proliferator-activated receptor (PPAR) gamma, coactivator 1 alpha (PGC-1α) increases mitochondrial biogenesis and reduces intracellular oxygen level (71). This provokes accumulation of HIF-1α and subsequent HIF-1 target gene expression. Likewise, overexpression of CHCD4, a homolog to an important yeast component of the mitochondrial disulfide relay system, regulating electron transfer to cytochrome c, increases oxygen consumption in cells. Again hypoxic accumulation of HIF-1α is elevated, while its knockdown decreases the ability of cells to accumulate and activate HIF (102). HIF-1α accumulation was not impaired when desferrioxamine or dimethyloxaylglycine were used instead of hypoxia. Both are inhibitors of PHD enzymes and their inhibitory activity is not influenced by cellular pO2. Furthermore, antioxidants did not reduce enhanced HIF-1α accumulation in cells overexpressing CHCD4, making a ROS-dependent mechanism unlikely. Rather the amount of oxygen consumed by mitochondria is altered in these studies linking this parameter to HIF-1α accumulation.
While the action of NO is easily explained with inhibition of cytochrome c oxidase under hypoxia, the ROS hypothesis would benefit from explaining how a decrease in oxygen availability increases ROS production by mitochondrial complex III, how the increase in ROS stabilizes HIF-1α either by affecting intracellular levels of antioxidants or other PHD cofactors, and whether a direct coupling between ROS formation and HIF-1α stability control exists. It is worth considering hypoxia/reoxygenation and/or ischemia/reperfusion episodes that are linked to ROS formation, most likely involving mitochondria (73). Return to ambient oxygen concentrations produces massive ROS, but is accompanied by rapid loss of HIF-α protein expression, which does not support an obvious link between mitochondrial ROS production and HIFα subunit stabilization. Important questions for the NO-driven spared oxygen hypothesis would be the identification of the NO source and its activity adjustment to hypoxia. There may be multiple oxygen sensors, such as PHDs, FIH, and mitochondria, which collectively promote HIF molecular responses in hypoxia. Furthermore, it will be essential to establish whether regulatory features proven for HIF-1α also hold true for HIF-2α. As HIF-2α responds to ROS like HIF-1α and considering that both isoforms are under the control of the same PHDs, it is reasonable assume overlapping regulatory features (26, 35).
The NO/O2 − Cross Talk at the Level of HIF-1α
The near diffusion controlled interaction between NO and O2 − implies that their cross talk adds another layer of complexity in achieving biological outcomes, that is, affecting expression of HIF-1α. The simultaneous generation of O2 − and NO by xanthine/XO and a NO donor increased oxidative intermediates, followed by dihydrorhodamine oxidation, with a decrease in steady-state NO concentrations and a proportional reduction of NO-evoked HIF-1α stabilization [reviewed in ref. (8)]. Results were corroborated by using the redox cycler dimethoxynapthoquinone to attenuate stabilization of HIF-1α by NO. The ability of ROS to attenuate NO-induced HIF-1α stabilization required proteasomal degradation, which implies that PHD activity is restored under these conditions. Whether tyrosine nitration of 26S proteasome components, which increases its proteolytic activity, adds to these ROS/NO effects remains to be established (100). It can be concluded that the primary consequence of O2 − and NO reaction is a change in the cellular phenotype due to an altered bioavailability of either NO or O2 −. The rate of NO or O2 − formation is critical because O2 − concentrations determine the steady-state concentrations of NO and vice versa [reviewed in ref. (98)]. O2 − and NO coformation alters the bioavailability of either radical, and thereby accounts for signaling, rather than the resultant chemistry of newly formed higher nitrogen oxides. This consideration may help to explain discrepancies among various studies on the ability of either NO or O2 − to stabilize HIF-1α. Variables reside in the relative flux rates of NO or O2 − and/or their coformation, which are under the control of the electron flow through the ETC, conditions that allow constitutive or iNOS to be active, or when NO is generated from nitrite under hypoxia. The unique feature of the reaction between O2 − and NO is its simplicity. Nevertheless, these considerations do not exclude the possibility that nitrosating species formed by NO and O2 − at a ratio of 3:1 may alter cysteine residues, which may affect stabilization of the HIF-1α protein (19).
HIF and Mitochondrial Metabolism
Hypoxia alters glucose metabolism in all cells, known as the Pasteur effect and HIF-1 accounts for these adaption (84). When reduced O2 availability diminishes mitochondrial ATP production, cells adjust by increasing glycolytic ATP, as shown in Figure 3. HIF-1 upregulates glucose transporters and induces glycolytic enzymes (38, 92). Increasing flux rates of glucose breakdown, two enzymes appear of particular importance. 6-Phosphofructo-1-kinase (PFK) defines the amount of glucose that enters the glycolytic pathway. A key allosteric regulator of PFK, and therefore of the glycolytic flux is fructose-2,6-bisphoshate (F-2,6-BP). HIF-1 increased F-2,6-BP levels, which stimulates the glycolytic flux (83). Levels of F-2,6-BP are maintained by a family of four bifunctional enzymes, 6-phosphofructo-2-kinase/fructo-2,6-bisphosphatase (PFKB)1–4, which have both kinase and phosphatase activities and which all are induced by HIF (65). Among them, PFKB3 produces high concentrations of F-2,6-BP and shows the strongest HIF inducibility. Moreover, HIF not only upregulates PFK, but also increases its activity by inducing PFKB3. Toward C3-metabolism, pyruvate kinase (PK) activity also defines the glycolytic flux. Humans and mice have two PK genes (PKLR and PKM2) and four isozymes (L, R, M1, and M2). PK-L and PK-R are expressed from alternative PKLR promoters in gluconeogenic tissues and erythrocytes, respectively. PK-M1 and PK-M2 are coded by alternatively spliced PKM2 transcripts. PK-M1 is confined to the muscle and brain, whereas PK-M2 is found in proliferating cells, including tumor cells of various tissues (23). HIF increases the transcription of the PKM2 gene locus, while expression of specific splice factors defines the expression of PK-M1 versus PK-M2 in hypoxic cells (20). This profoundly influences glycolysis as PK-M2 in opposite to PK-M1 limits the glycolytic flux and shifts metabolism toward the pentose phosphate pathway (PPP) thus, increasing the cellular NADPH level (see below) (23, 41).

Parallel to enhancing glycolysis, the entry of pyruvate into mitochondrial metabolism is attenuated by lactate dehydrogenase (LDH) that effectively converts pyruvate to lactate (Fig. 3). This reaction allows to export this metabolite via monocarboxylate transporters like MCT-1 or -4 (21) and also restores NAD+ level for ongoing glycolysis (30). LDH, MCTs, as well as pyruvate dehydrogenase kinase (PDK)-1 are HIF-target genes that are frequently upregulated under hypoxia. PDK-1 inactivates pyruvate dehydrogenase (PDH) by phosphorylation, and thereby prevents entry of pyruvate into the mitochondrial TCA cycle (51, 74). As NADH and other reductive equivalents cannot be used by the ETC, this is a key step in altering cellular metabolism toward anaerobic energy production.
Besides glucose/pyruvate, fatty acids imported into mitochondria and degraded via β-oxidation are the main supplier of acetyl-CoA and subsequent NADH for the ETC. Knockout experiments in mice suggest that HIF-2 decreases expression of PGC-1α and lowers PPAR transcripts that reduces β-oxidation (81). While HIF-1 mainly increases glycolysis and limits entry of metabolites into the TCA cycle, HIF-2 seems to minimize β-oxidation.
Additionally, HIF-1 directly modulates the ETC by switching COX subunit 4-1 to COX4-2 (32). As summarized in Table 1, HIF-1 activates transcription of COX4-2 and LON, a mitochondrial protease that is required for degradation of COX4-1. This subunit switch allows a more efficient O2 metabolism and thus, helps to optimize ETC fluxes under hypoxic conditions. In contrast, miRNAs upregulated by HIF, mainly miR210, represses the ETC by blocking expression of iron–sulfur cluster assembly proteins ISCU1/2 and thus, reduce expression of TCA enzymes, such as aconitase and ETC complexes (12). In addition, complex I subunit NDUFA4L2 is upregulated by HIF-1 and appears responsible for reduced complex I activity (89). Although switching COX4-1 to COX4-2 enhances, while switching NDUFA4 to NDUFA4L2 reduces O2 consumption both mechanisms prevent the production of H2O2 under hypoxia (32, 89). The same holds true for the other HIF-dependent adaptations, for example, upregulation of PDK-1 and ISCU1/2 (12, 51). These mechanisms are summarized in Table 1 and increase the viability and/or proliferation of cells under hypoxic conditions. Therefore, HIF represents a major transcription factor for adaptation of mitochondria to cellular hypoxia, shifting metabolites to still active ATP-producing pathways (glycolysis), at the same time reducing accumulation of mitochondrial NADH and ROS.
PDH, pyruvate dehydrogenase; COX, cytochrome oxidase.
HIF Mitochondria and Cancer Metabolism
Hypoxia enhances glycolytic ATP production, but also limits entry of glucose into the PPP thus, reducing NADPH production. NADPH is a cofactor for enzymes involved in fatty acid production and defense against oxidative stress. Besides NADPH, PPP delivers also precursors for DNA synthesis or its repair. Fatty acid production and DNA synthesis are mandatory for proliferating cells and thus, require appropriate adjustments. A close link between glucose entry into the PPP and expression of PFKB3 was shown comparing astrocytes to neurons. The later do not show PFKB3 protein expression because of its continuous proteasomal degradation [for details see (6)]. This limits glycolytic energy production and thus, renders them sensitive toward hypoxic cell stress. In contrast, astrocytes have the potential to increase PFKB3 protein, to raise F-2,6-BP, and are more resistant against cell stress accompanied by inhibition of the ETC. Overexpression of PFKB3 in neurons increases glycolytic ATP production, but induces oxidative stress and cell death as NADPH levels are reduced, and therefore their capacity to reduce GSSG is lost. Obviously, enhanced glycolysis is coupled to reduced PPP activity and thus, reduced NADPH production.
To balance glucose fluxes, cancer cells enhance production of PK-M2. As discussed, the expression of PK-M2 decreases glycolysis, and thereby channels glucose metabolites into biosynthetic intermediates to support anabolic growth. The expression of the PK-M2 splice variant is linked to the activity of c-Myc [v-myc myelocytomatosis viral oncogene homolog (20)]. c-Myc increases expression of RNA-binding proteins heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1/2) and polypyrimidine tract binding protein (PTBβ), that promote splicing of PK-M2 instead of PK-M1. Interestingly, it was shown that proliferation of cancer cells is enhanced by HIF-2α, but reduced by HIF-1α (37). This is linked to a negative influence of HIF-1α on c-Myc protein expression and activity. HIF-2α on the other hand enhances c-Myc activity. This led to the proposal that in hypoxic nontumor cells, which mostly express HIF-1α over HIF-2α, hypoxic activation of HIF-1 enhances the expression of PK-M1 and thus, allows extended energy production via glycolysis. In cancer cells that express higher HIF-2α levels, enhanced proliferation might be a result of enhanced PK-M2 expression with a shift toward PPP and production of NADPH and DNA precursors. PK-M2 may also function as a cellular sensor for oxidative stress. A critical cysteine residue (Cys358) of PK-M2 is oxidized and this oxidation inactivates the enzymes to enhance glucose metabolism through the PPP (2). The elevated production of NADPH then enables cells to regenerate GSH and to restore their redox balance.
The consequence of PDH inactivation by HIF-1-mediated upregulation of PDK1 and LDH is the limited entry of acetyl-CoA into the TCA. To circumvent this, glutamine is imported into mitochondria of cancer cells, converted to α-KG, and used to fuel TCA [for more details see (23)]. The aim of cancer cells is to produce citrate, which is then transported into the cytosol and used as a substrate for lipid synthesis. In addition to the breakdown of α-KG during TCA cycle is its conversion to isocitrate by mitochondrial IDH2. In contrast to IDH3, which preferentially produces α-KG, IDH2 and also cytosolic IDH1 are able to produce isocitrate in a NADPH-dependent reaction, known as reductive carboxylation. Hypoxic cancer cells maintain proliferation by reductive carboxylation catalyzed by IDH2; thus, replenishing their citrate pool for fatty acid synthesis (99). NADPH needed for this reaction in mitochondria can arise from the H+-transhydrogenase or malate dehydrogenase (malic enzyme). Metabolic changes are illustrated in Figure 4. In addition to mitochondrial IDH2, the cytosolic IDH1 also contributes to citrate production in hypoxic cells. This reaction is favored by the HIF-1-mediated upregulation of PDK-1, and therefore inactivation of PDH (64). Cells overexpressing HIF-1α due to pVHL-defects preferentially use this pathway for citrate production even under normal pO2. Inhibition of PDK-1 prevents this reductive glutamine metabolism, demonstrating the close relationship between HIF-induced PDH deactivation and glutaminolysis thus, linking HIF-1 to cancer cell proliferation. NADPH that is needed for this reaction can be produced by the malate enzyme, which uses malate exported from mitochondria to produce pyruvate and NADPH (22). A high rate of glutamine metabolism in cancer cells fulfills the need to produce citrate even under hypoxic conditions and additionally supplies NADPH necessary for the reaction.
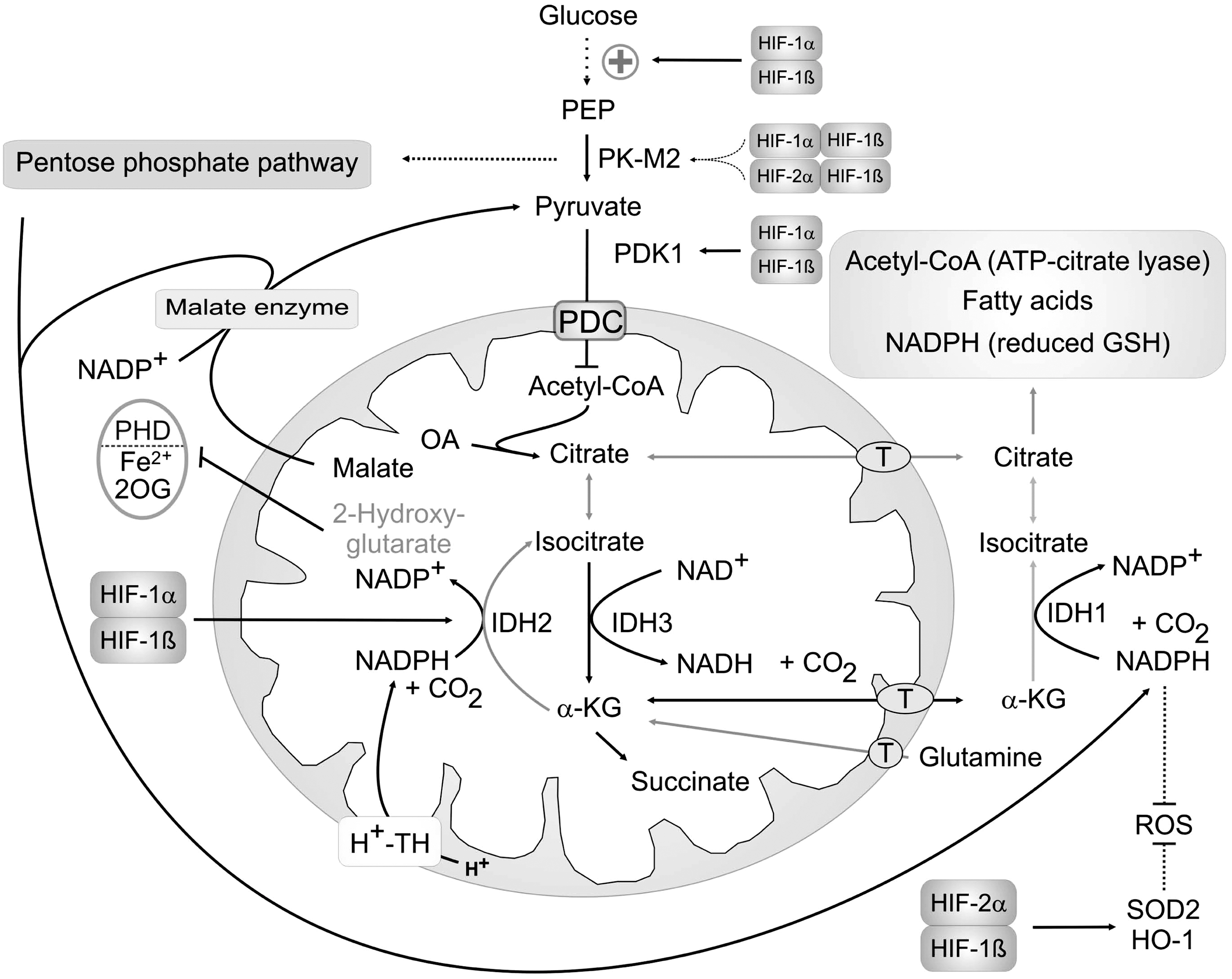
One consequence of the altered metabolism in cancer cells is an enhanced accumulation of HIF-1α in the presence of high glucose levels, which is not observed in noncancer cells (24). This HIF-1 activation even under normal pO2 is mediated by inhibition of PHDs by increased TCA intermediates (58). Mutations of TCA enzymes, such as fumarate hydratase or succinate dehydrogenase, also activate HIF-1. The products of these enzymes, fumarate and succinate, inhibit PHDs and thereby accumulate HIF-1α (7, 45, 70). PHD inhibition can be reversed by the addition of α-KG, showing that these mutations interfere with the product/substrate equilibrium of hydroxylases. As HIF activation further promotes altered mitochondrial metabolism, this is a feed forward pathway enhancing cancer cell proliferation.
Hypoxia and Autophagy
Hypoxia has not only a profound effect on mitochondrial metabolism, but also on mitochondrial morphology and mass. Reduction of mitochondrial mass is a consequence of reduced mitochondrial biogenesis. Although c-Myc is one of the transcription factors enhancing glycolytic pathway in normoxic cancer cells, it also enhances mitochondrial biosynthesis and mass (37). HIF-1α inactivates c-Myc by upregulating the c-Myc inhibitor MXI-1, but also by promoting c-Myc proteasomal degradation (104). Reduced c-Myc activity results in a loss of PGC-1β expression and thus, reduces respiration. In addition, activation of FoxO3A (forkhead box O3A) by HIF-1 antagonizes c-Myc gene induction, and thereby participates in reducing mitochondrial mass, oxygen consumption, ROS production, and enhances survival under hypoxia (46). For details see Figure 5.
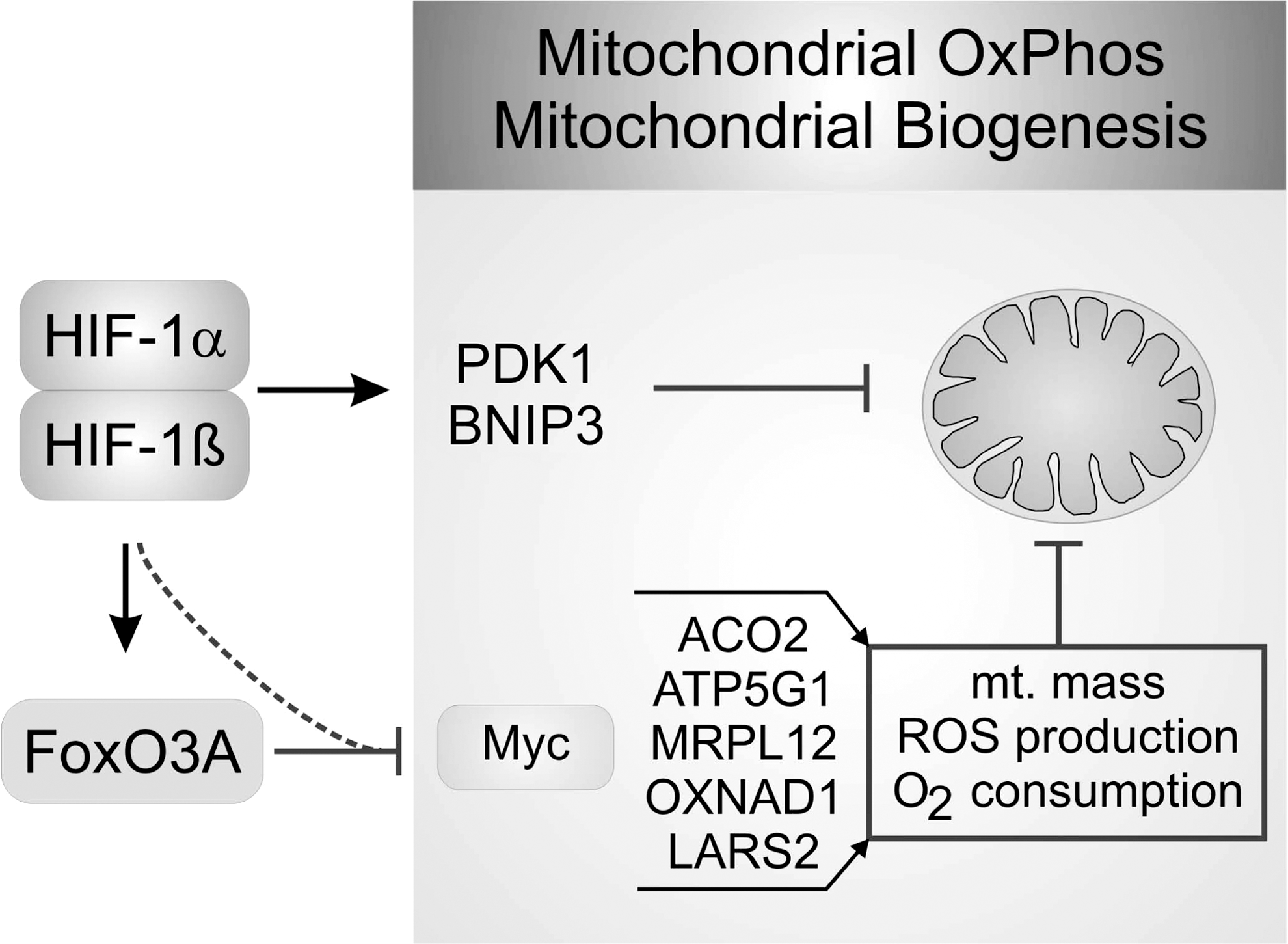
The second mechanism to reduce mitochondrial mass is to eliminate mitochondria by autophagy as shown in Figure 5. Autophagy occurs by the formation of autophagosomes, double-membrane vesicles that sequester organelles, proteins, or parts of the cytoplasm. Fusion of these autophagosomes with lysosomes results in the degradation of their content by lysosomal enzymes and recycles them as a source of energy. Cell survival is tightly linked to mitochondria that not only produce ATP, but also participate in activation of apoptosis. Intrinsic, but also extrinsic apoptosis pathways are initiated/amplified by outer mitochondrial membrane permeabilization and the release of cytochome c, among other proteins. This provokes the formation of the apoptosome, subsequent caspase 9 activation, followed by apoptotic cell death. Opening of the permeabilization pore is regulated by Bcl2 family members. Pore opening is controlled by the mitochondrial membrane potential, Ca2+ concentrations, or mitochondrial ROS and participates in hypoxic cell death. Thus, elimination of defect or spared mitochondria is crucial for cell survival.
Upregulation of the Bcl2 family members, BCL2/adenovirus E1B 19 kDa interacting protein 3 (BNIP3) or BINP3L, by HIF does not interfere with pore formation, but induces mitochondrial elimination via autophagosome formation (103). Enhanced BNIP3 expression recruits Beclin 1 and Atg5 (autophagy-related 5 homolog) to mitochondria by disrupting the Bcl2/Beclin 1 complex to eliminate mitochondria without inducing cell death (4). Recruitment of Beclin 1 to mitochondria is considered a crucial step in autophagosome formation that is identified by the appearance of LC3 on the autophagosome. FUN 14 domain-containing protein 1, an integral mitochondrial outer-membrane protein, interacts directly with LC3. Phosphorylation by Src kinases prevents this interaction and thus, inhibits autophagy induction. Under hypoxia, phosphorylation by Src kinases is reduced, allowing an enhanced interaction of FUNDC1 with LC3 and enhanced autophagosome formation (55). Additionally, Parkin was shown to translocate to mitochondria in cells treated with an uncoupler to chemically simulate hypoxia (27). Parkin suppresses mitochondrial fusion and also promotes mitochondrial-specific mitophagy (87). The induction of autophagy under hypoxic conditions delays the point of no return in apoptotic and necrotic cell death, and therefore represents an additional mechanism to increase survival of affected tissues (57). Considering the fact that maintaining ΔΨm consumes vast amounts of ATP under hypoxia when energy production is limited, the reduction of mitochondrial mass might represent a crucial mechanism to reduce ATP wastage, and therefore enhance viability.
Conclusion and Ongoing Questions
Hypoxia initiates cell and systemic adoptive responses, with the notion that HIF-1 and HIF-2 are crucial transmitting components. The discovery of HIF and the oxygen-regulated PHDs provided an intriguing direct connection between the lack of oxygen, post-translational protein modification, and HIF-transcriptional responses. This concept neglects the need of mitochondria to directly sense hypoxia, despite being the largest oxygen-consuming system in cells. Rather, mitochondria modulate hypoxic responses by adjusting the rate of intracellular oxygen consumption, thus allowing oxygen to be used by nonmitochondrial sources. There is, however, evidence that ROS, generated by mitochondria or NOX contribute to HIF regulation. However, a direct, highly dynamic and fully reversible coupling of ROS formation and HIF activation is missing, as uncertainties about transmitting systems exist. Whether the redox state of the cell fulfills these requirements is not known, but appears unlikely, as otherwise pro-oxidative perturbation should always be associated with hypoxic responses. For mitochondrial O2 − formation at complex III, it will be needed to define a mechanistic link how reduced oxygen availability accounts for ROS generation and to clarify how this signal is specifically transmitted to the HIF proteins. For NO, at least iNOS-derived NO, stabilization of HIF-1α is proven in cellular systems with the notion that either tumor cell iNOS or macrophage iNOS generate sufficient quantities of the radical to activate HIF. These data obtained primarily in the murine system need to be extrapolated to the human system with great caution, considering the controversy on the relevance of human iNOS, particularly in human macrophages. In addition, the notion that iNOS is basically inactive under 1% oxygen limits iNOS-derived NO in affecting the HIF system to conditions, for example, inflammation that would allow a high NO output system to be active. A relevant system to approach this question might be the tumor microenvironment. If correct, NO formation in a progressing tumor can limit the appearance of HIF with consequences for tumor growth and vascularization.
During the onset of hypoxia, adaptive responses may help to preserve respiration by inducing COX4.2, which enables mitochondria to continue consuming oxygen even when oxygen becomes limited. Under these conditions, hypoxia may also redirect Krebs cycle intermediates to guarantee citrate formation, which is essential for tumor cell replicating under reduced oxygen content. However, other HIF-responses are mastering these organelles to lose their function and to decline in terms of mass. The lower reductive power of the cell under hypoxia may require save guard systems to operate in parallel and a reduced electron flow in the ETC may lower the risk of ROS formation. At continued hypoxia, the demand of energy sources may then foster autophagy for recycling nutrient and synthetic components to ensure survival under chronic hypoxia. The additional benefit in reducing mitochondrial mass may reside in the fact that maintaining ΔΨm consumes substantial amounts of energy. Mitophagy simply eliminates a large energy consumer and spares ATP.
There appear to be gradual responses of mitochondria to hypoxia as summarized in Figure 6. In an initial phase, they may contribute to HIF activation and adopt to guarantee oxygen consumption. During hypoxia, controlling mitochondrial oxygen consumption by, for example, NO, may fine-tune the system, enabling hypoxic cells to shift back to normoxic metabolism. Prolonged hypoxic periods paralyze mitochondria to avoid the risk of harmful ROS production, which later may shift to their removal by autophagy to survive chronic periods of nutrient supply. When and how these responses are fully or in part reversible need to be proven in the future. HIF appears a perfect relay system to sense and fight mitochondrial hypoxic responses.

Footnotes
Acknowledgments
We apologize to researchers whose primary observations, that form the basis for our current knowledge in this field, could not be cited due to space limitations, or have been acknowledged indirectly, by referring to current reviews. The work was supported by grants from Deutsche Forschungsgemeinschaft (SFB 815, Excellence Cluster Cardiopulmonary System), Deutsche Krebshilfe (109599), Sander Foundation (2007.070.2), and TRIP (Translational Research Innovation Pharma).