
Abstract
Introduction
M
More than 90% of oxygen intake in aerobic organisms serves as an electron acceptor for hydrogen liberated from nutrients, a process that relies on a proper function of mitochondria.
The flux of oxygen to even the remotest cells and their mitochondria is crucial, and, therefore, regulated by a multitude of adaptive mechanisms in order to meet changes in oxygen supply, as observed in an acute phase after vessel occlusion (ischemia) (16) or under chronic conditions such as high altitude or in fast growing tumor tissue (158). A clear distinction has to be made with regard to the cellular counter-regulatory measures in response to acute, respectively chronic hypoxic conditions. After an ischemic insult, the tissue has to react in a time scale of seconds and minutes dependent on the energy demand of the organ, whereas the adaptation to prolonged periods of limited oxygen levels may take hours and days and typically includes newly synthesized enzymes such as hypoxia-inducible factor (HIF)-1 (109, 134), as reviewed in the subsequent article by Dehne and Brune. In contrast, a sudden block in oxygen supply requires a fast reacting sensor that can rapidly trigger counter-regulatory measures.
This review explicitly focuses on the acute cellular events in response to hypoxic conditions. Particular emphasis will be spent on the role of cellular redox signaling with reactive oxygen species (ROS) and reactive nitrogen species (RNS) as the key players (40, 144, 153). Based on extensive reviewing of literature data, we take the freedom to suggest a new hypothesis of mitochondria as the main sensors for ischemic conditions, including a free radical-based temporal, but reversible, “silencing” of mitochondria, and a reactivation in response to re-supply with oxygen.
We herein introduce the concept of mitochondria as the main sensor in ischemic hypoxia and then concentrate on the cytosolic response, leading to later events in the counter-regulation and compensation for decreases in oxygen tension. Many data and facts can be found as isolated information in the literature but are now placed within the framework of a unifying hypothesis with all the shortcomings of such an attempt. It may, however, encourage researchers either to prove or falsify such ideas.
An event outside the scope of this review is the phase of reperfusion injury. Due to space limitation, we only briefly point to the connections and otherwise refer to the vast literature on this topic (74, 143, 157). The same applies to the phenomenon of “preconditioning,” which is a direct consequence of the sensing and counter-regulation in hypoxia but with many clinical aspects that could not be incorporated here (21, 32, 67).
Sensing Hypoxia by Mitochondria
In spite of the great importance of ischemic conditions in medicine after stroke, infarction, or organ transplantation, the initial steps in sensing a decreased tissue supply of oxygen occurring within seconds and minutes are not firmly established (133). In part, this may be a consequence of the postulated existence of several sensors that have been described in different organs or species (17, 67, 114), but is mainly due to the involvement of short-lived ROS (42, 121). The lack of molecular O2 on the one hand and the formation of superoxide (
One long-recognized regulatory principle consists of sensing the decline of adenosine triphosphate (ATP)-levels and the formation of ADP/AMP/Pi (expressed as the energy charge) in the absence of mitochondrial consumption of oxygen (123). Indeed, the activation of AMP-activated protein kinase (PK) (142), the involvement of the ATP-dependent mitochondrial K+-channel (KATP-channel) (91), restrictions in ATP-dependent Ca2+-sequestration (110), or in Na+/K+ pump activity (48) by a lack of ATP can play an important role in the cellular answer to hypoxia. However, the time course involved in the dropping of ATP levels is far too slow to serve as a signal for a rapid response, as creatine-phosphate, adenylate kinase, and glycolysis provide an efficient buffer that, for example, in the ischemic heart, prevents a steep decline of ATP levels, typically leading to a linear decline over a period of up to 50 min (85). In the context of this review, all cellular effects directly linked to this decreasing energy charge are considered “secondary effects” in hypoxia that not only include mitochondria, but also events in the cytosol, as discussed in The Cytosolic Response in Counter-Regulating Hypoxia section.
Nitrite as a sensor for anoxia
One of the earliest physiological responses as consequence to decreasing blood oxygen supply occurring in the time range of seconds seems to be a vessel relaxation, possibly by an increased availability of nitric oxide (•NO) (83, 98). According to common knowledge, the obvious source for this vasorelaxant would be endothelial NO-synthase (NOS-3) (112). However, its activation would require an earlier rise in Ca2+ and would not solve the controversy of having less •NO release under conditions of lowered oxygen tension.
Another source of •NO would be the one-electron reduction of nitrite, which can occur via the interaction with partially deoxygenated hemoglobin under formation of a nitrosated cysteine in the beta chain, and the subsequent release of vasodilatory •NO (47, 58, 96). Body fluids contain nitrite in micromolar concentrations (78), but in the aorta, about 20 μM and in endothelial cells even up to 80 μM are constantly present (15). It took a while in the scientific community to recognize that such high levels could serve as a source for •NO, especially under the strongly reducing conditions found in hypoxic or even anoxic mitochondria (20, 72, 81, 88). In analogy to the reduction of nitrite to •NO by deoxyhemoglobin, deoxymyoglobin also can reduce nitrite which, especially in cardiomyocytes, is considered a process of oxygen sensing, as the newly formed •NO can negatively regulate cytochrome oxidase (4, 136, 150). Earlier reports suggested ubisemiquinone of the respiratory chain as an electron source (113), but also ferrous cytochrome c oxidase seems to be involved (73, 137) (Fig. 1).

When following the fate of reductively formed •NO in mitochondria, it is important to consider that not only ischemia, but also a minor decline in oxygen supply will lead to anaerobic segments in areas of the tissue that are remote from blood capillaries. This would result as a consequence of the high affinity of cytochrome c oxidase for O2 (14, 73). The binding of O2 to the ferrous heme-Cu1+ state of Complex IV of the respiratory chain is essentially irreversible, and, therefore, a steep gradient will form between the blood capillaries and the remotest part of the tissue, which may be up to 30 cell layers away. If •NO is generated in such anoxic areas, it can easily diffuse in the tissue (8), and due to the low oxygen concentrations under these conditions, it will have a long lifetime (oxidation of 2 •NO by O2 is a third-order reaction), allowing an exposition of adjacent cells and mitochondria in hypoxic penumbral areas by freely diffusing •NO. This cooperative behavior of anoxic cells to their hypoxic neighbors will allow •NO to bind to the reduced form of cytochrome c oxidase, and then, the affinity for oxygen decreases in a competitive way, thus sparing residual oxygen for signaling purposes (10, 11, 12, 106, 148). As a result of reversible •NO binding to Complex IV, the electron transferring components of the respiratory chain achieve a higher state of reduction even if nanomolar concentrations of oxygen are present (1, 126). It is one of the attractive features of this mechanism that with less oxygen being present, more nitrite is reduced to •NO, which will cause more Complex IV inhibition, followed by more
It is typically assumed that
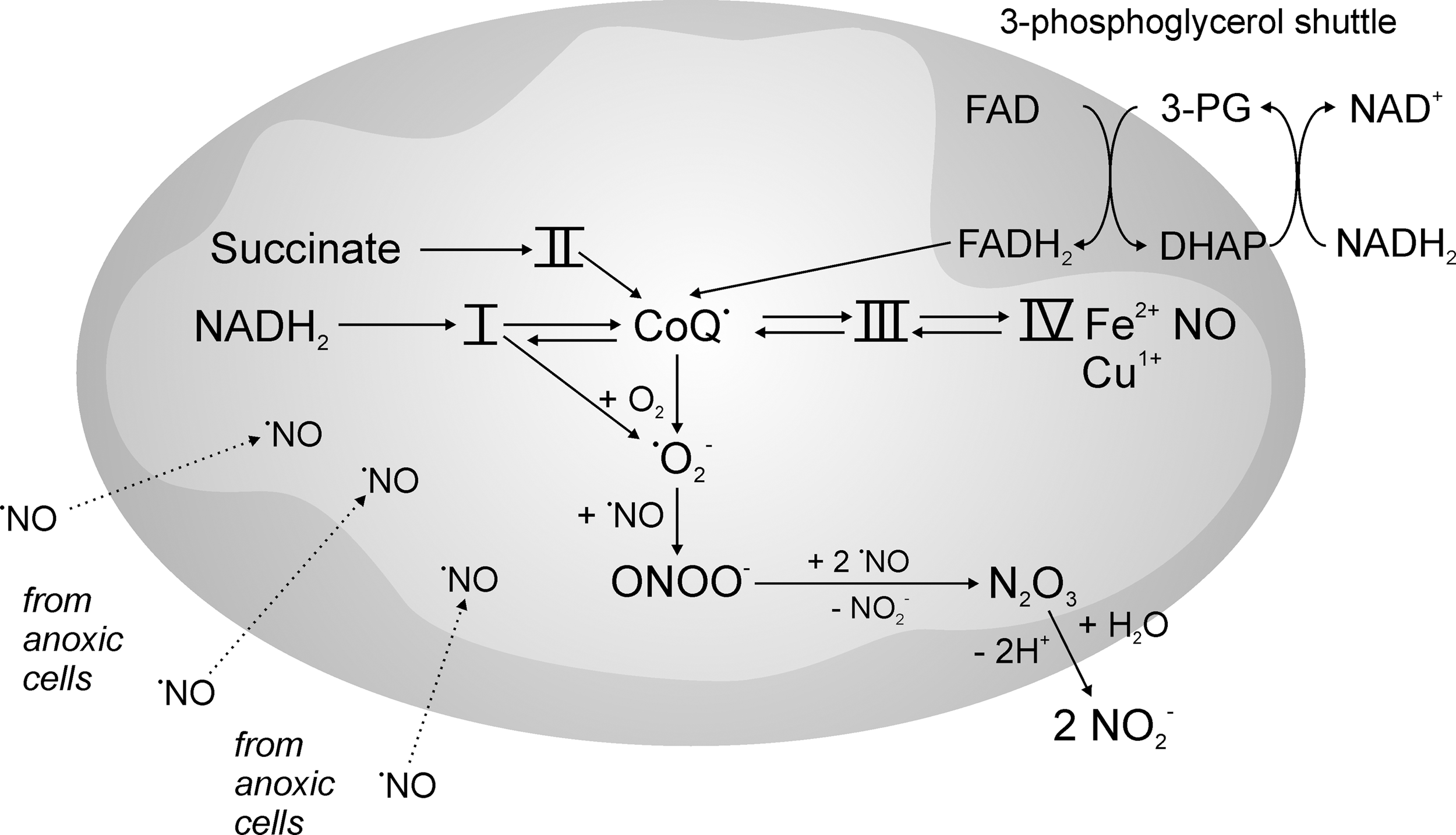
It is beyond knowledge which levels of peroxynitrite (ONOO−) can be reached under these conditions. One may argue that within the mitochondrial matrix, manganese SOD (SOD-2) might compete with •NO for
The answer to the question of how mitochondria sense acute hypoxic conditions would, therefore, lie in the formation of peroxynitrite from mitochondrial nitrite-reduction to •NO. This would subsequently cause reversible inhibition of cytochrome c oxidase, which finally leads to an increased availability of residual O2 for the 1e−-reduction to
This leads to the further question whether the formation of peroxynitrite could then be a possible mechanism for signal transduction to initiate the subsequent counter-regulatory mechanisms that fight hypoxia. Peroxynitrite is permeable to membranes (30) and can even leave mitochondria through anion channels to the cytosol (119), but it has to be considered that inside mitochondria, reducing conditions, maintained for example by glutathione (GSH) peroxidase, thioredoxins, peroxiredoxins (75, 90), or even by the reduced forms of respiratory chain components such as ubiquinone (UQ) (131), prevail. The outcome would be a futile circle in which ONOO− would be scavenged by reducing equivalents.
A hypothetical role for S-nitrosation in mitochondrial signaling
In biological systems, ONOO− usually reacts by two-electron oxidations or oxygen atom transfer. This changes when protonation forms peroxynitrous acid (ONOOH) with a pK of 6.6. In this acid, the O-O-bond is further weakened with the propensity for one-electron oxidations, releasing −OH as the driving force. Radical chain reactions are the consequence and are the basis for the toxicity of peroxynitrous acid (116):
There are several reports in the literature (28, 64, 129) indicating that characteristic reactions of peroxynitrite are abolished in the presence of •NO. Under those conditions, one can observe N-nitrosation of 2,3-diamino-naphthalene (36) and C-nitrosation of phenol (29). However, at the same time, a complete lack of zinc dithiolate center oxidation in aldehyde dehydrogenase (ALDH), which is characteristic for peroxynitrite, was detected (28, 139). This indicated that under excess of •NO, peroxynitrite was converted to a nitrosating species. When the same conditions were applied to NADP+-dependent isocitrate dehydrogenase-2 (ICDH-2), a maximum of S-nitrosation was obtained when the fluxes of •NO- and
Dinitrogen trioxide (N2O3) is a potent nitrosating agent. According to these reactions, any generation of
Mitochondria are known to be the preferred organelle for containing nitrosated thiols (29, 36), and
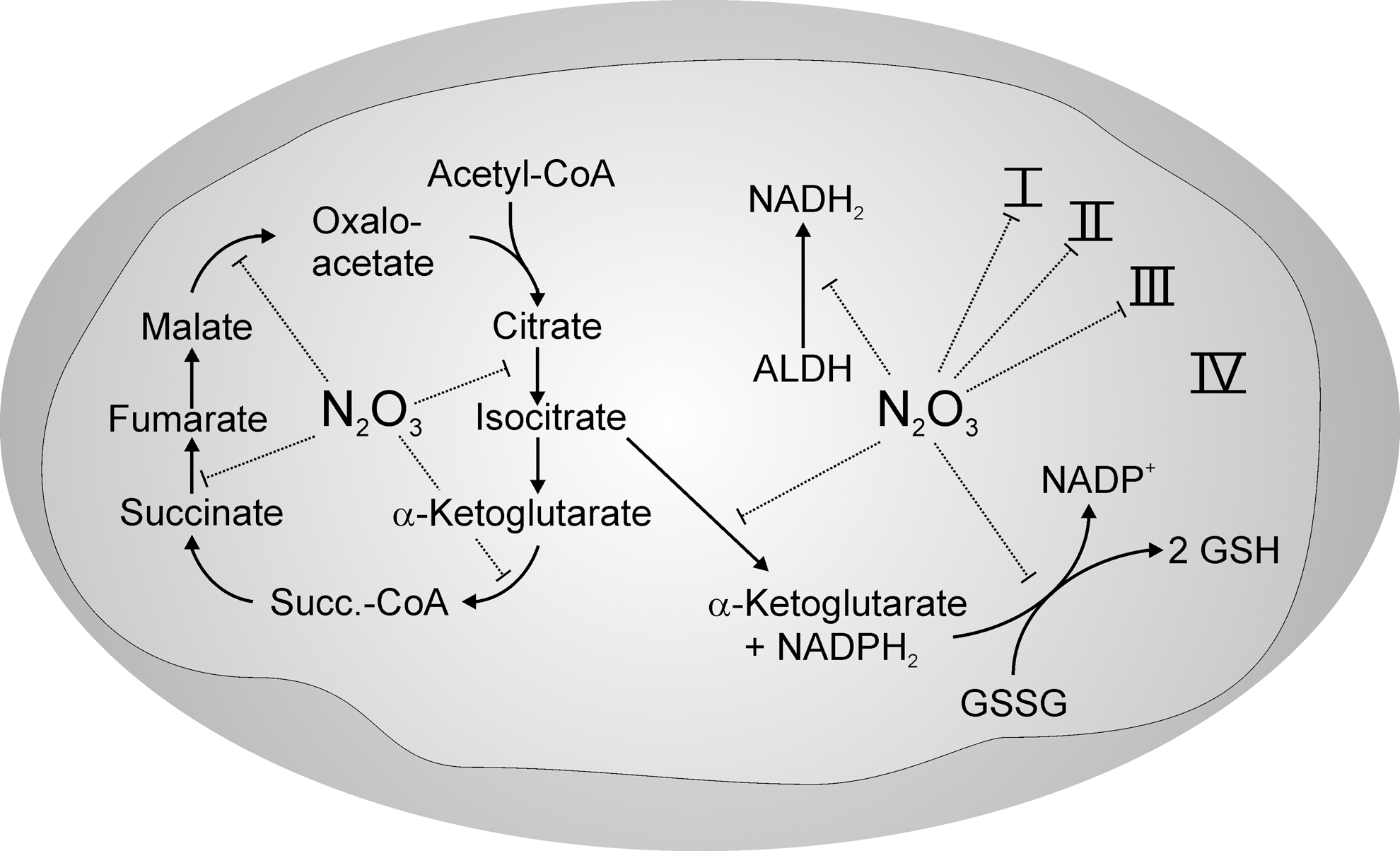

With regard to the function of S-nitrosation, all enzyme activities listed in Figure 4, except those of thioredoxin (Tx), were inhibited by S-nitrosation. As a result, the Krebs cycle would be blocked at several points, restricting the generation of β-nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2). Even ALDH-2 would not be able to provide NADH, and this also applies for the beta-oxidation of fatty acids (92). As a final consequence, no electrons are left for feeding into the respiratory chain. By the inhibition of ICDH-2 (29, 76, 155), β-nicotinamide adenine dinucleotide phosphate (NADPH) synthesis in mitochondria also ceases and blocks reduction of the oxidized forms of GSH, Tx, and peroxiredoxins, which themselves are S-nitrosated (7, 132) (Fig. 3). Although this situation seems to exactly match the definition of oxidative stress, it has to be reminded that S-nitrosation is easily reversible and, hence, can be considered a redox-regulated mechanism that may serve to keep mitochondria in a protected (“silenced”) oxidized state before reoxygenation hits the respiratory chain, as argued by Chouchani et al. (24). Indeed, the treatment of mice with a mitochondria-directed nitrosation agent in a model of ischemia/reperfusion protected them from ischemic insults (76, 115). Hence, S-nitrosation would prevent a situation in which the respiratory chain exists in a reduced state, vulnerable to autoxidation of the UQ-pool, the presence of iron ions released from aconitase, or the decomposition of other iron enzymes (18, 122). Not only would essential thiolate groups be protected from radical-based unspecific oxidations, but also the generation of ROS and RNS and, hence, oxidative modifications of mtDNA would be prevented by the lack of mitochondrial reducing equivalents (94). Since peroxynitrite preferentially reacts with not only components of the respiratory chain in the reduced state such as ubisemiquinone, but also with ubiquinol (75, 79), conditions in which components of the respiratory chain are reversibly maintained in an oxidized state could be regarded as an endogenous redox regulatory process in order to transiently protect these complexes from an irreversible modification as, for example, evoked by the action of peroxynitrite.
In summary,
Signaling to the intermembrane space
For the concept of redox regulation of mitochondrial function, it is of great importance whether S-nitrosation is restricted to the mitochondrial matrix space or whether it would extend to the cytosol and from there to the nucleus in order to regulate gene expression of enzymes involved in progressive stages of the cellular response to hypoxia. Since N2O3 is a diffusible gas, it can easily cross mitochondrial membranes, and, hence, it can reach the cytosol, but in contrast to •NO, it is effectively hydrolyzed and inactivated to nitrite (8). The cytosol contains glyceraldehyde-3-phosphate dehydrogenase (GAPDH), that, with its very reactive thiolate group, is a substrate for S-nitrosation and inhibition (52). Since glycolysis is fully active under anoxic conditions, GAPDH S-nitrosation can be excluded, which allows the conclusion that S-nitrosation is rather unlikely to have significant effects in the cytosol within the range of the cellular response to hypoxia, although it may extend to the mitochondrial intermembrane space (IMS).
When peroxynitrite diffuses out of the matrix space, it will meet one of its most sensitive targets, creatine kinase (CK) (70). This octameric enzyme bridges the IMS by anchoring to the adenine nucleotide transporter (ANT) in the inner, and to the voltage-dependent anion channel in the outer membrane (49). Peroxynitrite is able to oxidize reactive dithiol motifs in CK under the formation of a disulfide, with changes in quaternary structure and the inhibition of activity (5). In a possible connection with peroxynitrite action on CK may be the fate of mitofilin as an abundant coiled protein that also spans both mitochondrial membranes with an impact on the structure of cristae (63, 147). Mitofilin has thiolate groups that can be S-nitrosated, as shown in Figure 5, but according to earlier results in our laboratory (9), it binds to a phenylarsine affinity column which is selective for vicinal dithiols. Another enzyme present in the IMS is Cu,Zn-SOD (SOD-1), which is inactive in its dithiol state (61). It can be reactivated by H2O2 and inactivated in the presence of Tx/thioredoxin reductase (TxR) (62). Such data point to an action of a potent oxidant in the IMS for which peroxynitrite is a likely candidate (Fig. 5).
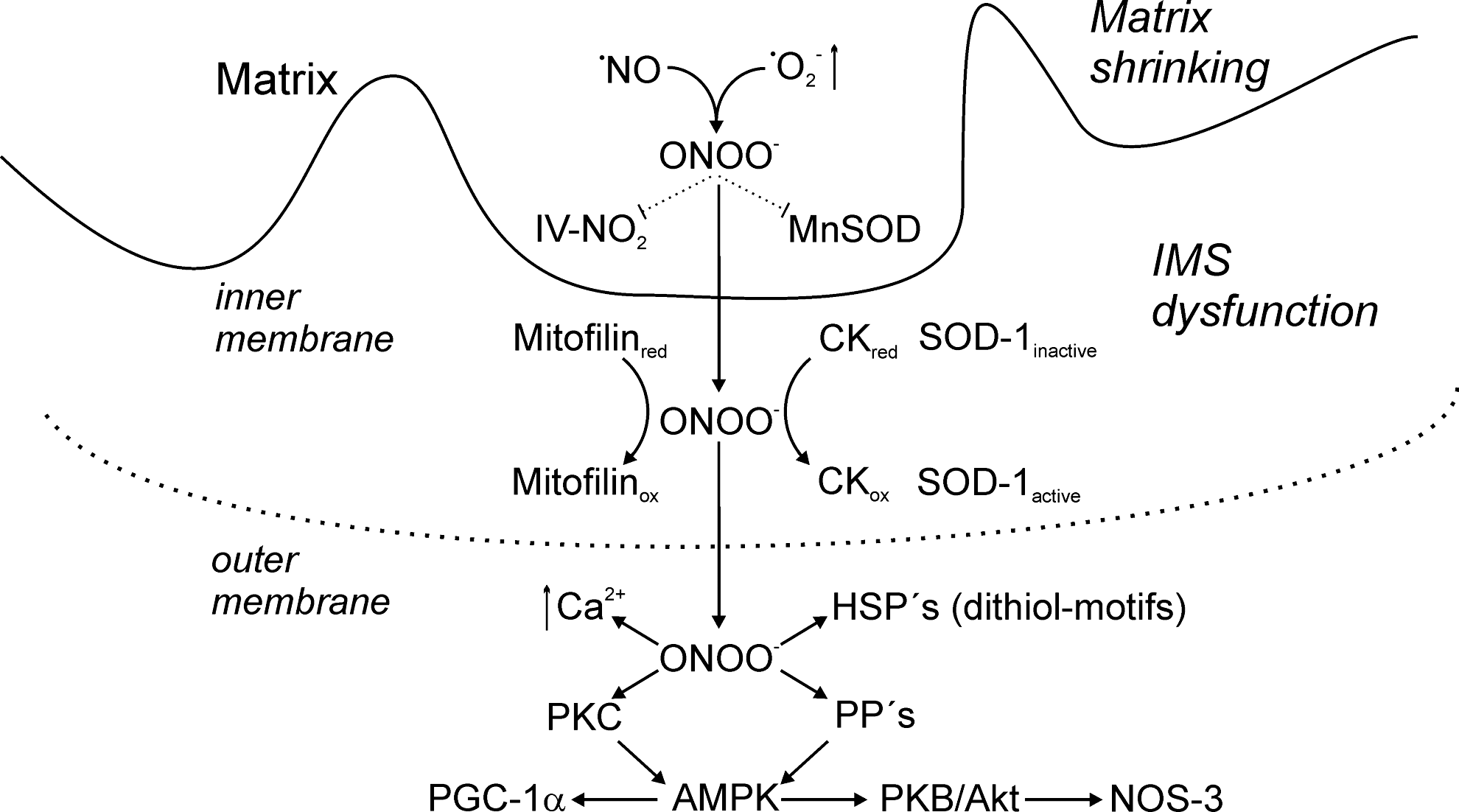
According to a seminating hypothesis by the group of Garlid (33), the oxidation of CK appears to affect the whole structure of the megapore between the two mitochondrial membranes and, thus, destroys the communication network between cytosol and mitochondria. It could be speculated that the biological function of this inhibition would lead to an expansion of the IMS and a concomitant decrease of mitochondrial matrix volume. Eventually, the action of peroxynitrite on the dithiol state of CK and other intermembrane proteins would expand the IMS and shrink the matrix (66).
The Cytosolic Response in Counter-Regulating Hypoxia
In the absence of functional mitochondria, the glycolytic pathway should take over the energy supply of an almost anoxic cell. For tissues with a high energy demand as the brain, this can represent an insurmountable challenge and will not be successful in the long run, as glycolysis would have to be up-regulated multiplicative to replace only part of the aerobic oxidative phosphorylation (2). As soon as ADP and inorganic phosphate accumulate, the glycolytic rate, dependent on the cell type, increases, but this up-regulation is limited by the inhibitory action of NADH on GAPDH and of acidosis originating from inevitably rising lactate levels.
After nitrite has served as an alternative •NO source, the resulting peroxynitrite would emerge as a mediator released by mitochondria in order to transmit the signal of hypoxia to the cytosol. As one of the earliest responses in the cytosol, an up-regulation of intracellular Ca2+ levels as a permissive signal for many situations of stress can be observed due to redox control of Ca2+ pumps and release mechanisms from intracellular stores (35, 105, 146). Moderate Ca2+ levels as well as calmodulin will enforce not only glycolysis and glycogen utilization but also other stress-activated pathways.
With the help of adenylate kinase, ADP can be converted to AMP and ATP, and then, AMP can act as a powerful stimulus for AMP-activated kinase (53). This enzyme is a master switch for the glucose-dependent energy supply and initiates a plethora of events (19, 99). These include the direct and the transcriptional activation of glucose transporters, the stimulation of 6-phosphofructo-2-kinase for the formation of fructose-2,6-bisphosphate as the rate-limiting metabolite of glycolysis (27, 86), and a down-regulation of the ATP-consuming pathways.
In addition, protein phosphorylations, as the most abundant posttranslational modification of enzymes, are often redox regulated with the general rule that PKs are usually activated and phosphatases are often inhibited through thiol group oxidation (31, 43, 44, 60, 68, 103). In case of PKC epsilon and PKC zeta, the thiolate groups are complexed by zinc, which compensates the electrostatic repulsion with the negatively charged ONOO−. Since kinases are often activated by other upstream kinases, the cascades can become complex. This is even enforced by synergistic effects of oxidation and phosphorylation, which are often observed (71). Likewise, IP3 receptor triggers release of Ca2+ not only in response to oxidation but also upon phosphorylation by mediated PKC. Since calcineurin is effectively dephosphorylating the receptor, this Ca2+-dependent enzyme is an important determinant, especially as it is inhibited by superoxide (93, 97).
The increase in the AMP/ATP ratio, however, does not occur immediately due to the buffering action of creatine phosphate and glycolysis, suggesting that more rapid regulations of this key enzyme in energy control should exist (39). Indeed, the formation of peroxynitrite under hypoxia seems to be such a pathway that involves activation of protein kinase C zeta (PKC zeta) which subsequently regulates LKB-1-dependent AMP activated PK (154). Not much is known about the inhibition of phosphatases for Thr 172 dephosphorylation, but PP2A and PP2C are likely candidates (23, 152).
Readmission of Oxygen
A biochemical approach to the topic of hypoxia would not be complete without discussing the situation arising when oxygen is readmitted to the previously hypoxic and partially anoxic tissue. This situation reflects the clinical setting of reflow or reoxygenation; however, both expressions are usually connected with pathophysiological injury, happening after sudden increases in oxygen pressure as derived from surgery or clot lysis. Such injuries result as a consequence of an extended and severe oxidative damage and/or inadequate counter-regulation, and ultimately to a switch to cell death with an involvement of ROS and RNS production by leukocytes, NOS-2, and xanthine oxidase (89). These late events also include cellular Ca2+ overload (57), opening of the permeability transition pore (51, 77), and breakdown of adenosine to hypoxanthine/xanthine, which serve as a substrate for xanthine oxidase (145).
In the context of this review, we concentrate on the less spectacular, but more relevant aspect for normal physiological situations, when the re-admission of oxygen leads to a reversal of transiently inactivated mitochondria in order to reactivate normal ATP formation from oxidative phosphorylation. The postulated silenced and oxidized state of mitochondrial respiratory complexes can be considered an endogenous means to protect proteins, and especially mtDNA from oxidative damage, but the necessary switch back to aerobic metabolism requires a reversal of the inactivation process, that is, S-denitrosation. This evokes a demand for reductive mechanisms, but, at the same time, raises the question on where, in the oxidized state, these reducing equivalents might come from and how the increased oxygen tension can be sensored to initiate the process of mitochondrial reactivation.
In contrast to the oxidized and inactive state of mitochondria, the cytosol is highly loaded with glycolytically generated NADH, and also the pentose phosphate cycle is activated in order to provide NADPH (151). Can these reducing equivalents be used to reverse the peroxynitrite mediated oxidations in the IMS and the S-nitrosation of enzymes in the matrix space? So far, there is no explicit answer in the literature, but sufficient data are available that allow the following hypothetical approach.
Opening of the KATP-channel
With regard to the question of mitochondrial reactivation by transferring reducing equivalents to the matrix space, the opening of the mitochondrial KATP-channel in the inner membrane seems to be of utmost importance (33, 34). This channel is kept closed under normoxia and adequate ATP levels, and opens under hypoxia and a deficit in ATP (104, 107). By opening with pharmacological tools such as diazoxide (37), a small influx of K+ can be observed, accompanied by a minor decrease in the inner transmembrane potential. As a consequence, an osmotic swelling and also a stabilization of cristae as well as a reorganization of mitochondrial structure was observed (102). The KATP-channel was observed to be activated by nitroxyl, S-nitrosothiols, and H2O2 and inhibited by NADPH and lipoic acid (26, 127). PKC seems to mediate most of the stimulating effects (128, 149).
Most striking for the concept of redox regulation in hypoxia is the burst of superoxide that is directed to both sides of the inner mitochondrial membrane (11, 140). It originates from complex I or from autoxidation of the ubisemiquinone pool (100), but the question on the source of reducing equivalents if S-nitrosation is still blocking their main pathways of generation in the matrix inevitably arises. In contrast to mitochondria, the cytosol by its up-regulated glycolysis generates an excess of NADH, which is unable to enter the mitochondria. However, in order to shuttle electrons from NADH into mitochondria, NADH can reduce dihydroxyacetone phosphate to glycerol-3-phosphate by cytosolic glycerol-3-phosphate-dehydrogenase and, subsequently, glycerol-3-phosphate gets reoxidized (25). This enzyme is located on the IMS side of the inner membrane and in its reduced state, it transfers electrons to the coenzyme Q (CoQ)-pool (120). A prerequisite for these events is an opening of the KATP-channel to initiate the superoxide burst (3, 107). Under those conditions, Complexes III and IV of the respiratory chain are still being blocked, as otherwise oxygen reduction to water by cytochrome c oxidase could proceed (Fig. 6). The explanation of
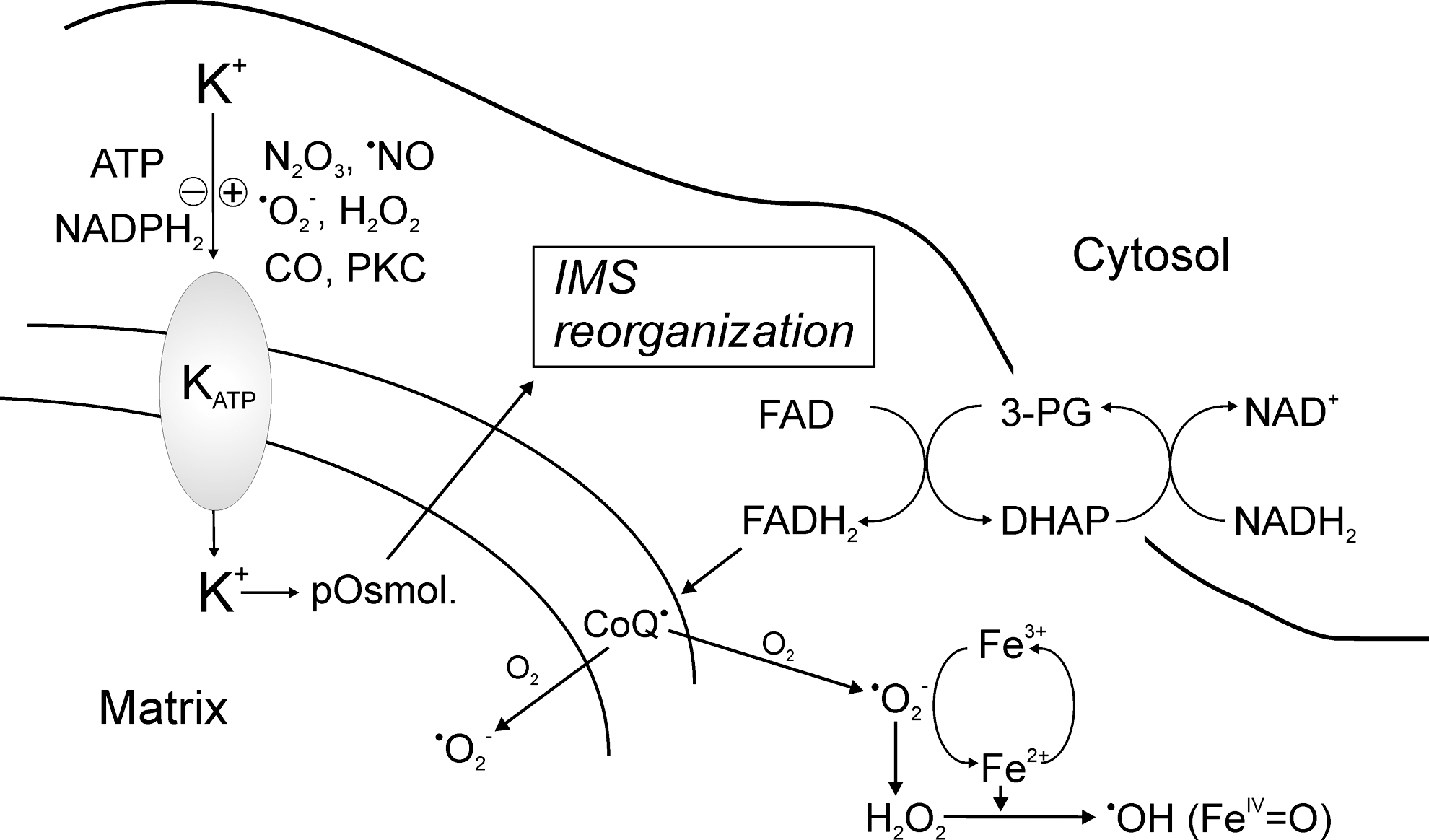
What remains unclear is the mechanism by which the opening of the KATP-channel causes the GP-shuttle to transfer electrons to CoQ. In view of the structural changes by KATP-channel opening occurring in the IMS that affect cristae morphology and evoke shrinking of the IMS, a connection between both events seems possible. Of note, the restoration and reorganization of the IMS would also require the reduction of the peroxynitrite-mediated disulfide formation. This can be achieved by the Tx/TxR system present in the IMS (56) and by NADPH from the pentose phosphate cycle. In this regard, it is remarkable that Tx is the only protein in Figure 4, which by S-nitrosation is activated through the nitrosation of Cys 69 (50, 55). This leads to the question as to by which mechanism the S-nitrosated state of matrix enzymes can be converted back to their active thiol state.
S-denitrosation by superoxide anion
It is evident that the
The chemistry of this reaction has been investigated in detail (65). Interestingly, when nitrosoglutathione (GSNO) was treated with

With regard to a physiological function of this reaction, it has been argued that its rate constant is too slow to compete with the dismutation of
After the entire set of S-nitrosated proteins has been converted back to the thiolate state, the normal function of mitochondria would be re-established and O2 can again serve as an electron acceptor for cytochrome c oxidase. We refer to the abundant literature on the Tx system in restoring the dithiol status of those proteins that had been oxidized by peroxynitrite as postulated (95, 101). If this sequence of events could be confirmed, then the superoxide burst would function as a sensor for the readmission of oxygen and trigger mitochondrial reactivation.
Conclusions and Outlook
The description and hypothetical interpretation of biochemical events associated with sensing and fighting hypoxia just provided are based on a vast, but certainly not complete literature on this topic. Since O2 is the main player in this life-threatening situation, it is evident that not only redox regulation is necessary for our understanding but also the network of oxidation/reduction and the ROS and RNS species involved is complex and its chemistry is not sufficiently known.
We conclude that mitochondria can act as sensors for transiently decreased oxygen concentrations in the tissue and that nitrite serves as an acceptor for accumulating electrons at the UQ pool of the respiratory chain. The resulting freely diffusible •NO radical would subsequently interact with cytochrome c oxidase, and, thus, decrease its affinity to O2 that would allow an increase in the ubisemiquinone pool even in hypoxic cells. It is a critical issue of the unifying hypothesis that the resulting
The next step in the redox regulation of hypoxia starts with peroxynitrite, passing the inner mitochondrial membrane and forming disulfides from Zn-stabilized dithiol groups of CK and mitofilin, both supposed to take part in the connection between the inner and outer membrane and remodeling the architecture of the IMS by conformational changes. This leaves mitochondria in a noncommunicating inactive state, with the cytosol now being responsible for ATP supply. Peroxynitrite, when passing from the IMS to the cytosol, would cause a rise in cytosolic Ca2+ and stimulation of phosphorylation cascades, mainly PKC isozymes, supporting cytosolic activation and glucose supply.
A real challenge of the redox hypothesis becomes apparent when reflow and re-oxygenation would allow oxidative phosphorylation to be re-established. This requires that in a reversed order, first the cytosol-mitochondrial communication through the IMS needs to be reverted and then, reconstitution of the reducing pathways by a process of S-denitrosation takes place. Although no details are known, the opening of the KATP-channel seems to be crucial and could first allow the reduction of CK and mitofilin by Tx/TxR and then the reduction of the UQ-pool by the reorganized glycerol-3-phosphate shuttle as an essential electron pathway linking cytosolic NADH with the UQ-pool. The following burst of

All options are subject to experimental approaches as are also the other discussed assumptions of the redox–regulatory concept of sensing and fighting hypoxia. The scientific community is welcome to prove or disprove the postulated unifying hypothesis.
Footnotes
Acknowledgment
This work was supported by the Deutsche Forschungsgemeinschaft (Sonderforschungsbereich 969).