
Abstract
Introduction
Physiological B-cell signaling via BCR and TNFRSF receptors, both cysteine-rich receptors, generates hydrogen peroxide (H2O2) as a second messenger (11, 50) via nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and involves redox-controlled protein modifications that are crucial for growth and apoptosis regulation. In CLL, signaling through BCR (36) and TNFRSF members, for example, TNFRSF1A (TNFR1, CD120a, and p55), TNFRSF1B (TNFR2, CD120b, and p75) (17, 19, 32), TNFRSF13C (BAFFR), TNFRSF17 (BACI) (33), TNFRSF8 (CD30) (16), and TNFRSF5 (CD40), seems to be operational for prolonged survival.
Innovation
Tumor necrosis factor receptor 1 (TNFR1) and TNFR2 have numerous cysteine residues in the extracellular domain that are targets for redox conformational modulations. Oxidative stress was previously shown to promote ligand-independent and enhanced ligand-dependent TNFR signaling. Other studies have revealed that membrane protein disulfide isomerase (PDI) redox regulates the structure/function of membrane proteins, including b-integrin, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, and metalloproteinase ADAM17/TNF-α-converting enzyme (TACE). In this study, we discovered an unrecognized physical and functional association of TNFRs with PDI complexed to thioredoxin-1. These complexes were abundant in leukemic B-cells of chronic lymphocytic leukemia origin, but very low expressed in normal B-cells. Our study points at possible pharmacological/therapeutical interventions that may block an ongoing leukemic clonal expansion as indicated by the efficient cessation of autocrine growth-promoting TNF and viability exerted by PDI inhibitors.
Redox modulation of proteins is efficiently carried out by the Trx superfamily, including protein disulfide isomerase (PDI) and Trx. Trx1 is a small ubiquitous 12-kDa protein with a -Cys-Gly-Pro-Cys- (CXXC) active-site motif, and it is regenerated/reduced by the selenoprotein Trx-reductase 1 (TrxR1); and NADPH. The Trx system has multiple functions [reviewed in ref. (29)]. These include (i) gene regulation by transcriptional factor redox modulation, for example, glucocorticoid receptor and nuclear factor kappa B (NFκB); (ii) apoptosis control by structural binding to apoptosis-signaling kinase 1 (ASK1); and (iii) growth stimulation by membrane (cytokine) receptor dithiol/disulfide modulation and direct antioxidant capture of ROS (e.g., H2O2). In addition to its intracellular functions, Trx is released in full-length (41) and truncated forms (42) under physiological conditions of oxidative stress caused by a variety of stimuli such as mitogens, inflammatory signals, burns (1), viral infections, for example, human immunodeficiency virus (HIV) (18), and influence from stromal cells in the microenvironment (8).
The cell surface redox environment is essential for control of thiol–disulfide exchange in cell surface proteins affecting the 3D structure and function. PDI (also termed PDIA1 and P4HB), is a 57-kDa protein of 508-amino-acid length belonging to a family of molecular chaperones and soluble oxidoreductases that act in the endoplasmic reticulum to promote disulfide bond formation and efficient folding of the nascent protein (7, 25). PDI members are also identified at the surface membrane of cells, that is, lymphocytes and platelets. Cell surface PDI can catalyze reduction of disulfide bonds in cell surface proteins. Proteomic studies on cancers, including CLL, have revealed an unusually high surface membrane expression of PDI (22, 47). PDI was shown to mediate invasive properties in human gliomas via integrin-dependent cell adhesion (22). Whether PDI is related to the progression with overexpression in aggressive versus indolent type of CLL is not known.
In this study, we have investigated potential interactions between the redox-active proteins Trx1 and PDI, their physical association, and functional control of TNFRs on CLL cell surface membranes, which may promote TNF autocrine signaling and survival. Prompted by reports showing that membrane Trx1 and PDI are responsible for reduction of a disulfide bond in CD4 that allows HIV-1 envelope fusion (18), and studies showing that PDI redox regulates integrins and cell migration (22), we analyzed thiol–disulfide redox modulation of the cysteine-rich surface membrane TNFR1 and TNFR2.
Results
Membrane PDI, TNFR1, TNFR2, TNF, and Trx1 expression on CLL cells
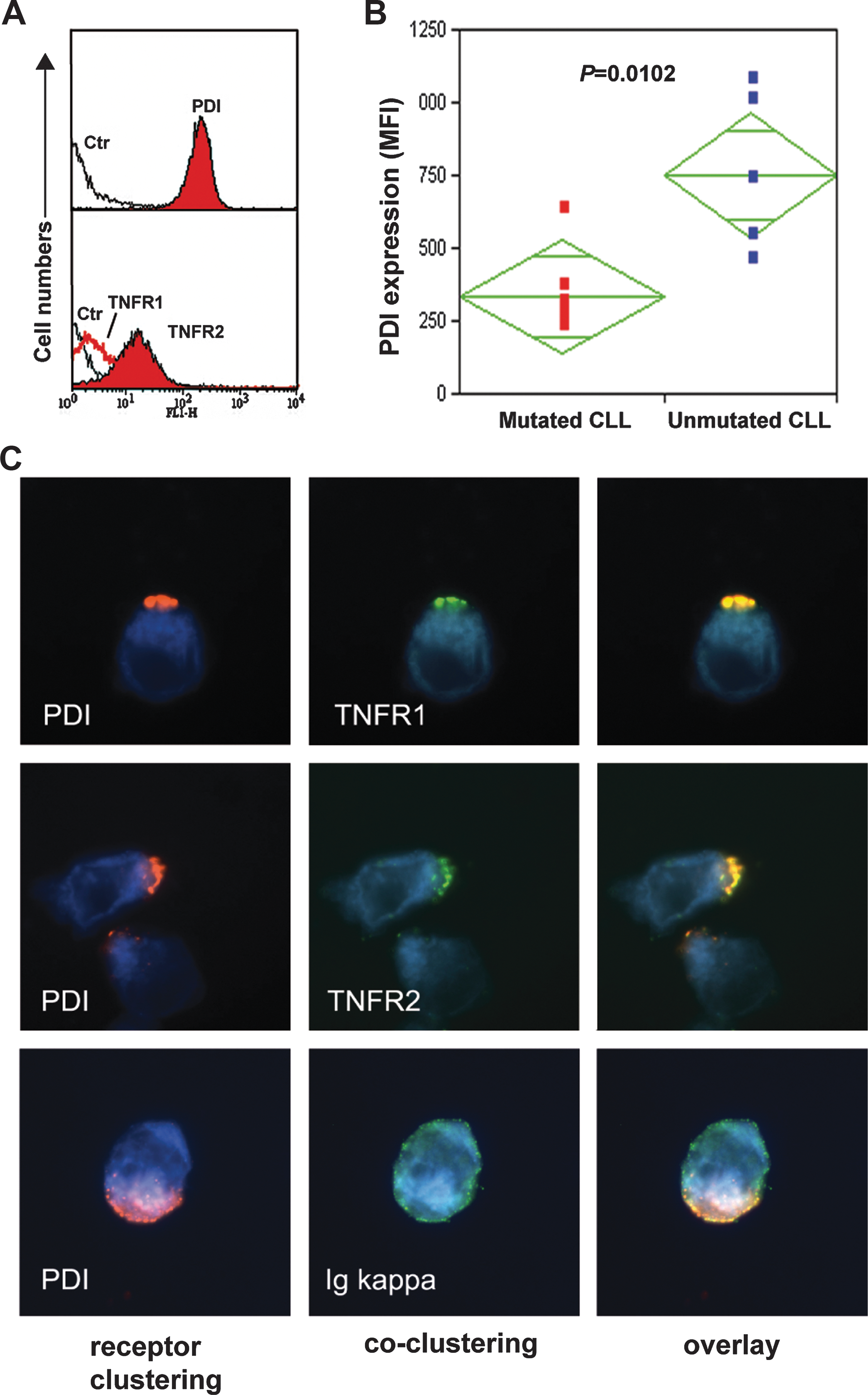
Robust expression of membrane PDI was detected on all (n=21, Table 1) primary CLL cells (Fig. 1A, upper flow histogram, and B). These findings corroborate previous report showing that B-CLL cells (mean of 25 patients cases) express 10-fold higher surface membrane protein thiol (SH-) groups and significantly higher PDI compared with control healthy donor peripheral blood mononuclear cells (PBMCs) (47). TNFR1 was expressed at a lower level compared with TNFR2 (Fig. 1A, lower flow histogram). CLL cells were positive also for membrane TNF, but negative for membrane Trx1. CLL patients expressing immunoglobulin heavy-chain variable (IGHV)-unmutated genes in their neoplastic B cells are known to have more aggressive disease progression compared with patients with IGHV-mutated genes (15, 24). We analyzed whether the PDI membrane expression in a panel of five IGHV-unmutated and six IGHV-mutated CLL cases differed between the two subtypes of CLLs. A significantly higher PDI membrane expression was observed in the IGHV-unmutated cases (p=0.0102; Fig. 1B).
Age, year at sampling.
ID-23 has a 9-codon-long CDR3, but with IGHJ3 usage.
PD, progressive disease; SD, stable disease; PR, progressive relapse; UM, unmutated; M, mutated; IGHV, immunoglobulin heavy-chain variable; CLL, chronic lymphocytic leukemia.
Direct molecular interaction between membrane PDI and TNFR1/TNFR2
Membrane PDI clustering was induced on CLL cells by cross-linking PDI with anti-PDI antibodies (Abs). PDI clusters (membrane caps) were formed, and noteworthy, TNFR1 clusters were also induced by PDI crosslinking. Identical patterns with a pixel-to-pixel match were found, indicating a direct molecular interaction (Fig. 1C, top panel). PDI also interacted with TNFR2 in a similar manner as shown in overlay digital images (Fig. 1C, mid panel). Control anti-kappa light chains, however, did not cocluster with PDI, and appeared with a dispersed dot-like pattern, proving absence of molecular interaction between the kappa light chain and PDI (Fig. 1C, bottom panel).
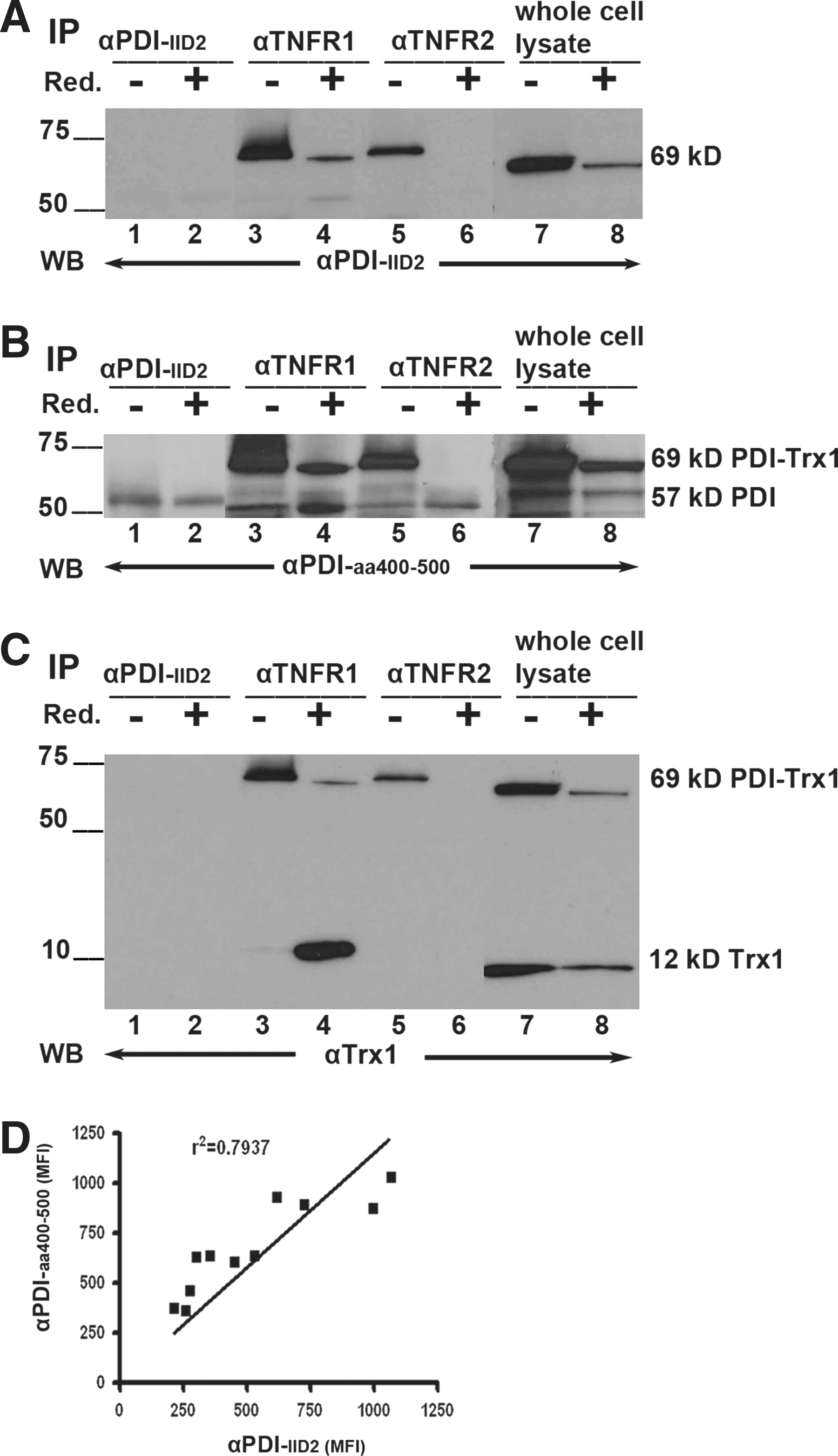
The physical association between PDI and TNFR1 and TNFR2 was validated by co-immunoprecipitation (IP) analysis. Cell lysates of 232B4 CLL were precipitated with or without pretreatment with Reductacryl® beads (solid-phase dithiothreitol regenerated by sodium borohydride) by αTNFR1 and αTNFR2 Abs. The conformation-sensitive αPDI-IID2 monoclonal Ab (mAb) that detects native (in IP), but not denatured, 57-kDa PDI (in western blot [WB]) was used as a positive control. The precipitates were analyzed in sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) followed by WB using either the αPDI-IID2 mAb or a sequence-specific anti-PDI Ab (αPDI-aa400-500). Noteworthy, we detected a novel 69-kDa PDI variant that coprecipitated with αTNFR1 (Fig. 2A, lane 3; and B, lane 3) and αTNFR2 (Fig. 2A, lane 5; B, lane 5). The expected 57-kDa PDI protein was precipitated by αPDI-IID2 and detected in WB by the sequence-specific αPDI-aa400-500 Ab (Fig. 2B, lanes 1–8). αTNFR1 and αTNFR2 precipitated both the 69-kDa and 57-kDa PDI entities (Fig. 2B, lanes 3 and 5). Whole lysates also contained both entities (Fig. 2B, lanes 7 and 8).
The metalloproteinase Adam 17 (also termed TNF-α-converting enzyme [TACE]) has been reported to be redox activated by PDI (49). We analyzed whether it formed complexes with PDI. However, no complex formation was observed with TNFR1, TNFR2, or with TNF in co-IP experiments (data not shown).
We investigated the nature of the PDI-TNFR1 and PDI-TNFR2 interactions/bindings, whether these were covalent disulfide bonds, for example, reduction sensitive. The cell lysates were pretreated with an efficient reducing agent in the form of freshly NaBH4-regenerated Reductacryl beads for 10 min followed by alkylation with iodoacetamide. The 69-kDa PDI variant was indeed reduction sensitive, and it disintegrated partly by the pretreatment (Fig. 2A, B, lanes 4 and 8) or disintegrated completely (Fig. 2A, B, lane 6). The 69-kDa PDI was, however, stable by the conventional SDS-PAGE sample buffer containing 1% β-mercaptoethanol (β-ME) (Fig. 2A, B lanes 3, 5, and 7). The 57-kDa PDI entity was present both in pretreated/reduced and nonreduced IP, but it was enhanced in prereduced samples compared with nonreduced (Fig. 2B, lane 4 vs. 3; and B, lane 6 vs. 5). It is noteworthy that the reduction–alkylation procedure did not affect the epitopes that were recognized by the precipitating Abs, since the 57-kDa PDI band showed identical intensities/signals in WB (Fig. 2B, lanes 1 and 2).
Based on previous reports that Trx1 binds to membrane TNFR superfamily members, for example, TNFRSF8/CD30 (43), we hypothesized that extracellular Trx1 may be involved in TNFR1/2–PDI membrane interactions. Indeed, when reprobing the polyvinylidene difluoride (PVDF) WB membrane for Trx1, we found that the anti-Trx1 mAb recognized both a 69-kDa entity and the expected 12-kDa monomeric form of Trx1 in whole-cell lysates, but it did not recognize 57-kDa PDI (Fig. 2C, lanes 7 and 8). Unexpectedly, anti-TNFR1 coimmunoprecipitated 12-kDa Trx1 in spite of stringent Reductacryl pretreatment (Fig. 2C, lane 4), indicating that a trimeric reduction-resistant TNFR1-PDI-Trx1 complex resides in the CLL plasma membranes. Regarding the TNFR2, the 12-kDa Trx1 was absent (Fig. 2C, lane 6), indicating that the TNFR2-PDI-Trx1 was less stable and completely reduced by Reductacryl IP-pretreatment. The molecular weight of the 69-kDa PDI entity strongly indicates that it consists of a 57-kDa PDI plus a 12-kDa Trx1 protein associated into a reduction-sensitive complex.
The two anti-PDI Abs used in the above IP and WB experiments (Fig. 2A–C) were compared to each other with regard to their binding efficiency to native surface membrane PDI in flow cytometry of eleven CLL patient B-cell samples. Their binding capacities (mean fluorescence intensity [MFI] values) showed a strong correlation (r 2=0.7937; Fig. 2D).
The 69-kDa PDI-Trx1 membrane complex is abundant in leukemic CLL cells versus healthy donor cells
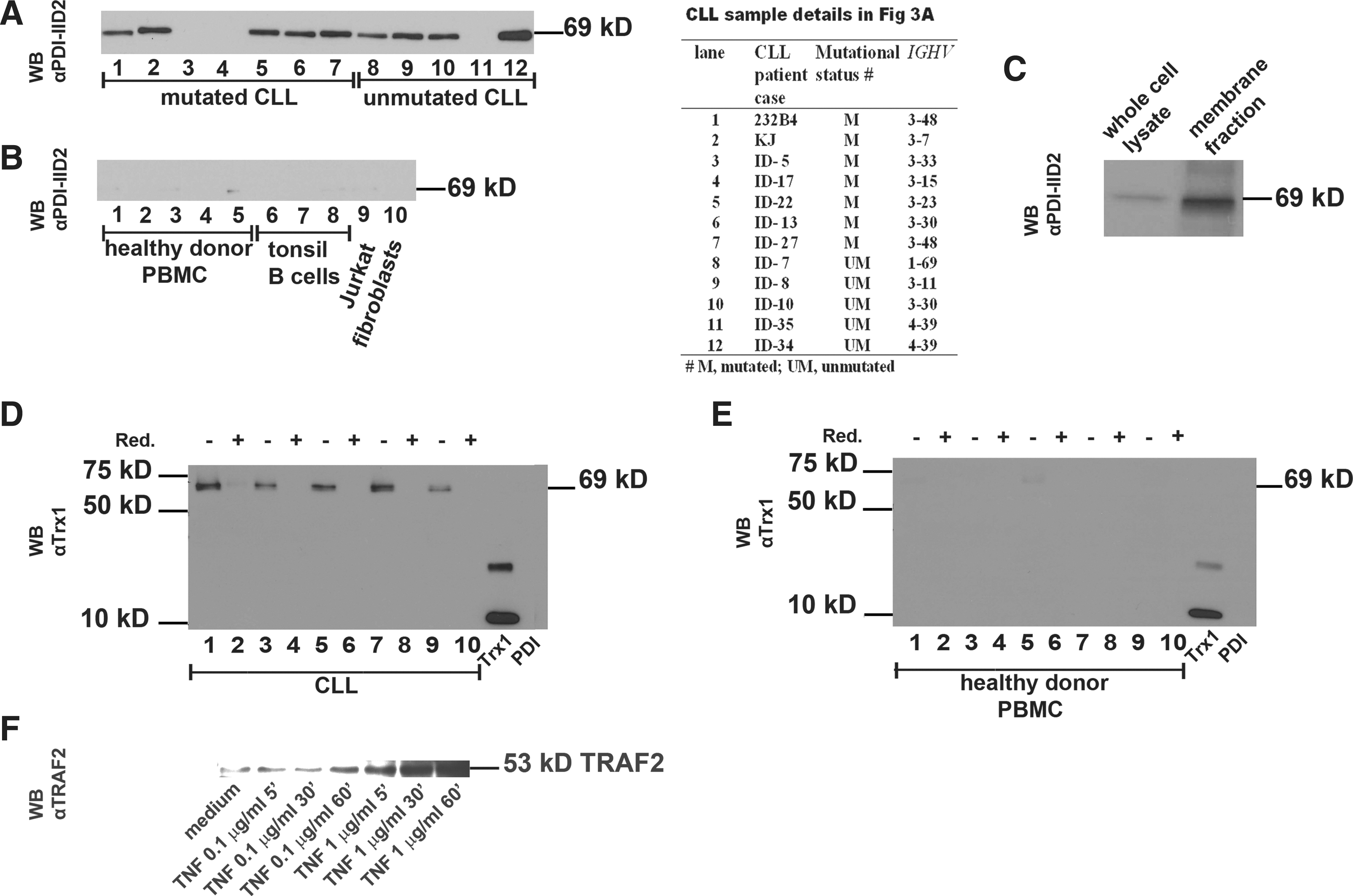
Twelve CLL whole-cell lysates were analyzed in WB with αPDI-IID2 (Fig. 3A). Patient IGHV mutational status is detailed in the Figure 3A Table inset. In a parallel gel in the same electrophoresis experiment, 10 control samples were investigated: 5 healthy donor B-cell samples (Fig. 3B, lanes 1–5), 3 tonsil (activated) B-cell samples (Fig. 3B, lanes 6–8), Jurkat T-cell line, and human fibroblasts (Fig. 3B, lanes 9 and 10, respectively). The 69-kDa band was found in 9 of 12 CLL patients. Expression was found in unmutated as well as mutated (4/5 unmutated, and 5/7 mutated). The protein quantity in the WB-bands (Fig. 3A), however, did not recapitulate fluorescence-activated cell-sorter (FACS) results in Figure 1B, which most likely reflects different levels of expression at the surface membrane (FACS) and in whole-cell lysates (WB). The 69-kDa entity was expressed very dim in three of five healthy donor controls, in one (activated) of three tonsil B-cell samples, in Jurkat T-cells, but not in fibroblasts (Fig. 3B). The three tonsil samples represent high-density (resting) B-cells (lane 6), medium-density, preactivated B-cells (lane 7), and low-density, activated B-blasts (lane 8). Healthy donor B-cell preparations (n=5) were isolated from PBMCs. In addition we found that T-cell depletion (by anti-CD3) could not induce higher expression levels of 69 kDa after 48 h of activation with phorbol 12-myristate 13-acetate (PMA)/ionophore (data not shown). The membrane localization of the 69-kDa PDI-Trx1 complex was validated by live cell membrane biotinylation and analysis in WB (Fig. 3C), showing preferential expression on the exofacial side of cell membrane, whereas the cell lysate showed weak expression only. Further analysis of five CLL patient samples and five healthy donor blood cells revealed that the 69-kDa complex (analyzed by anti-Trx1) was abundant in CLL cells (Fig. 3D, E), and it was reduction sensitive. Anti-Trx1 did not cross-react with 57-kDa recombinant PDI. Taken together, the difference in PDI-Trx1 expression between 12 CLL samples and 10 control samples was significant: 9/12 CLL expressed the PDI-Trx1 complex abundantly, whereas control samples were very weak with a ∼20-fold lower level of expression (Fig. 3A, B, D, E).
Functional role of PDI-Trx1 in TNFR1/2 signaling
To address the functional consequences of the observed TNFR1/TNFR2 association with PDI-Trx1 and possible effects on receptor signaling activity determined as autocrine TNF-release, we first analyzed as a preparatory step the signaling competence of TNFR1/2 by analysis of TNFR-associated factor 2 (TRAF2), which is required for signaling transduction. We also analyzed potential signaling dependence on Trx1.
The TNFR signaling competence was determined by analysis of TRAF2 expression in CLL cells exposed for 15 min, 30 min, and 60 min with increasing doses of human TNF. A basal low level of TRAF2 was found in 232B4 CLL cells. Escalating exposure (dose and time) from 100 ng/ml for 15, 30, 60 to 1 μg/ml of TNF for 15, 30, and 60 min induced a dose-dependent increase of TRAF2 (Fig. 3F). The signaling dependence on Trx1 in the TNF autocrine loop was investigated by addition of 1–4 μg/ml of recombinant human Trx1 (0.085–0.34 μM) to 1-h PMA-preactivated CLL cells for 72 h. Figure 4A shows a significant dose–response increase of TNF release induced by Trx1 (p<0.0001). Parallel kinetic analysis showed a TNF increase from 20 pg/ml at 24 h to 58 pg/ml at 48 h, reaching 75 pg/ml at 72 h (p<0.0001) upon exposure to 0.085 μM Trx1. The viability also significantly increased from 88% to 94% in the presence of Trx1 for 72 h (Fig. 4A). In parallel experiments, we found that inhibition of Trx1 by addition of monoclonal anti-Trx1 Abs in the CLL cell line 232B4 significantly reduced, in a dose-dependent manner, the viability as compared to the isotype IgG1 control (p=0.001) (Fig. 4B).
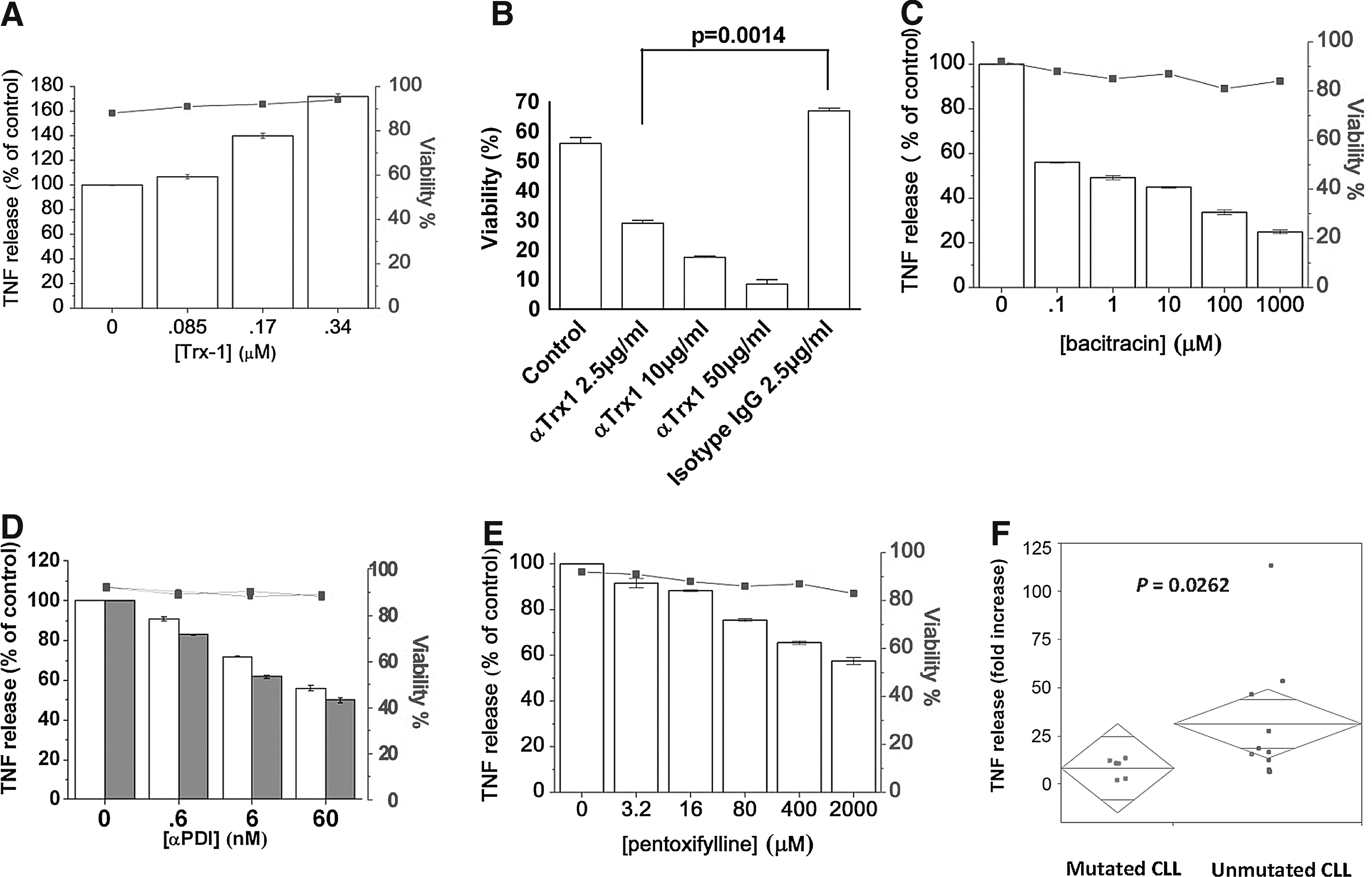
The functional role of PDI-Trx1 in TNFR1/TNFR2 signaling was analyzed by inhibition of PDI activity. The effect of the PDI inhibitor bacitracin (0.1 μM to 1.0 mM) on TNF autocrine signaling was significant with a 44%–73% inhibition of TNF release (p<0.0001) (Fig. 4C). The viability of the cells was surveyed and showed a significant decrease (Fig. 4C, upper solid line). The bacitracin effect was verified by two different anti-PDI mAbs [clones C1D11 and IID2, both previously shown to inhibit PDI enzymatic activity (47)], which were tested in the 0.1–10 μg/ml range. Both mAbs were found to exert a significant inhibition of TNF release (Fig. 4D). As a methodological control, we used pentoxifylline (a cyclic nucleotide phosphodiesterase inhibitor that interferes with cGMP and cAMP, inhibiting synthesis of TNF) in the range 3.2–2000 μM (Fig. 4E). Furthermore, to analyze whether the level of PDI expression correlated with CLL severity (unmutated cases with high PDI vs. mutated case with low PDI), we investigated the effect of ROS induction on TNF release (which indirectly promotes growth). We triggered ROS production by a 72-h PMA/ionophore exposure and observed significantly higher release in IGHV-unmutated versus mutated cases, p=0.0262 (Fig. 4F).
Discussion
There are two novel findings in this study: first, PDI interacts physically and functionally with TNFR1 and TNFR2 at the exofacial cell surface membrane of CLL B cells. Secondly, the membrane PDI attached to the TNFRs forms a covalent complex with Trx1, and this complex is abundantly expressed in most unmutated and mutated CLL cases in contrast to healthy donor B cells that express very low amounts.
The close proximity of the TNFRs with membrane PDI was revealed in membrane receptor-clustering experiments, and the actual binding was verified in a co-IP analysis. This molecular interaction seen in leukemic cells, and not in cells from healthy donors, adds a novel aspect to the intricate TNF-TNFR interaction recently suggested in detailed structural studies to form large aggregates (>1000 kDa) from TNFR–dimer or trimer complexes (30). Oxidative stress was shown to promote ligand-independent and enhanced ligand-dependent TNF signaling, also induced by the disulfide oxidase DsbA (34), which can serve as a bacterial mimic of eukaryotic PDI (37).
Functionally, TNFR signaling and activation were determined in our study by an analysis of autocrine TNF release. The TNF release was interrupted by PDI inhibitors bacitracin and anti-PDI mAbs, which are known from previous studies to inhibit PDI enzymatic disulfide isomerase activity (47). Enhanced TNF release was induced by Trx1 addition, and the level of TNF release correlated with increased/improved viability, whereas decreased TNF induced by bacitracin or anti-PDI correlated with decreased viability. These observations strongly support previous reports that the NFκB-driven TNF pathway is autostimulatory in CLL (14, 17, 20). The findings also point at the importance of the NFκB pathway in the pathogenesis of CLL: the 13q14 deletion, which is the most frequent genetic abnormality in CLL, involves DLEU7, which is an inhibitor of NFκB (35).
Our results show that PDI interacts with TNFR1, and TNFR2 raises the intriguing possibility that in addition to the classical ligand–receptor model of TNF signaling, an additional level of control of TNF signaling may reside at the cell membrane level, wherein PDI-Trx1 can act as a redox sensor of the extracellular environment. The open (reduced) or closed (oxidized) conformation of the TNFRs (34) can be regulated by the extracellular GSH/GSSG, cystine/cysteine, and ROS level. Several PDI-controlled functions could operate synergistically: (i) PDI may be instrumental in regulating the cysteine-rich domain regions (CRDs) of TNFR1 and TNFR2, as suggested by Ozsoy et al. (34), at the CRD1 domain, which is essential for the formation of homotypic, ligand-independent receptor complexes through the preligand assembly domain (6), at the ligand-binding CRD2/CRD3, or at the CRD4 domain. (ii) PDI has a close functional association with NAD(P)H oxidase (NOX) on the external cell membrane (26). (iii) PDI also redox regulates the activity of ADAM17/TACE (49), which enzymatically cleaves TNF, TNFRSF members, and diverse membrane receptors and adhesion molecules. PDI inhibitors were shown to prevent ADAM17 activity (49). Recent report on cell migration in human gliomas has identified PDI (PDIA6) as instrumental in redox regulation of β-integrins (22), findings that underscore that PDI is a potent membrane disulfide reductase cleaving disulfide bonds at the exofacial cell surface and regulates the extracellular protein redox status (27), in contrast to its intracellular function where it catalyzes disulfide bond formation, for example, disulfide isomerase function.
We propose a model for PDI-Trx1-TNFR1/2 interactions at the cell surface. It is convincingly reported that TNFR-TNF ligation (Fig. 5, step 1) generates significant levels of ROS (12, 45). The ROS species are derived from several NOX family members, also present at the membrane of B-cells and responsible for H2O2 generation (Fig. 5, step 2), not only from the TNFR superfamily members but also from Toll-like receptors (TLR) (4) and BCRs (38). TNFR/TNF ligation activates transcription factor NFκB (Fig. 5, step 3), leading to synthesis of pro-TNF (Fig. 5, step 4) followed by a redox/PDI-controlled TACE/ADAM17-induced release of mature TNF trimers (49) (Fig. 5, step 5). The pathway is under complex control by ROS, and both proapoptotic and antiapoptotic programs can be activated by ROS action on NFκB (44). Most interestingly, results by Capasso et al. (11) show that the voltage-gated proton channel HVCN1 is associated with the BCR complex, and it generates ROS in high concentrations. Further studies are necessary to understand its role in redox regulation of the TNFRSF receptors, which are abundantly expressed on CLL B-cells, including several NFκB-signaling TNFRSF members: TNFRSF1A/TNFR1, TNFRSF1B/TNFR2, TNFRSF5/CD40, TNFRSF8/CD30, TNFRSF13B/BAFF, and TNFRSF13/APRIL. The BCR-elevated peroxide tonus (38) may synergize with the TNFRSF members and TLR in TNFR oligodimerization and ligand-independent or enhanced ligand-dependent signaling (34). Furthermore, in addition to autocrine-produced TNF, stromal cells (Fig. 5) present in the microenvironment of the CLL proliferative centers supply neoplastic B-lymphocytes with TNF, Trx1, and recently discovered cysteine (necessary for GSH synthesis) that together protect the leukemic cells from apoptosis (3). Important recent studies show that stromal cells modulate the redox status of CLL cells and promote survival (51). Primary CLL cells exhibit a limited ability to transport cystine for GSH synthesis owing to low expression of the highly specific amino acid antiporter system Xc. Stromal cells, however, express the Xc transporter and effectively import cystine and convert it to its reduced form: cysteine, which is then released into the microenvironment for uptake by CLL cells via a neutral amino acid transport system. Zhang et al. (51) showed that cystine did not promote CLL survival unless reduced to cysteine by β-ME. In vivo, there is no β-ME. In contrast stromal cells produce Trx1, which operates as a cellular equivalent to β-ME and effectively reduces cystine to cysteine (8). The elevated level of GSH (shifting the GSH:GSSG ratio to a more reducing condition) enhances leukemia cell survival and protects them from drug-induced cytotoxicity (51). We suggest that the reducing environment, found in the BM and lymph node proliferative compartment, is generated by stromal cell-released cysteine, GSH, Trx1, as well as TrxR, as previously shown to be released from stromal cell, macrophages, and activated B-cells (46). This extracellular released cysteine importantly influences the extracellular redox tonus (5), and favors a condensed (reduced) PDI conformation that allows a TNFR1/TNFR2-confomation compatible with ligand binding, signaling, and proliferation. In the peripheral compartment (blood circulation), on the other hand, there are no stromal cells, and the extracellular redox environment is more oxidative (as it is in in-vitro cultures), resulting in a ligand-closed conformation with inactive, resting cells.

Our present results taken together with the frequent CLL genetic deletion of the NFκB inhibitor DLEU7, which tunes up the pathway, we suggest that the PDI-Trx1 complex may redox regulate ligand-independent signaling and enhance TNF ligand-dependent signaling, and possibly also signaling by other ligands of the TNFRSF members. The robust expression of PDI on B-CLL cells is in agreement with previous data from Täger et al., who found that high surface membrane PDI expression on CLL cells (higher than on normal B cells) correlated with high surface thiol (-SH) expression (47). PDI inhibition resulted in increased protein -SH-groups on the CLL surface membrane due to inhibition of -SH consumption via sulfhydryl disulfide (2SH→S-S) interchange reactions catalyzed by the PDI. The increased PDI expression indicates a crucial role in -SH-mediated protection and resistance against cytotoxic, ROS-generating drugs such as vinblastine and cisplatin (47). However, PDI has multiple targets, and the intracellular/ER functions of PDI are different from those of exofacial PDI (2). On the surface membrane, PDI is retained in the membrane by galectin-9 and redox regulates cell migration, integrin function, HIV entry (9, 22), TACE/ADAM17 (49), and NOX (26). This study shows that it is the membrane PDI and not total PDI content per se that is crucial, since high membrane PDI level significantly correlates with a more aggressive disease progression (IGHV-unmutated cases). Interestingly, high-titer anti-PDI auto-Abs were induced in an acute myeloid leukemia patient who achieved a complete response after vaccination with irradiated autologous granulocyte macrophage-colony stimulating factor-secreting tumor cells in a setting of nonmyeloablative allogeneic BM transplantation (21).
In summary, we describe in this study a new level of TNF-regulation, in which membrane TNFR1 and TNFR2 are redox controlled at the exofacial cell surface by the PDI-Trx1 complex that amplifies NFκB-driven TNF autostimulatory release. These complexes may contribute to the observed survival benefit in the more aggressive IGHV-unmutated CLL cases, which have more PDI compared with the indolent IGHV-mutated cases. We present evidence showing that increased PDI levels in unmutated CLL are associated with higher TNF-release, whereas decreased PDI, by PDI inhibitors, including anti-PDI, blocks autostimulatory TNF release. Our study points at possible pharmacological/therapeutical interventions that may block an ongoing clonal expansion.
Materials and Methods
Cell separation
Peripheral blood was collected from CLL patients at the Hematology Clinic, the Linköping University Hospital, Linköping, Sweden (Table 1). Lymphocytes were isolated by density-gradient centrifugation (Ficoll-Hypaque; GE Healthcare, Uppsala, Sweden) and stored in liquid N2 until assayed. The study protocol was approved by the ethics committee of the Linköping University Hospital, including written informed consent from the patients (Dr. No. 02–459).
Ab reagents and CLL cell line
All Abs used in this study, their origin, and designation, are presented in Table 2. 232B4-CLL is an authentic lymphoblastoid cell line, derived from a male patient with an IGHV3-48 mutated B-CLL clone, with a verified neoplastic genotype and phenotype (48). Cells were grown at 37°C in 5% CO2 atmosphere in an RPMI 1640 medium (Invitrogen, Paisley, United Kingdom), supplemented with 10% fetal calf serum (FCS; Invitrogen), 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM
Ab, antibody; ELISA, enzyme-linked immunosorbent assay; IP, immunoprecipitation; mAb, monoclonal antibody; Trx, thioredoxin; TNFR, tumor necrosis factor receptor; TRAF2, TNFR-associated factor 2; WB, western blot; IF, immunofluorescence.
TNF autocrine release
CLL cells (1×106 in 1 ml) were cultured in 24-well plates in an RPMI 1640 medium, 10% FCS, and antibiotics at 37°C in a 5% CO2 atmosphere. Cells were stimulated by PMA (25 ng/ml; Enzo Life Sciences, Inc., Farmingdale, NY), ionophore (300 ng/ml; Calbiochem/Merck, Darmstadt, Germany), and human Trx1 (1–4 μg/ml; IMCO Corporation, Ltd., AB, Stockholm, Sweden). Recombinant Trx1 was reduced by Cleland's Reductacryl-immobilized beads (Calbiochem) before each experiment. Inhibition of PDI activity was analyzed by addition of αPDI-IID2 mAb and αPDI-C1D11 mAb. Bacitracin and pentoxifylline used for inhibition studies were purchased from Sigma-Aldrich Chem. Co. (St. Louis, MO). Cultures were continued for 24–72 h. Viable cell numbers were assessed by the Trypan blue dye exclusion method. At the end of cultivation, supernatants were collected by centrifugation and stored at −70°C until assayed in TNF enzyme-linked immunosorbent assay (ELISA). Each experiment was performed three to five times. Recombinant TNF was purchased from R&D Systems (Minneapolis, MN).
TNF ELISA
Sandwich ELISA was performed according to standard procedure using αTNF-28401.111 (4 μg/ml) as catcher Ab and biotinylated αTNF-biotin as an indicator Ab, followed by streptavidine–horseradish peroxidase (HRP; Invitrogen) and 3,3’, 5’5-tetramethylbenzidine substrate (Sigma-Aldrich). Optical density was recorded at 450 nm.
FACS analysis
Cells were tested with or without preactivation with 25 ng/ml PMA and 300 ng/ml ionophore (1 h). Cell viability was assessed by Trypan blue dye exclusion before onset of the experiment. FACS analysis was performed with 0.5×106 cells. Cells were incubated with a primary mAb or isotype IgG, anti-mouse IgG-biotin (Dako, Glostrup, Denmark; No. k5001), and FITC-conjugated streptavidin (F0422; Dako). For each analysis, 10,000 cells were acquired and analyzed using CellQuest software proversion v4.0.1 (CellQuest). Kolmogorov–Smirnov test was used for statistical analysis of significance.
Receptor clustering and cocapping
TNFR coclustering analysis was performed as previously described (31). In brief, cells were washed and resuspended to 1×106/ml in phosphate-buffered saline (PBS) containing 2% bovine serum albumin (capping buffer). αPDI-IID2 (1 μg/100 μl capping buffer) was added to the cells for 30 min at 37°C. The cells were washed twice in the capping buffer at 37°C. Anti-mouse IgG-Alexa-Fluor® 594 (1 μg/100 μl) was then added as a second Ab and incubated for 1 h at 37°C. The cells were washed twice in ice-cold PBS (0°C) containing 0.02% NaN3 and centrifuged at 200 g. Mouse serum (diluted 1:10) was added for 10 min, as a blocking reagent, followed by an additional washing step. Then, the cocapping Ab, αTNFR1-biotin (0.1 μg/100 μl), or αTNFR2-biotin, or anti-kappa (as a negative control), was added in ice-cold PBS with 0.02% Na-azide, for 20 min at 0°C. After two additional washing steps, an Alexa-Fluor-488 streptavidin-conjugate (0.1 μg/100 μl) was incubated for 20 min 0°C, followed by two washes. Adhesion slides (Bross; Marienfeld GmbH, Lauda-Königshofen, Germany) were washed in cold distilled water and PBS. Cells were adhered for 15 min and fixed in 4% paraformaldehyde for 5 min on ice. Coverslip glasses were mounted face-down in a SlowFade Light Antifade kit (Molecular Probes/Invitrogen) and sealed by nail varnish. Samples were analyzed in a Zeiss Axiovert 200M inverted microscope (Carl Zeiss, Heidelberg, Germany). Images were collected and analyzed using AxioVision 3.1 Software. Huygens software (SVI, Hilversum, The Netherlands) was used for deconvolution image restoration.
Coimmunoprecipitation
232B4 cells (250×106) were lysed in 12 ml cold HEPES lysis buffer (50 mM HEPES, pH 7.6, 150 mM NaCl, 5 mM EDTA, 1% NP40) containing a cocktail of protease inhibitors (Roche, Basel, Switzerland) for 30 min on ice. The lysates were passed several times through a 26-gauge needle to disperse any large aggregates. Lysates were centrifuged for 10 min at 20,000 g at 4°C, and supernatants were collected. Protein concentration was determined using bicinchoninate methods. The lysates were divided into two aliquots: one was reduced by Reductacryl-immobilized beads (Calbiochem) according to the manufacturer's protocol; the second aliquot was kept nonreduced. Free
Cell surface labeling
Viable 232B4 CLL cells were biotinylated at the cell surface membrane using the EZ-link sulfo-NHS-SS-biotin cell surface protein isolation kit (Pierce Biotechnology). Labeled proteins were then analyzed in WB for PDI expression.
Footnotes
Acknowledgments
We thank Drs. G. Spyrou, M. Björnstedt, M. Los, and R. Ihnatko, for valuable comments on the manuscript, and Dr. Michael Täger for the kind gift of anti-PDI hybridomas. The excellent technical assistance of Anita Lönn, Inga-Lill Scherling, Bita Sahaf, and Ana María Barral is gratefully acknowledged. This work was supported by the grants from the Swedish Research Association and the Swedish Cancer Society B04-17XC. GSD.
Author Disclosure Statement
No competing financial interests exist.