
Abstract
Introduction
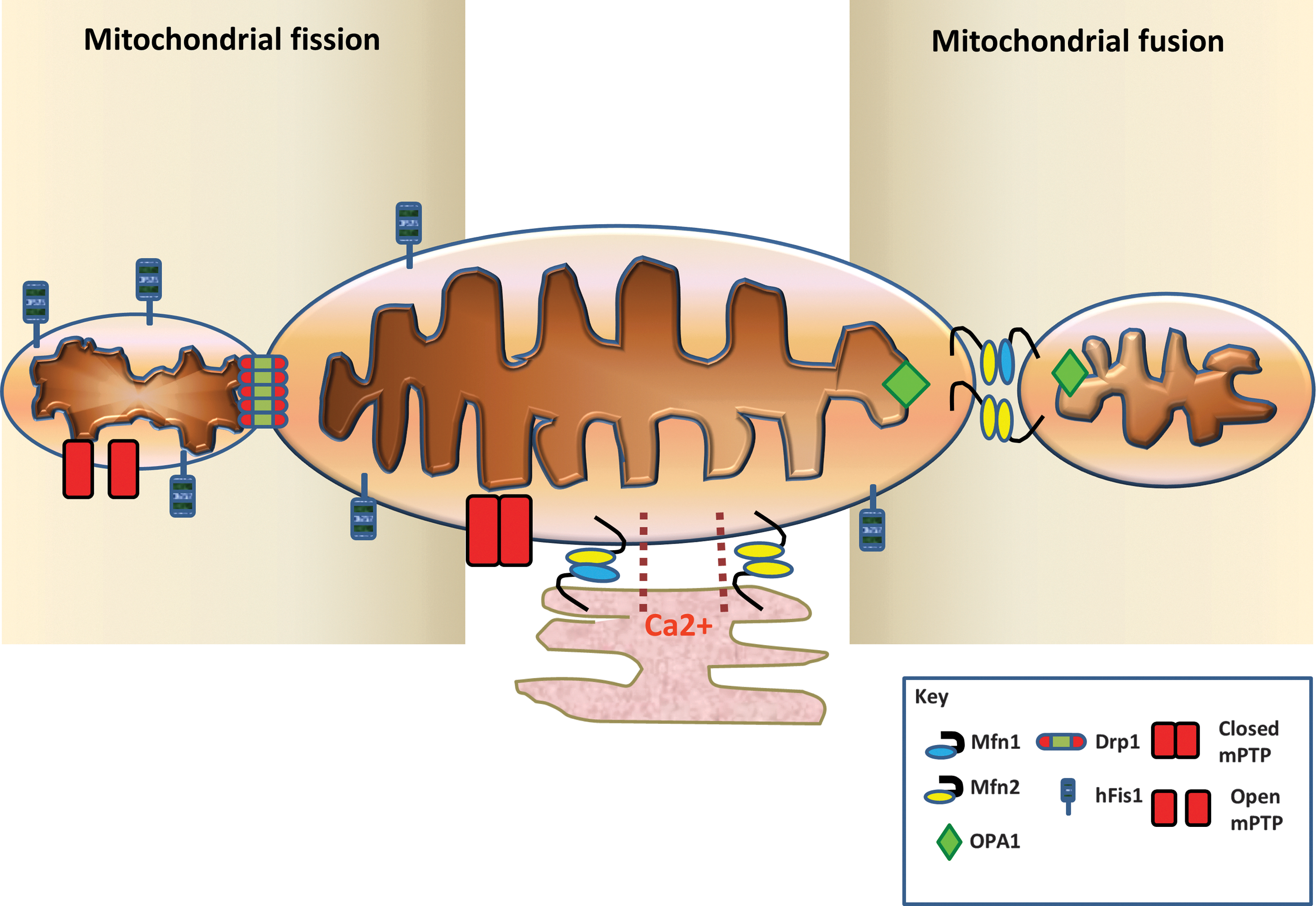
Mitochondrial Fission and Fusion
Changes in mitochondrial dynamics in mammalian cells are under the control of a group of evolutionary conserved mitochondrial fusion proteins (mitofusins 1 and 2 [Mfn1 and 2], and optic atrophy-1 [OPA1]) and mitochondrial fission proteins (dynamin-related peptide 1 [Drp1], and mitochondrial fission protein 1 [Fis1]) (Table 1, Fig. 2). A number of additional mitochondrial fission proteins have recently been discovered including, amongst others, mitochondrial fission factor (MFF) (34, 77) and mitochondrial dynamics proteins of 49 and 51 kDa (MiD49/51) (78, 111). The movement and distribution of mitochondria within the cell are also dependent on the interplay of the mitochondrial fission and fusion proteins with the Miro/Milton/kinesin complex, which mediates the interaction of mitochondria with the cytoskeletal components of cell to allow movement along the microtubules (66). Interestingly, all of these mitochondrial proteins are present in the heart and vascular system, but their role there has only begun to be elucidated. In the following sections, an overview will be given of the mitochondrial fission and fusion machinery (for a more detailed account, please see the following reviews [12, 25, 64, 90]).
Mitochondrial fusion
The process of mitochondrial fusion between individual mitochondria requires the coordinated fusion of their respective outer and inner mitochondrial membranes, a process which is governed by three large GTPases, the mitofusins and OPA1 (64). Fusion of the outer mitochondrial membranes is under the regulation of the outer membrane proteins, Mfn1 and 2, whereas fusion of the inner mitochondrial membranes is under the control of the inner membrane protein, OPA1 (Fig. 2) (64). In yeast, there is an additional mitochondrial fusion protein called Ugo1 that has been reported to synchronize both OMM and IMM fusion by binding to both Mgm1 (the yeast orthologue of OPA1) and Fzo1 (the yeast orthologue of Mitofusin), although its mammalian counterpart has not yet been identified (Table 1) (94).
Mitofusins 1 and 2
The first mitochondrial fusion gene to be discovered was in Drosophila in 1997 by Hales and Fuller (39). Its name, fuzzy onions (Fzo), referred to its role in mediating the mitochondrial fusion required for the formation of the Nebenkern (a spherical structure containing coiled-up mitochondria that resembles an onion slice when imaged in cross-section) in the developing spermatid (39). The Fzo gene was found to encode a novel large transmembrane GTPase of which yeast (Fzo1p) and mammalian homologues (Mitofusins 1 and 2) were subsequently discovered in 1998 (86) and 2001 (92), respectively. Mitofusins are present in all tissues, but the expression of Mfn1 and Mfn2 mRNA varies depending on the tissue with high levels present in the heart compared to other tissues (91). In the heart, the expression of Mfn2 protein was found to be higher than that for Mfn1 protein (91).
In 2001, Santel and Fuller (92) first discovered two human genes encoding the first known mitochondrial fusion proteins in mammalian cells, mitofusin 1 and 2 (Mfn 1 and 2). These proteins, which are large GTPases, reside in the outer mitochondrial membrane, tether mitochondria together, and mediate the process of outer membrane fusion, a process which requires GTP hydrolysis, although the exact details of this process remain unclear. Both mitofusins are transmembrane GTPase proteins and comprise a COOH-terminal that contains a transmembrane domain and a coiled-coil domain 2 (also called heptad-repeat domain or HR2) (91). The NH2-terminal part of the mitofusin proteins contains the GTP-binding domain and another coiled-coil domain (HR1) (61, 91). In the first step of outer mitochondrial membrane fusion, the HR2 domains of Mfn1 or Mfn2 on one mitochondrion are believed to form dimers or complexes with Mfn1 or Mfn2 on adjacent mitochondria, a process which tethers two adjacent mitochondria together (Fig. 2) (61). Mfn1 has a greater tethering efficiency than Mfn2, and this is due to its higher GTPase activity (52).
Whole body genetic ablation of Mfn1 and Mfn2 results in embryonic lethality due to a major placental defect (15). Mutations in human Mfn2 are responsible for the autosomal dominant neurodegenerative disease Charcot-Marie-Tooth type 2A, a peripheral sensorimotor neuropathy (113).
Pleiotropic actions of Mfn2
Human Mfn2 has a number of pleiotropic actions, many of which are independent of its function as a pro-fusion protein (these are further discussed in the relevant section) (23). At the time of its initial discovery as a mammalian mitochondrial fusion protein in 2001, it had been independently discovered in muscle from Zucker obese rats (named mitochondrial assembly regulatory factor or MARF) (6), and in vascular smooth muscle cells of rats with genetic hypertension (named hyperplasia suppressor gene or HSG) (16). Some experimental studies have also implicated Mfn2 as a modulator of the mitochondrial apoptotic pathway (see Mitochondrial morphology and apoptosis). More recently, Mfn2 has been demonstrated to be present on endoplasmic reticulum (ER) where it tethers ER to mitochondria, participating in calcium signaling by establishing a calcium microdomain (22). The close juxtaposition of ER to mitochondria has been demonstrated to be required for IP3 signaling and calcium-induced apoptotic signaling– whether this is relevant to the heart remains to be determined.
Optic atropy-1
Optic atrophy-1 (OPA1) is the human homologue of the yeast mitochondrial fusion protein, Mgm1, first discovered in yeast in 1992 (57). OPA1 is expressed throughout the body, but is present in largest quantities in the retina, brain, testis, liver, heart, skeletal muscle, and pancreas (1). Mutations in the gene were first identified in 2000 to be associated with the human neurogenerative condition, autosomal dominant optic atrophy (24). The OPA1 protein is a large GTPase, which is present in the inner mitochondrial membrane, and whose main function is to promote mitochondrial fusion of the inner mitochondrial membrane (24).
Structurally it comprises an NH2-terminal mitochondrial import sequence, transmembrane domain, coiled-coil domain, GTPase domain, a middle domain, and a GTPase effector domain at the COOH-terminus. The hydrophobic segments constitute the transmembrane domain that has been predicted to anchor or associate OPA1 with the mitochondrial membranes. The GTPase domain is required for mitochondrial fusion (74). OPA1 requires the presence of Mfn1 to induce mitochondrial fusion (20).
Post-transcriptional processing of OPA1 is rather complex (64). As an oversimplification, alternative splicing at exons 4, 4b, and 5b generates eight human isoforms of OPA1. All the 8 human OPA1 isoforms have the NH2-terminal mitochondrial import sequence, which is removed by the mitochondrial matrix protease (mitochondrial processing peptidase) upon import into mitochondria. The OPA1 isoforms are then constitutively cleaved at site 2 (S2 in exon 5b) by the intermembranous AAA (ATPase associated with diverse cellular activities) protease, YME1L, which generates the short forms of OPA1 or S-OPA1 (this includes the OPA1 isoforms 3, 5, 6, and 8), which are soluble and reside in the intermembranous space. The remaining OPA1 isoforms (1, 2, 4, and 7) do not normally undergo cleavage and constitute the long form of OPA1, which has the transmembrane domain, and is anchored into the inner mitochondrial membrane. Under normal conditions, both the S- and L-OPA1 in equal amounts are required for mitochondrial fusion and an imbalance in the respective levels of S- and L-OPA1 can impact on the pro-fusion activity of OPA1 (64).
In this regard, under conditions of mitochondrial membrane depolarization, ATP deficiency and apoptosis, the L-OPA1 isoforms undergo inducible cleavage by OMA1 (28, 46) at site S1 to generate S-OPA1 isoforms, which removes the pro-fusion activity of OPA1, resulting in mitochondrial fragmentation. Through this process, OPA1 plays a role in ‘tagging’ depolarized fragmented mitochondria for removal by mitophagy (103). Independent of its pro-fusion activity, the S- and L- forms of OPA1 have been shown to form oligomers and keep cristae junctions tightly closed, thereby preventing the cristae remodeling required for the cytosolic release of cytochrome C during apoptosis (33).
Mitochondrial fission
The process of mitochondrial fission or division is hugely complex and is only beginning to be unraveled. It can be divided into several stages that include the initial constriction of the mitochondria, the recruitment of cytosolic Drp1 to mitochondria, the assembly of a scission complex on the outer mitochondrial membrane, the actual scission event, and finally the disassembly of the scission complex (Fig. 2) (64).
Dynamin-related protein 1
In 1998, several independent groups first identified dynamin-related peptide 1 (Drp1), a mammalian homologue of yeast and Drosophila, as a protein capable of inducing mitochondrial division in mammalian cells (97, 108), an action which is related to the yeast orthologue, Dnm1p (8). Drp1 is mainly cytosolic, and translocates to scission sites on the outer mitochondrial membrane to induce fission or division of mitochondria. After the initial constriction of the mitochondria, the yeast Drp1 orthologue, Dnm1, has been demonstrated to self-assemble into a helical structure that encircles the dividing mitochondria and mediates the final constriction and scission process in a GTP-dependent manner (64).
Structurally, Drp1 is made up of a GTPase, the central domain, and the GTPase effector domain or assembly domain (97). On activation, the Drp1 oligomerizes and constricts the mitochondrial scission site, a process that requires GTPase. The GTPase effector domain is required for regulation of GTPase activity and for mitochondrial targeting (96). Because Drp1 does not have a transmembrane domain, for it to bind to the outer mitochondrial membrane it requires a docking receptor on the OMM which may or may not be hFis1 (107). In yeast, the adaptor proteins, Mdv1 and Caf4, have been reported to mediate the binding of Drp1 to hFis1 (36, 101). However, the homologous proteins in mammalian cells have yet to be identified. Two other components of the mitochondrial fission machinery have recently been discovered, mitochondrial fission factor (MFF) (34, 77) and mitochondrial dynamics proteins of 49 kDa (MiD49) and 51 kDa (MiD51) (78); whether these factors facilitate the binding of Drp1 to the OMM remains to be determined. Drp1 has been recently demonstrated to be regulated by a number of post-translational modifications such as phosphorylation and nitrosylation (see later section on cardioprotection) (reviewed in Ref. 90). Drp1 is essential for embryonic development and synapse formation as mice deficient in Drp1 die in utero at embryonic day 11.5, confirming that mitochondrial division is essential for normal embryonic development (53, 104).
Human mitochondrial fission protein 1 (hFis1)
Mammalian fission protein 1 (hFis1) was first identified in 2001 by its homology with Fis1p, the yeast ortholog (55, 107). It is a small protein of 17 kDa and 152 amino acids whose primary function is to promote fission of mitochondria and peroxisomes. It is ubiquitously expressed and is detected throughout the mitochondrial network where it is inserted into the outer mitochondrial membrane. It inserts into the outer mitochondrial membrane via its COOH-terminal part, which contains an α-helix, a transmembrane domain, and a COOH-terminal tail exposed to the intermembrane space. The NH2-terminal part of the protein contains four distinct regions with five α-helices (26, 98), with the first α-helix of rat Fis1 critical for oligomerization and for its fission activity (56). The next four α-helices make up two tetratrico-peptide repeat peptides (TPR1 and TPR2), which are involved in protein–protein interactions required for fission but are not required for Fis1 oligomerization (56).
Overexpression of Fis1 in cells induces mitochondrial fragmentation that can result in Drp1-dependent cytochrome c release and apoptosis (63). In contrast, inhibiting Fis1 can protect against apoptotic cell death (63). Fis1 can also induce apoptotic cell death independent of Drp1 and can promote mitochondrial fragmentation without inducing apoptotic cell death (2). Finally, Fis1 is required to induce mitochondrial fission in senescent long mitochondria to facilitate their removal by mitophagy (109). However, there are experimental studies which question the obligatory role of hFis1 in Drp1-mediated mitochondrial fission. hFis1 localizes throughout the MOM in contrast to the punctate localization of Drp1, and mitochondrial recruitment of Drp1 is not affected by hFis1 knockdown (63, 97A, 106A). Similarly, neither mitochondria-associated Drp1 nor mitochondrial fission is affected by hFis1 overexpression (98). These contradictory observations on hFis1 may suggest that, although Fis1 is required for the mitochondrial fission, it is not a limiting factor in the mitochondrial fission process and mitochondrial recruitment of Drp1 is regulated by other factors (see below).
Mitochondrial fission factor
In 2008, whilst undertaking a siRNA screen for novel proteins that affect mitochondrial morphology in Drosophila cells, Gandre-Babbe and van der Bliek (34) discovered another outer mitochondrial membrane protein that mediates both mitochondrial and peroxisome fission, called CG30404/Tango11—its human homologue was named mitochondrial fission factor (MFF). It is highly expressed in heart, kidney, liver, brain, muscle, and stomach, and at low levels in other tissues and has a carboxy-terminal transmembrane domain that anchors it into the OMM (34). Ablation of MFF in HeLa cells using siRNA-induced mitochondrial fusion, partially prevented uncoupler-induced mitochondrial fission and reduced cytochrome c release from mitochondria during apoptosis, suggesting a potential interplay with Drp1 (34). A subsequent study by Otera et al. (77) demonstrated that MFF was required for Drp1 recruitment to mitochondria, and that MFF interacted with Drp1 to mediate mitochondrial fission in a manner which is independent of hFis1. Whether MFF, for which there is no yeast orthologue, plays a similar role to yeast Mdv1 and Caf4 in mediating the mitochondrial fission process, is unclear at present.
Mitochondrial dynamics proteins
To complicate things further, two additional components of the mitochondrial fission machinery have been identified in the last year. During a random cellular localization screen of uncharacterized human proteins, Palmer et al. (78) noticed that the expression of human Smith–Magenis syndrome chromosome region candidate gene 7L protein (SMCR7L) induced the appearance of elongated mitochondria. The authors renamed this protein and its closely related homologue, SMCR7, mitochondrial dynamics proteins of 49 kDa (MiD49) and 51 kDa (MiD51), respectively (78). These authors demonstrated that MiD49/51 formed foci and rings around mitochondria, and directly recruited Drp1 to the mitochondria. In contrast, siRNA ablation of MiD49/51 reduced translocation of Drp1 to mitochondria and resulted in mitochondrial fusion (78). Interestingly, the overexpression of MiD49/51, paradoxically, resulted in an abnormal form of mitochondrial elongation, a finding which was attributed to and associated with the sequestration of Drp1 at mitochondria resulting in unopposed fusion (78).
Of note, another experimental study also published in 2011, investigated the effects of mitochondrial elongation factor 1 (MIEF1, independently identified as MiD51) on mitochondrial dynamics and also observed recruitment of Drp1 to mitochondria and elongation of mitochondria (111). However, these authors interpreted the findings to suggest that MIEF1/MiD51 was inducing mitochondrial fusion rather than fission (111). The major discrepancies between these two experimental studies were the results of the siRNA knockdown. Zhao et al. (111) demonstrated that siRNA knockdown of MIEF1/MiD51 resulted in mitochondrial fragmentation, whereas Palmer et al. (78) found that siRNA knockdown of both MiD49 and MiD51 induced mitochondrial elongation. Clearly, further studies are required to elucidate the actual role of MiD49/MiD51 in mitochondrial dynamics and its interplay with the other mitochondrial fission proteins, Drp1, hFis1, and MFF.
Mitochondrial motility
Mitochondria play an essential role in intracellular calcium signaling and directing energy supply. This function depends on the ability of mitochondria to move up and down microtubules in a calcium-sensitive manner, particularly in the neurons. Mitochondrial motility is under the control of a calcium-sensitive complex, composed of Miro, GRIF1-OIP106 (the mammalian orthologues of Milton), and the motor proteins kinesin or dynein, which regulates mitochondrial trafficking along microtubules.
The mitochondrial Rho (Miro) proteins are Ras GTPases, which are evolutionary conserved from yeast to man (30). Mammalian cells have two types of Miro (1 and 2), which comprise 2 GTPase domains one at each end, a transmembrane domain at the C terminus (which anchors the protein to the outer mitochondrial membrane), and Ca2+-binding EF-hand motifs (which confer calcium sensitivity) (30 –32). Grif-1 and OIP106, which are the mammalian homologues of Milton (a Drosopholia protein which has no yeast equivalent) (9A), are cytosolic proteins that bind both Miro and kinesin heavy chains (KHC) to mediate mitochondrial trafficking (35, 88).
In H9c2 cardiac cells and primary cortical neurons, genetic ablation of the Miros arrested mitochondrial movements, whereas the overexpression of Miros increased mitochondrial motility at low cytosolic calcium levels (93). This effect of Miro1 on mitochondrial motility was dependent on its GTPase activity, which in turn was required to recruit Grif-1 (69). In hippocampal neurons, Miro has also been demonstrated to interact with KHC and dynein motors, suggesting that Miro is required for both anterograde and retrograde mitochondrial transport along the microtubule (69, 89). Mitochondrial motility is influenced by cytosolic calcium concentrations with movements reduced in areas of high cytosolic calcium concentrations within the cell. It has been suggested that in the presence of elevate cytosolic calcium levels, the calcium-sensitive EF-hands on Miro bind directly to the KHC, preventing the interaction of the latter with the microtubule (69, 93, 106).
The mitochondrial motility proteins have also been reported to influence mitochondrial morphology. Saotome et al. (93) demonstrated in H9c2 cells that overexpressing of Miro1 and Miro2 induced mitochondrial elongation, whereas ablating Miro resulted in Drp1-mediated mitochondrial fragmentation. This effect of Miro on mitochondrial shape was shown to be dependent on GTPase but not the EF-hands.
Although mitochondrial motility is important in the H9c2 cardiac cell line (93), its relevance to the adult heart in which the interfibrillar mitochondria are tightly packed between myofibrils is unclear. Perhaps for the peri-nuclear and subsarcolemmal mitochondria, which are less restricted in their movements, mitochondrial motility may be more important, in which case the function of the above mitochondrial motility proteins in the adult heart would be interesting to investigate.
Mitochondrial Morphology in Vascular Cells
Mfn2 and vascular smooth muscle cells
In 2004, Chen et al. (16) were the first to implicate Mfn2, as a novel hyperplasia suppressor gene (HSG), capable of inhibiting vascular smooth muscle cell (VSMC) proliferation in a variety of vasculo-proliferative conditions. This pleiotropic effect of Mfn2 was independent of its ability to promote mitochondrial fusion. These authors reported that HSG expression was reduced in VSMCs harvested from spontaneously hypertensive rat arteries and angioplasty balloon-induced endothelial injury in arteries from rats and in murine arteries from ApoE-knockout mice (16). HSG overexpression reduced VSMC proliferation induced by serum, blocked angioplasty balloon-induced neointimal VSMC proliferation, and inhibited restenosis in rat carotid arteries, effects that were found to be dependent on the binding of Mfn2 to Ras and the subsequent inhibition of Ras-Raf-MAPK-Erk1/2 signaling, one of the major growth factor responsive signaling pathways in mammalian cell proliferation (16).
In a subsequent study, the same research group found that the overexpression of Mfn2 inhibited VSMC proliferation triggered by oxidative LDL, and also reduced atherosclerotic formation, beneficial effects which were shown to be dependent on the downregulation of the MEK1/2-Erk1/2 and P3IK-Akt pathways (38). With respect to a potential mechanism for this anti-proliferative effect of Mfn2, the same authors have gone on to show that overexpressing Mfn2 promotes apoptotic cell death in VSMC using both in vitro and in vivo experimental models (see Mitochondrial morphology and apoptosis) (37).
Most recently, the same researchers have implicated PKA in the regulation of Mfn2 in its anti-proliferative actions on VSMCs in culture, and neointimal hyperplasia and restenosis in a rat carotid artery balloon injury model (112). These authors demonstrated that after mutating a PKA-specific phosphorylation site (Ser442) on Mfn2, anti-proliferative effects were blocked (112).
Whether Mfn2 has a similar role to play in cardiomyocytes has been investigated by Fang et al. (29), who have examined the role of Mfn2 in several different in vitro and in vivo models of cardiac hypertrophy, and demonstrated reduced mRNA expression of Mfn2 associated with increased Erk1/2 expression in phenylephrine-treated neonatal rat cardiomyocytes, spontaneous hypertensive hearts, mice with total aortic constriction, and β2-TG mice. Interestingly, Mfn2 was not found to be downregulated in hypertrophied hearts with 15 weeks of total aortic constriction, nor in hypertrophied noninfarcted myocardium post MI (29). However, protein levels of Mfn2 were not assessed and the significance of these findings remains unclear.
Mitochondrial morphology and endothelial cells
In the context of diabetes, it has been reported that in rat coronary and retinal endothelial cells, hyperglycemia induced mitochondrial fragmentation, mitochondrial ROS production, heterogeneity in mitochondrial membrane potential, reduced oxygen consumption, and increased propensity to apoptotic cell death (70, 102, 110). Whether this mechanism of mitochondrial dysfunction underlies the vascular abnormalities underlying diabetic vascular disease is unknown. Interestingly, the same authors have recently reported that hyperglycemia induces mitochondrial fragmentation and mitochondrial dysfunction in neonatal rat cardiomyocytes and this effect is associated with O-GlcNAcylation of OPA-1 (71). The role of Mfn1 and Mfn2 in mitochondrial function and angiogenic potential has been recently investigated in cultured human embryonic vascular endothelial cells (68). Lugus et al. (68) found that treatment with VEGF-A activated both Mfn1 and Mfn2 in human embryonic vascular endothelial cells, and that ablating Mfn1 and Mfn2 reduced the angiogenic response, reduced cell survival following serum deprivation, increased the susceptibility to hydrogen peroxide-induced mitochondrial membrane depolarization. Interestingly, in cells deficient in Mfn1, VEGF-A failed to activate Akt and eNOS, suggesting an impairment in endothelial signaling (68).
Mitochondrial Dynamics and the Heart
Many of the experimental studies investigating the phenomenon of mitochondrial dynamics have been confined to noncardiovascular cell lines and neuronal cells. However, more recently, a number of studies have begun to investigate the role of mitochondrial dynamics in the heart. Many of these experimental studies have been performed using immortal cardiac cell lines (H9c2, HL-1 cells), vascular cells (VSMC, endothelial cells), or neonatal cardiomyocytes, in which the mitochondria are distributed throughout the cytoplasm in a reticular network and are unrestricted in their movements. However, in the adult heart, mitochondria are not arranged in such a reticular network and their movements are heavily restricted. Therefore, the relevance of mitochondrial dynamics to the adult heart may not appear so obvious. However, the machinery for regulating mitochondrial dynamics is present in the adult heart—suggesting that adult cardiac mitochondria have retained their ability to undergo fission and fusion, processes that would be deemed essential for quality control of mitochondria and mitochondrial turnover. Although initial experimental studies investigating mitochondrial morphology in the cardiovascular system have been confined to immortal cardiac and vascular cell lines, more recent work has begun to investigate the role of the mitochondrial fission and fusion proteins in the adult heart.
Relevance of mitochondrial dynamics to the adult heart
In the adult heart, mitochondria measure 1–2 μm in length and are arranged in 3 distinct populations (Fig. 3) (3, 50): (a) Interfibrillar mitochondria (containing lamelliform cristae) which are arranged in rows alongside the myofibrils are presumably the main producers of ATP generation for contractile function and calcium-signaling; (b) Subsarcolemmal mitochondria (containing tubular cristae) which are arranged in clusters just beneath the sarcolemma may predominantly be involved with ion channel function or signaling pathways; and (c) Perinuclear mitochondria which are arranged in clusters adjacent to the nuclei and are presumably involved with nuclear transcription.

Interestingly, the possibility that mitochondria in the adult heart may undergo fission and fusion was first observed in 1972 (100). In the rat heart, mitochondria have a high turn-over rate (half-life estimated at 5.6–6.2 days, a process that would be expected to involve mitochondrial fission (100). In contrast, giant or mega- mitochondria have long been recognized to be present in some cardiomyopathies and appear to be associated with mutations in mitochondrial DNA (4, 62, 99). How these giant mitochondria are formed is unclear, although it may be due to combined mitochondrial growth and fusion of adjacent mitochondria (99). Interestingly, the formation of giant mitochondria appears to be limited to the interfibrillar mitochondria as opposed to the subsarcolemmal population: the reason for this difference in susceptibility is unknown.
Even under basal conditions it is possible to observe elongated mitochondria in the adult heart that most likely resulted from end-to-end mitochondrial fusion. Using electron microscopy, we have shown that a proportion of interfibrillar mitochondria are elongated extending up to 3–4 sarcomeres (6–8 μm) in length (Fig. 4) (76).

Mitochondrial morphology and apoptosis
Mfn2 has been implicated as a modulator of apoptotic cell death but whether its actions are pro-apoptotic or anti-apoptotic appears to vary with the experimental model, cell type, and may also relate to the many pleiotropic actions of Mfn2. For example, using COS-7 cells, Neuspiel et al. (73) demonstrated the overexpression of Mfn2 protected against apoptotic cell death and mPTP opening. In cerebellar granule neurons, Jahani-Asl et al. (54) found that Mfn2 protected against apoptotic cell death induced by DNA damage, oxidative stress, and K+ deprivation. In contrast, Mfn2 has been shown in noncardiac cells to form a functional unit with Drp1 and BAX at OMM scission sites and to mediate apoptotic cell death (51, 59).
Consistent with a potential pro-apoptotic effect of Mfn2, Shen et al. (95) demonstrated that hydrogen peroxide increased the expression of Mfn2 and augmented apoptotic cell death in neonatal rat cardiomyocytes. Furthermore, Mfn2 overexpression was found to suppress Akt activation and increase apoptotic cell death in neonatal cardiomyocytes (95). In H9c2 cardiac cells, siRNA knockdown of Mfn2 protected against oxidative stress-induced apoptosis (95). Collectively, the data from this experimental study suggested that Mfn2 may be is a major determinant of oxidative stress-induced cardiomyocyte apoptosis (95). In support of these findings, it was recently been demonstrated the genetic ablation of Mfn2 in the adult murine heart, also protected against apoptotic cell death, suggesting a potential pro-apoptotic role for Mfn2 (79).
In vascular smooth muscle cells (VSMC), Mfn2, which has an anti-proliferative effect (see Mrf2 and Vascular Smooth Muscle Cells), has also been reported to be a major determinant of apoptotic cell death (37). These authors found that triggers of apoptosis such as hydrogen peroxide, staurosporine, forskolin, and serum deprivation increased Mfn2 expression in VSMC. In contrast, silencing Mfn2 protected the cells against apoptotic cell death (37). The pro-apoptotic effect of Mfn2, which was found to be independent of its action on mitochondrial morphology, was reported to be mediated through the downregulation of the PI3K-Akt pathway and executed through the mitochondrial pathway of apoptosis (37).
In contrast with these findings suggesting that Mfn2 is pro-apoptotic, Parra et al. (82) found that, in neonatal cardiomyocytes, treatment with ceramide-induced changes associated with apoptosis (mitochondrial fragmentation, Drp1 localization with hFis1, mitochondrial membrane depolarization, cytochrome C release, and cell death) and these effects were exacerbated when Mfn2 was ablated, suggesting that Mfn2 may exerts an anti-apoptotic effect against ceramide.
Mitochondrial morphology and myocardial ischemia-reperfusion injury
During myocardial ischemia, cardiac mitochondria are subjected to abrupt biochemical changes characterized by hypoxia, sodium, calcium and phosphate overload, acidic conditions, and ATP depletion. These effects are exacerbated at the onset of myocardial reperfusion with the re-energization of the mitochondrial electron transport cycle generating oxidative stress, restoration of the mitochondrial membrane potential which drives calcium into mitochondria, a rapid recovery of physiological pH, factors that act in concert to induce the opening of the mitochondrial permeability transition pore (mPTP) (40, 45). The opening of the mPTP at the onset of myocardial reperfusion uncouples mitochondrial oxidative phosphorylation, resulting in ATP depletion and cardiomyocyte death (40, 45) and preventing its opening at this time by pharmacologically (42, 43) or genetic inhibition of mitochondrial cyclophilin D (7, 65, 72, 83), a regulatory component of the mPTP, has been shown to reduce myocardial infarct size in both the experimental and clinical settings. Therefore, preventing mitochondrial dysfunction and mPTP opening during a sustained episode of myocardial ischemia reperfusion is a powerful cardioprotective strategy. Emerging studies suggest that modulating mitochondrial dynamics may influence the susceptibility to IRI by preventing mitochondrial dysfunction and mPTP inhibition (76).
Experimental studies suggest that changes in mitochondrial dynamics occur in the heart in response to acute ischemia and reperfusion. Brady et al. (9) were the first to demonstrate that 2 hours of simulated ischemia induced mitochondrial fragmentation in over 90% of HL-1 cells (a murine-derived atrial cell line). These changes persisted 5 hours into simulated reperfusion, and interestingly, treatment with the pharmacological p38MAPK inhibitor, SB203580, reverted mitochondria into an elongated phenotype, suggesting that in this setting p38MAPK may have contributed to the fragmented morphology induced by simulated ischemia (9). Chen et al. (17) also found that simulated ischemia induced mitochondrial fragmentation in H9c2 cells (a cardiac myogenic cell line), a process that was associated with a reduction in OPA1 protein expression. Ablating OPA1 in cells induced mitochondrial fragmentation and enhanced the mitochondrial fission during simulated ischemia, suggesting that a reduction in OPA1 may contribute to mitochondrial fragmentation during ischemia (17). We have recently confirmed that mitochondrial fragmentation occurs in HL-1 cardiac cells subjected to simulated ischemia and that this process can be prevented in cells transfected with Drp1K38A, the dominant negative mutant of Drp1, suggesting that mitochondrial fragmentation during myocardial ischemia is a Drp-1-dependent process (76). Finally, we have also demonstrated that this change in mitochondrial morphology occurs in the adult heart, demonstrating that in vivo acute coronary occlusion for 20 min induced mitochondrial fragmentation in the adult murine heart as imaged by electron microscopy (Fig. 5) (76).

The mechanism through which myocardial ischemia induces mitochondrial fragmentation is unclear. The absence of oxygen halts oxidative phosphorylation, resulting in a collapse in mitochondrial membrane potential that could provide the signal for Drp1 translocation to the mitochondria. Whether mitochondrial calcium overload or oxidative stress generated during myocardial ischemia contributes to the observed change in mitochondrial morphology is unknown. In this regard, experimental studies have reported that calcium overload induced mitochondrial fragmentation in both neonatal and adult rat cardiomyocytes (48). It is interesting to note that calcium-induced activation of calcineurin in HeLa cells has been shown to dephosphorylate and activate Drp1, resulting in mitochondrial fragmentation (13). In this regard, Wang et al. (105) have recently demonstrated that during myocardial ischemia, levels of miR-4199 decrease, resulting in the activation of calcineurin and the subsequent dephosphorylation and activation of Drp1, the result of which was mitochondrial fragmentation. The regulation of Drp1 by phosphorylation and dephosphorylation is an example of its post-translationaI modification, with a number of experimental studies suggesting that Drp1-mediated fission activity can be inhibited by PKA by phosphorylation (14, 21).
Recently, Liu and Hajnóczky (67) investigated the changes in mitochondrial dynamics during simulated ischemia and reperfusion in H9c2 cells. Following mitochondrial fragmentation, they observed the formation of ‘donut’ shaped mitochondria, a process which was dependent on mPTP opening to induce mitochondrial swelling, the result of which was detachment from the cytoskeleton and anomalous fusion events forming a ‘donut’-shaped mitochondria (67). Interestingly, these ‘donut’ mitochondria were found to be resistant to further cell swelling and their offspring were able to regain mitochondrial membrane potential, suggesting that this altered fusion state may be a protective response against cellular stress (67). We have confirmed the formation of these ‘donut’-shaped mitochondria in HL-1 cardiac cells subjected to simulated ischemia (Fig. 6). Whether this phenomenon occurs in adult cardiac mitochondria is not clear.

Mitochondrial morphology and cardioprotection
The observation that changes in mitochondrial morphology during myocardial ischemia could be prevented led our research group to investigate the modulation of mitochondrial morphology as a novel therapeutic intervention for protecting the heart against acute IRI (76). In this regard, we found that transfecting HL-1 cardiac cells with mitochondrial fusion proteins, such as Mfn1 and Mfn2 as well Drp1K38A, the dominant negative Drp1 mutant, induced mitochondrial elongation, delayed the opening of the mPTP, and improved cell survival following simulated IRI (76). These findings suggested that acutely inhibiting mitochondrial fission or promoting mitochondrial fusion could protect the heart against an episode of lethal IRI. Furthermore, the acute pharmacological inhibition of Drp1 using mdivi-1, a small molecule inhibitor of Drp1, also delayed mPTP opening and reduced cell death induced by acute IRI in HL-1 cardiac cells and adult murine cardiomyocytes (76). Finally, in vivo pre-treatment of the adult murine heart with a single intravenous bolus of the Drp1 inhibitor reduced MI size and limited the extent of mitochondrial fragmentation in the adult heart as imaged by electron microscopy (76). These findings suggest that acute inhibition of mitochondrial fission during an episode of lethal myocardial ischemia-reperfusion injury may be a novel therapeutic strategy for cardioprotection.
The mechanism through which acutely inhibiting mitochondrial fission or promoting mitochondrial fusion delays mPTP opening and protects the cardiomyocyte against IRI is unclear. Whether the elongated mitochondria are more tolerant of myocardial ischemia and are able to preserve ATP levels and accommodate greater levels of oxidative stress and calcium before provoking mPTP opening is one possible explanation that needs to be investigated. Whether inhibiting calcineurin activation during myocardial ischemia protects the heart against IRI by preventing the activation and translocation of Drp1 to mitochondria remains to be directly demonstrated. It is interesting to note that experimental studies have previously observed cardioprotection with pharmacological inhibition of calcineurin, although in that case the cardioprotection was attributed to the preserved phosphorylation of pro-survival kinases (11).
Although Chen et al. (17) had found that ablating OPA1 exacerbated mitochondrial fragmentation during simulated ischemia in H9C2 myoblasts, the overexpression of OPA1 in these cells did not appear to reduce cytochrome C release or decrease apoptotic cell death induced by simulated IRI, although it did reduce the extent of mitochondrial fragmentation. Wang et al. (105) found that mice overexpressing miRNA-4199 were protected against IRI and underwent less adverse remodeling post-MI and this protective effect was associated with less mitochondrial fragmentation during IRI. Importantly, in that study it was found that ablating Drp1 in the adult rat heart using siRNA reduced MI size following IRI (105), confirming the role of Drp1 as a target for cardioprotection in the adult heart. Interestingly, it has been reported that the pharmacological inhibition of Drp1-mediated fission can also protect the kidney against acute IRI, suggesting that this mechanism of cell death is present in other tissues (10).
Interestingly, Papanicolaou et al. (79) investigated the effect of Mfn2 deficiency in the adult murine heart, and the results were quite interesting. Genetic ablation of Mfn2 in the adult murine heart had the following effects: mildly enlarged subsarcolemmal mitochondria, decreased levels of Drp1, mitochondrial membrane depolarization in the absence of any change in mitochondrial respiration, delayed mPTP opening (to either calcium or oxidative stress), less susceptibility to acute IRI, and reduced MI size post-IRI. These findings suggest that in the adult murine heart, Mfn2 appears to play a detrimental role and promotes mPTP opening (79). In contrast, siRNA ablation of Mfn2 in neonatal murine cardiomyocytes induced mitochondrial fragmentation, increased the susceptibility to mPTP opening, and augmented cell death post-IRI (79). Although Mfn2 appears to protect against mPTP opening in neurons (73), HL-1 cells (76), and neonatal cardiomyocytes (79), it appears that in the adult murine heart it promotes mPTP opening (79). The reason for Mfn2 promoting mPTP opening in the adult murine heart is difficult to comprehend. These conflicting findings suggest that the role of Mfn2 in the heart may be cell specific, and may alter with mitochondrial cardiomyocyte maturation. The same authors have demonstrated that in adult mice lacking Mfn1 the hearts are structurally and functionally normal, have smaller cardiac mitochondria, and are protected from reactive oxygen species (ROS)-induced death (80). The effect of simultaneous ablation of both Mfn1 and Mfn2 in the heart on the susceptibility to acute IRI remains undetermined.
In summary, it is important to appreciate that modulating mitochondrial dynamics may have cell-specific effects and the effects may vary with development such as with Mfn2. However, as a target for cardioprotection, both pharmacological and genetic inhibition of Drp1, in both the HL-1 cardiac cell line or the adult rat heart, have been reported to protect the heart against acute IRI. Whether the cardioprotection can be elicited by inhibiting the other mitochondrial fission proteins remains to be determined.
Mitochondrial morphology and cardiac hypertrophy
The mitochondrial fusion protein, OPA1, has been recently investigated in the adult heart in the context of chronic pressure-overload. Piquereau et al. (84) demonstrated that mice partially (50%) deficient for OPA1 (OPA1+/−) were more susceptible to developing cardiac hypertrophy (2-fold increase) and impaired LV ejection fraction, 6 weeks following transversal aortic constriction (TAC), when compared to wild-type mice. The mechanism underlying this effect was unclear, although it was noted in this study that mitochondria were larger with the presence of clusters of fused mitochondria and altered cristae (84). Interestingly, it was also noted that the susceptibility to MPTP opening was attenuated in cardiomyocytes deficient in OPA1 (84), a feature in common with Mfn1 and Mfn2 deficiency (79, 81), confirming that the role of the mitochondrial fusion proteins in the adult heart may differ to cardiac cell lines and neonatal cardiomyocytes (41).
Mitochondrial morphology and heart failure
Ischemic cardiomyopathy
Chen et al. (17) have investigated the changes in mitochondrial morphology in heart failure using a post-MI rat heart failure model and human dilated and ischemic cardiomyopathy samples. These authors found that the expression of OPA1 protein was decreased in ischemic heart failure samples, a finding that was associated with the presence of small fragmented mitochondria when compared to control samples, as visualized by electron microscopy (17). Whether the observed reduction in OPA1 protein expression can explain the changes in mitochondrial morphology in ischemic heart failure remains to be determined. Knowlton's research group has recently demonstrated that mice heterozygous for OPA1 developed an age-related cardiomyopathy (unpublished data).
Inherited cardiomyopathy
Dorn et al. (27) have recently investigated the role of the mitochondrial fusion proteins MARF (mitochondrial assembly regulatory factor) and OPA1 in the Drosophila fly heart tube. The Drosophila fly is known to have only one mitochondrial outer membrane fusion protein and this is called MARF (ortholog of mammalian mitofusin). These researchers demonstrated that silencing cardiac-specific MARF and OPA1 resulted in a change in mitochondrial morphology (increased heterogeneity and 30% reduction in mean mitochondrial size) and a cardiomyopathy (characterized by dilatation of the heart tube and severe contractile impairment) (27). Interestingly, the MARF-deficient cardiomyopathy could be rescued by overexpressing either of the human mitofusins (Mfn1 or Mfn2) or superoxide dismutase 1, implicating impaired mitochondrial fusion and oxidative stress in the pathogenesis of the MARF-deficient cardiomyopathy. Further experimental studies are now required to investigate the physiological role of MARF and OPA1 in the Drosophila fly heart tube (27). In this regard, in the MARF-deficient heart tubes there was no change in the arrangement of endoplasmic reticulum (ER), sarcoplasmic reticulum human embryonic vascular endothelial cells, human embryonic vascular endothelial cells or T-tubules, suggesting that in the Drosophila fly heart tube MARF does not tether mitochondria to the ER, as has been shown for Mfn2 in mammalian cells (22).
Ashrafian et al. (5) have described a novel mutation in the Drp1 gene (C452F) which gives rise to an autosomal dominant form of dilated cardiomyopathy in the python mouse. Although the homozygous mutation is embryonically lethal, the heterozygous form survives until adulthood and develops a severe dilated cardiomyopathy after 11 weeks which was associated with reduced content of mitochondrial respiratory enzymes and cardiac ATP depletion (5).
As mentioned earlier, giant mitochondria have been described in a variety of cardiomyopathy, including mitochondrial cardiomyopathy (58). The mechanism for the formation of these giant mitochondria is unknown but it has been suggested that the enlargement of the mitochondria may be due to compensatory mechanisms for the functional impairment induced by the mitochondrial DNA mutation or the result of end-to-end fusion of adjacent mitochondria (50).
The roles of Mfn1 and Mfn2 have recently been investigated in the adult heart: the absence of both Mfn1 and Mfn2 resulted in embryonic lethality (18). Conditional cardiac ablation of Mfn1 and Mfn2 resulted in mitochondrial fragmentation, defects in mitochondrial respiration, and a severe dilated cardiomyopathy, confirming a critical role for Mfn1 and Mfn2 in cardiac development and in the development of heart failure in the adult heart (18).
Mitochondrial morphology and cardiac development
Changes in mitochondrial function are necessary to able to supply the changing metabolic demands of the developing embryonic heart (reviewed in Ref. 85). Emerging evidence suggests that at each of these critical stages of development, changes in mitochondrial dynamics are needed to facilitate the evolving metabolic demands of the heart.
Embryonic development of the heart
In the early phase of embryogenesis (up until mouse embryonic day E8.5), the embryo relies upon diffusion for the delivery of oxygen and nutrients and the removal of waste products. During this initial period, and because of the low environmental oxygen tension (<10 mmHg O2), it is believed that for the most part, the heart relies upon anerobic glycolysis to provide its energy requirements (85). As the placenta develops and the circulation matures from E10 onwards, the increased environmental oxygen tension (10–30 mmHg O2) permits oxidative phosphorylation, thereby providing the energy required by the heart to support the embryonic circulation by day E13.5 (85). From E13.5 to birth, both proliferative and hypertrophic growth of the heart occur in order to sustain the energy requirements of the growing fetus.
It has recently been demonstrated that during this phase of embryonic development, changes in mitochondrial dynamics occur in order to sustain the required changes in heart metabolism. At this early stage of embryonic heart development (day E9.5), it has been demonstrated that the mitochondria: are predominantly fragmented (rounded and dilated), are localized to the perinuclear region with little association with the immature contractile apparatus, and have smooth IMM that lack cristae (49, 85). The immature appearance of the mitochondria at these low oxygen tensions would be consistent with the dependency on anerobic glycolysis for energy production. However, there is a significant change in mitochondrial morphology and function by day E13.5, at which time the mitochondria have become more elongated and branched to form an interconnected network aligned adjacent to the developing contractile machinery. The mitochondria appear more mature with more developed cristae and have an increased membrane potential and produce less ROS compared to E9.5 mitochondria. These findings are consistent with some degree of oxidative phosphorylation taking place as oxygen levels increase following development of the placental circulation. However, at this stage the predominant substrate is lactate and to a lesser extent glucose. Compared to adult cardiomyocytes, the mitochondria are smaller and are not as regularly aligned alongside the myofibrils. At this stage, the heart is still reliant mainly upon anerobic glycolysis for energy, and although the mitochondria appear more organized, they are not fully aligned with the sarcoplasmic reticulum and other contractile machinery (85).
The regulatory mechanisms that control this change in mitochondrial metabolism in the embryonic heart at this time are unknown, although a recent experimental study has suggested that mitochondrial cyclophilin D (CypD), a regulatory component of the mPTP, may contribute to this adaptive process (49). The mPTP is a nonselective channel of the IMM that has been demonstrated to be a critical determinant of cell death, as its opening uncouples oxidative phosphorylation and causes mitochondrial swelling resulting in cell death. Hom et al. (49) postulate that the mPTP is open in isolated embryonic cardiomyocytes at E9.5 and that it is closed at E13.5. The physiological role for mPTP opening and closing during early embryonic cardiac development is not clear from this study. Prolonged opening of the mPTP at E9.5 would be expected to induce mitochondrial swelling and cell death. Furthermore, if mPTP opening and closing were such critical events for cardiac development, then one might expect mice lacking CypD to have an abnormal cardiac phenotype, which has been shown to not be the case (7, 72).
Postnatal development of the heart
At birth there is an abrupt increase in oxygen tension and over the first 7 days of post-birth there is a metabolic switch from anerobic glycolysis and lactate metabolism to fatty acid oxidation which is more or less complete after the first week post-birth. Under physiological conditions, the adult heart predominantly utilizes fatty acids as its favored substrate (>90%) although it can metabolize other substrates. During the first week of life, the mitochondria continue to increase in both size and number, as well as continue to fuse and form the organized mitochondrial network, which, in cardiomyocytes, is characterized by strict alignment with the contractile machinery (85). The regulatory mechanisms controlling this change in mitochondrial morphology and function in the first week of post-birth life are unclear.
Mitochondrial dynamics and cardiac differentiation
Interestingly, it appears that similar changes in mitochondrial dynamics and function are necessary to supply the metabolic demands of the stem cell as it undergoes cardiac differentiation (reviewed in Ref. 87). It has been demonstrated that for the embryonic stem cell (ESC) to differentiate into a cardiomyocyte, there needs to be a metabolic switch from anerobic glycolysis (which is sufficient to supply the energy requirements of the ESC) to mitochondrial oxidative phosphorylation (to provide for the increased energy demands of the differentiated beating cardiomyocyte) (19). Crucially, this change in mitochondrial metabolism has been reported to require an alteration in mitochondrial morphology from fragmented rounded mitochondria (lacking cristae) present in the ESC, to an elongated interconnected mitochondria (with well-developed cristae) closely aligned with the myofibrils of the differentiated contractile cardiomyocyte (19). This change in mitochondrial metabolism was found to be associated with alterations in the expression of the mitochondrial fission (Drp1) and fusion (Mfn2) proteins, as well as expected changes in the metabolic transcriptome (19).
A recent experimental study confirmed this requirement for a change in mitochondria morphology in the cardiac differentiation of the ESC, and also demonstrated that the mitochondrial fusion proteins, OPA1 and Mfn2, are critical to this process (60). Kasahara and Scorrano (60) demonstrated that the presence of OPA1 and Mfn2 are required by the ESC as it undergoes cardiac differentiation to promote mitochondrial elongation, which in turns limits capacitative calcium entry, reduces calcineurin activity, and suppresses Notch signaling.
These findings suggest that modulating mitochondrial dynamics may provide a novel therapeutic strategy for facilitating the cardiac differentiation of stem cells.
Mitochondrial Dynamics As a Therapeutic Target for Cardiovascular Disease
The investigation of mitochondrial dynamics in cardiovascular health and disease has identified novel targets for therapeutic intervention. Mfn2, which has been reported to exert antiproliferative effects on the vascular smooth muscle cell, may provide a future pharmacological target for the treatment of atherosclerotic disease and/or coronary restenosis. The findings from the initial experimental studies investigating mitochondrial dynamics in the setting of myocardial ischemia-reperfusion injury, suggest that inhibiting the Drp1-mediated mitochondrial fission induced by myocardial ischemia may be a therapeutic strategy for protecting the heart against acute IRI. In theory, this could be achieved by the transient pharmacological inhibition of Drp1 or other components of the mitochondrial fission machinery such as hFis1, MFF, or MiD49/51. Given the essential role that both mitochondrial fission and fusion play in the adult heart in terms of mitochondrial quality control and turnover, one might expect that the chronic suppression of mitochondrial fission or the chronic activation of mitochondrial fusion may have detrimental effects on the heart. The experimental studies suggest that changes in mitochondrial morphology from mitochondrial fragmentation in undifferentiated stem cells to a more elongated and interconnected mitochondrial network are required to support the metabolic demands of cardiac differentiation. Therefore, it may be possible to pharmacologically modulate mitochondrial dynamics by promoting mitochondrial fusion in order to facilitate the stable differentiation of stem cells.
Conclusions
Emerging data suggest that changes in mitochondrial dynamics may be relevant to cardiovascular health and disease. Changes in mitochondrial dynamics have been implicated in vascular smooth cell proliferation, cardiac development and differentiation, cardiomyocyte hypertrophy, myocardial ischemia-reperfusion injury, cardioprotection, and heart failure. Initial experimental studies have confirmed that mitochondrial dynamics are relevant to the adult heart despite the distinctive arrangement of mitochondria in this organ. Investigating the role of mitochondrial dynamics in cardiovascular system should result in the identification of novel therapeutic targets for treating patients with cardiovascular disease, the leading cause of death and disability globally.
Footnotes
Acknowledgments
This work was supported by the British Heart Foundation Grant Number FS/06/023. This work was undertaken at UCLH/UCL, which received a proportion of funding from the Department of Health's NIHR Biomedical Research Centres funding scheme.
Author Disclosure Statement
No competing financial interests exist.