
Abstract
Introduction
Innovation
Antioxidants are an important defense system in fibrotic and inflammatory diseases, and the use of new antioxidant strategies may be useful as therapeutic tools or in the research setting to identify new pathways important in the pathogenesis of disease. The antioxidant superoxide dismutase mimetic, Mn(III)tetrakis(N-ethylpyridinium-2-yl)porphyrin (MnTE-2-PyP), exhibits important properties, as it has been shown to target extracellular ROS, which we propose are central to pulmonary vascular remodeling. In this study, we demonstrated a protective effect of MnTE-2-PyP on the regulation of the extracellular matrix component HA, which is not only a key matrix component, but also can itself promote inflammation. We then focused on a newly recognized pathway, the NACHT, LRR, and PYD domain-containing protein 3 inflammasome, which is responsible for caspase-1 activation and interleukin 1-beta (IL-1β) and IL-18. This inflammatory system is central to a number of chronic inflammatory diseases, but has not been well explored in human pulmonary arterial hypertension or animal models of pulmonary hypertension. Overall, our study is novel in its focus on extracellular events and the identification of new pathways amenable to therapeutic interventions.
MnTE-2-PyP belongs to a class of catalytic antioxidants and functions as an SOD mimetic. SOD antioxidant enzymes scavenge O2 •− by catalyzing the dismutation of two O2 •− radicals into hydrogen peroxide and oxygen (12, 26). As a metalloporphyrin, MnTE-2-PyP is a metal ion chelator that mimics the dismutation of O2 •− by alternate reduction and oxidation reactions between MN(III) and MN(II) within its extensive conjugated ring system (36). Pharmacokinetic studies of metalloporphyrins show that this compound is distributed at moderate levels to the lungs (AUC=26.5 μ*h/ml) and heart (AUC=14.8 μg*h/ml) in mice that were given a single intraperitoneal dose of 10 mg/kg (42). Maximum concentration is reached within 45 min and a slow elimination phase allowed a half-life of 60 to 135 h. Using these characterizations, we have developed a dosing regimen to study the effects of the SOD mimetic in chronically hypoxic mice. MnTE-2-PyP is an attractive agent to test, as SOD mimetics have been shown to effectively block oxidative stress in acute lung injury and attenuate bleomycin-induced pulmonary fibrosis, a model also used to study secondary PH (7, 34). While SOD mimetics can scavenge both intracellular and extracellular O2 •− (43, 49), we specifically investigated the potential contributions of extracellular protection by analyzing two extracellular targets of O2 •−, the extracellular matrix (ECM) component hyaluronan (HA) and an inflammatory pathway in macrophages, the NALP3 inflammasome.
The ECM is susceptible to oxidative damage and changes in the ECM contribute to the development of PH by regulating structural properties of the vessel and promoting inflammation (17, 20, 27, 37, 40). Extracellular antioxidant enzymes such as extracellular superoxide dismutase (EC-SOD) protect against oxidative damage to the ECM (13, 28). To test whether MnTE-2-PyP can protect against events in the extracellular vascular compartment, we chose to test one important matrix component and a potentially related inflammatory pathway. One relevant target of extracellular O2 •− is HA, a key component of the ECM (13, 49, 50). Fragmented HA is detected in the lung fluid and serum of patients with idiopathic pulmonary arterial hypertension (PAH) (1, 35), suggesting that oxidative damage to the vascular ECM may be a contributing factor in hypoxic PAH.
Chronic inflammation is also now well recognized to contribute to PH, and is both regulated by ROS and contributes to ROS production. Relevant to this study, pro-inflammatory cytokines interleukin 1-beta (IL-1β) and IL-18 have been implicated in lung diseases, including PAH (4, 19, 25, 41). It has recently been determined that the NALP3 inflammasome is central to the activation of IL-1β and IL-18 (22, 23, 45). This protein platform that includes the NOD-like receptor, NALP3 (NACHT, LRR, and PYD domain-containing protein 3), is a component of the innate immune system that regulates the activation of caspase-1 and subsequently IL-1β and IL-18 (48). The NALP3 inflammasome has not been previously characterized in models of PH and, based on its role in processing of IL-1β and IL-18, serves as a rationale to study targets of extracellular O2 •− potentially amenable to antioxidant treatment with AEOL 10113.
Based on the utility of MnTE-2-PyP to target extracellular O2 •−, we hypothesized that treatment with MnTE-2-PyP would protect against the development of chronic hypoxic pulmonary hypertension (CHPH), vascular remodeling, and inflammation.
Results
SOD mimetic attenuates CHPH
We first established that the SOD mimetic, MnTE-2-PyP, protected against CHPH in mice. Phosphate-buffered saline (PBS)-treated mice exposed to hypobaric hypoxia for 35 days developed increases in right ventricular systolic pressure (RVSP) (Fig. 1A). Treatment with the SOD mimetic attenuated the hypoxia-induced elevation in pulmonary arterial pressures. The SOD mimetic also blunted the hypoxia-induced increase in RV hypertrophy (Fig. 1B).
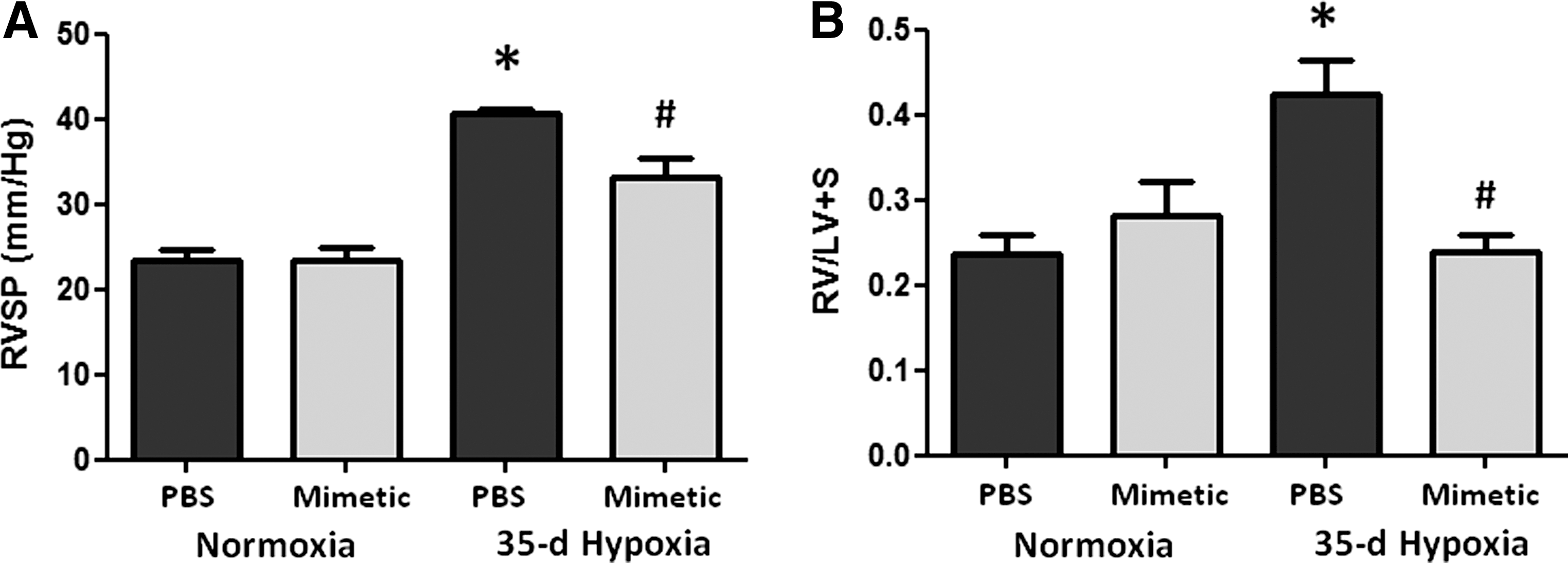
SOD mimetic attenuates pulmonary vascular remodeling
An established feature of CHPH is pulmonary vascular remodeling. We therefore evaluated cell proliferation in the vessel wall, muscularization of small vessels, and HA content, a key ECM component in the adventitia susceptible to oxidative fragmentation.
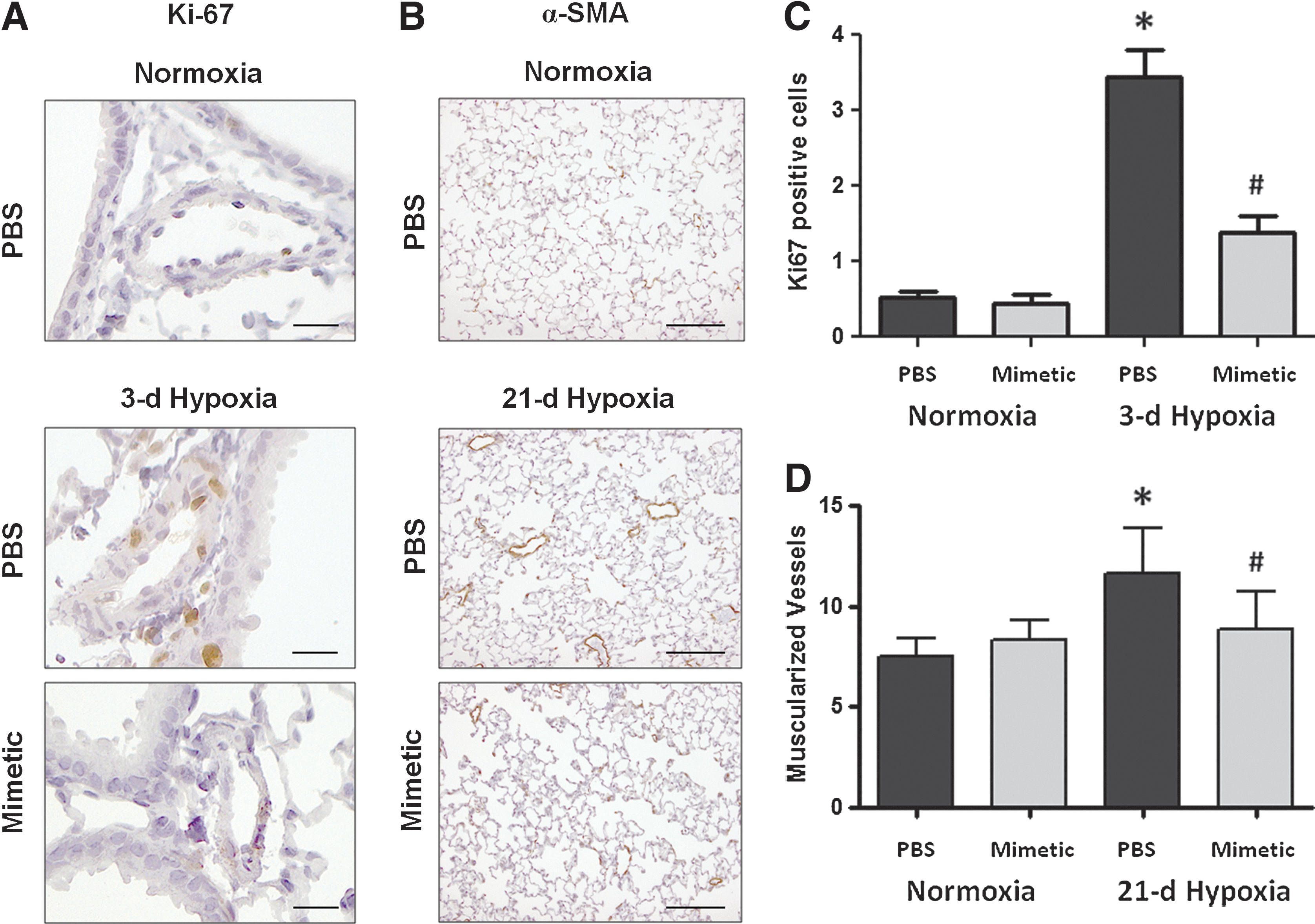
Pulmonary artery cell proliferation and muscularization
Our previous studies have shown a transient increase in pulmonary artery cell proliferation at early hypoxic time points (3 days) followed at later time points by small vessel muscularization (21 or 35 days) (32). Ki67 nuclear staining and alpha smooth muscle actin (α-SMA) staining were used as markers to quantify the number of proliferating cells and the number of muscularized vessels at the relevant time points, 3 and 21 days, respectively (Fig. 2A, B). In response to a 3-day hypoxic exposure, mice had increased numbers of proliferating cells within the medial and intimal layers of similar-sized pulmonary arteries (50–200 μm) compared to normoxic controls (image taken at 20× magnification). Hypoxia-exposed mice treated with SOD mimetic had less proliferating cells in the pulmonary arteries (Fig. 2C). The development of muscularized small (<50 μm) pulmonary vessels was also seen in response to 21 days of chronic hypoxia in PBS-treated mice. Treatment with the SOD mimetic attenuated the hypoxia-induced development of muscularized vessels (Fig. 2D).
Modulation of HA content
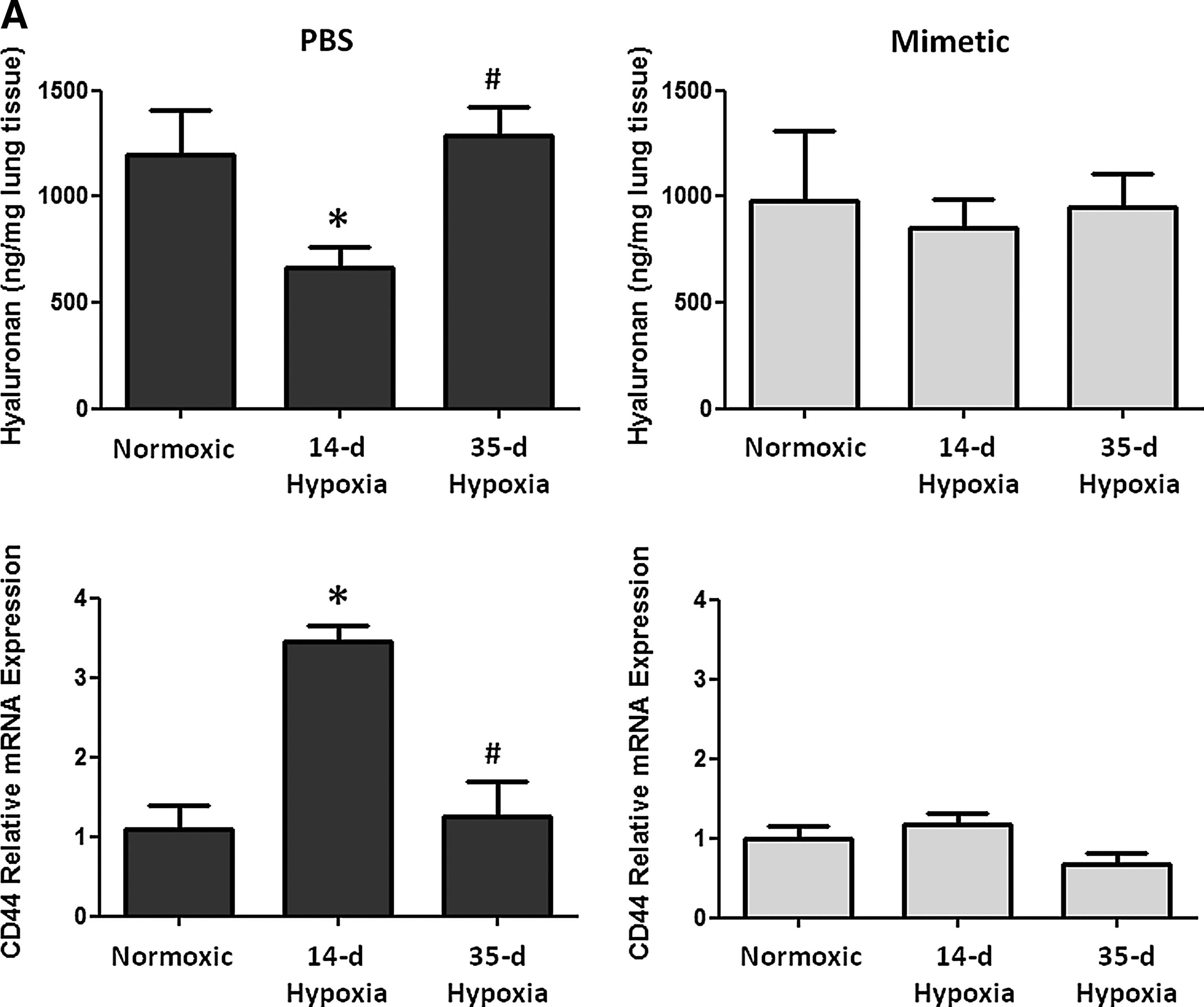
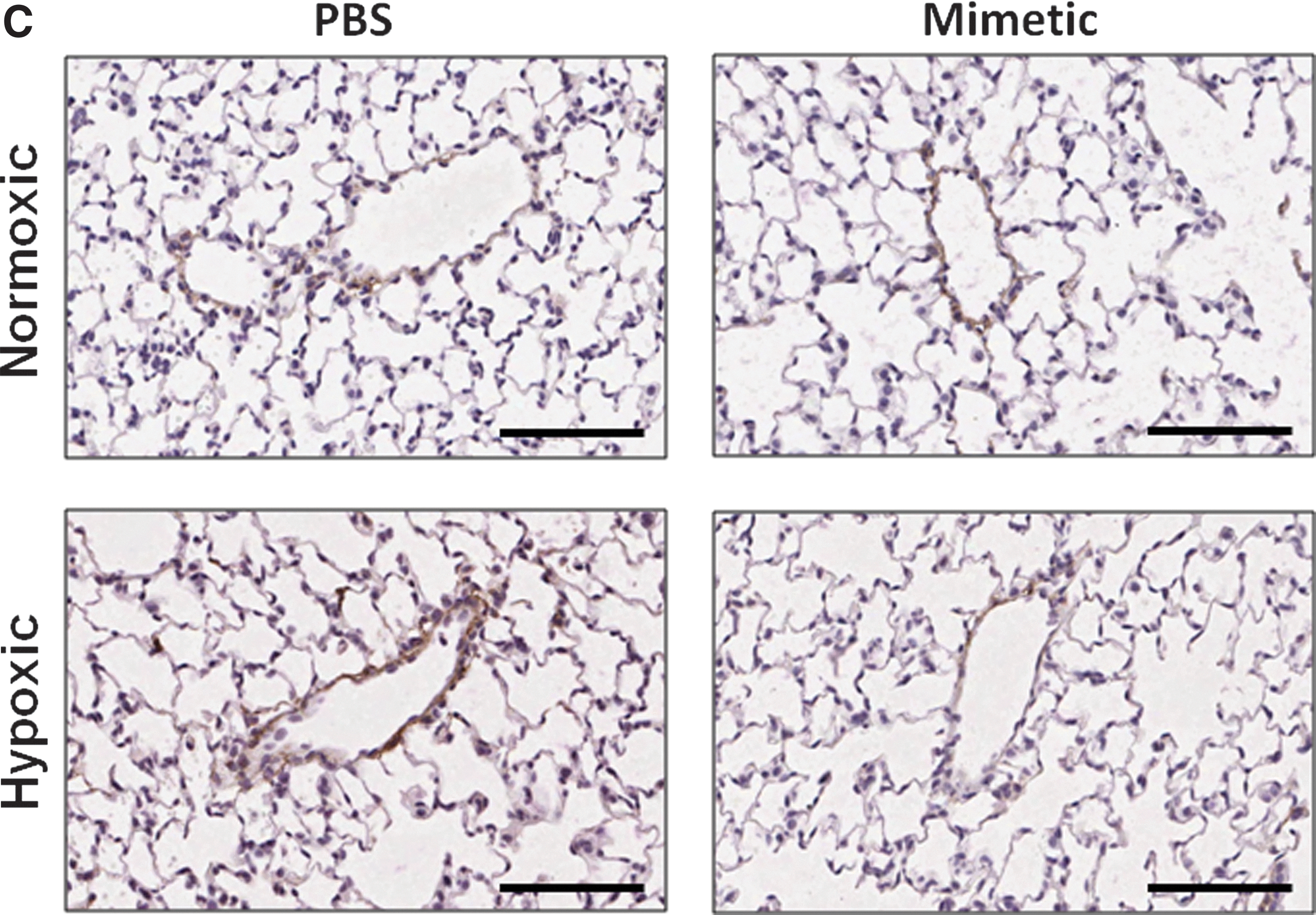
Oxidative stress in the lung can cause deposition and fragmentation of several components of the ECM, contributing to matrix remodeling (13, 28, 32, 49). Since HA content and activity is dependent upon its oxidative fragmentation, its synthesis and degradation, and its main target receptor, CD44, we measured HA content and transcript expression of the HA receptor, CD44 (Fig. 3A), HA synthases (HAS1 and HAS2), and hyaluronidases (Hyal1 and Hyal2) (Fig. 3B). We observed time-dependent changes in the PBS-treated hypoxic mice in each of these measurements, while none of these parameters changed at any time point in hypoxic mice treated with the SOD mimetic. In PBS-treated groups, hypoxia caused an initial decrease in HA content followed by increased content after longer hypoxic exposure. Hypoxia also caused an initial increase in CD44 and hyaluronidase expression followed by decreased expression after longer hypoxic exposures. In SOD mimetic-treated groups, there were no significant changes in HA content or gene expression upon hypoxic exposure, except for decreased HAS1 transcript. Visualization of HA content in lung sections showed that HA was localized within the pulmonary arteries of normoxic and hypoxic mice (Fig. 3C).

SOD mimetic attenuates hypoxia-induced NALP3 inflammasome activation
We tested the impact of the SOD mimetic on a recently recognized inflammatory pathway, the NALP3 inflammasome-dependent activation of IL-1β and IL-18.
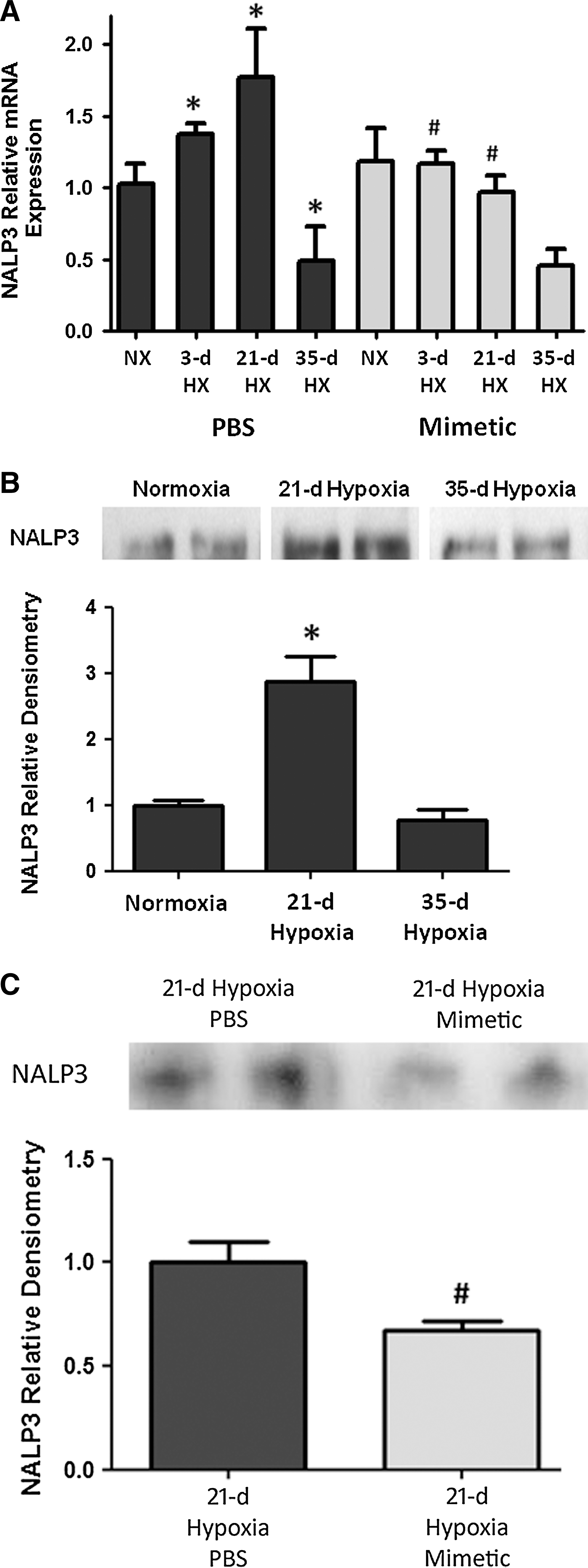
NALP3 expression
In whole lung homogenates, there was increased NALP3 transcript expression as early as after a 3-day hypoxic exposure, with a further increase at 21 days of hypoxic exposure, followed by a subsequent decline in expression by 35 days of hypoxic exposure (Fig. 4A). In hypoxia-exposed mice that were treated with SOD mimetic, there was up to two-fold attenuation of NALP3 transcript expression. Accordingly, immunoprecipitation analysis of NALP3 protein showed a significant increase after 21 days of hypoxia (Fig. 4B). Comparison of the NALP3 protein at the 21-day hypoxic time point showed less NALP3 expression in SOD mimetic-treated mice (Fig. 4C). Protein expression at the 35-day hypoxic time point was also analyzed, but no differences between the groups were seen (data not shown). However, despite differences in NALP3 expression only noted up to 21 days, differences in bound caspase-1 protein and IL-1β and IL-18 were seen at the 35-day hypoxic time point.
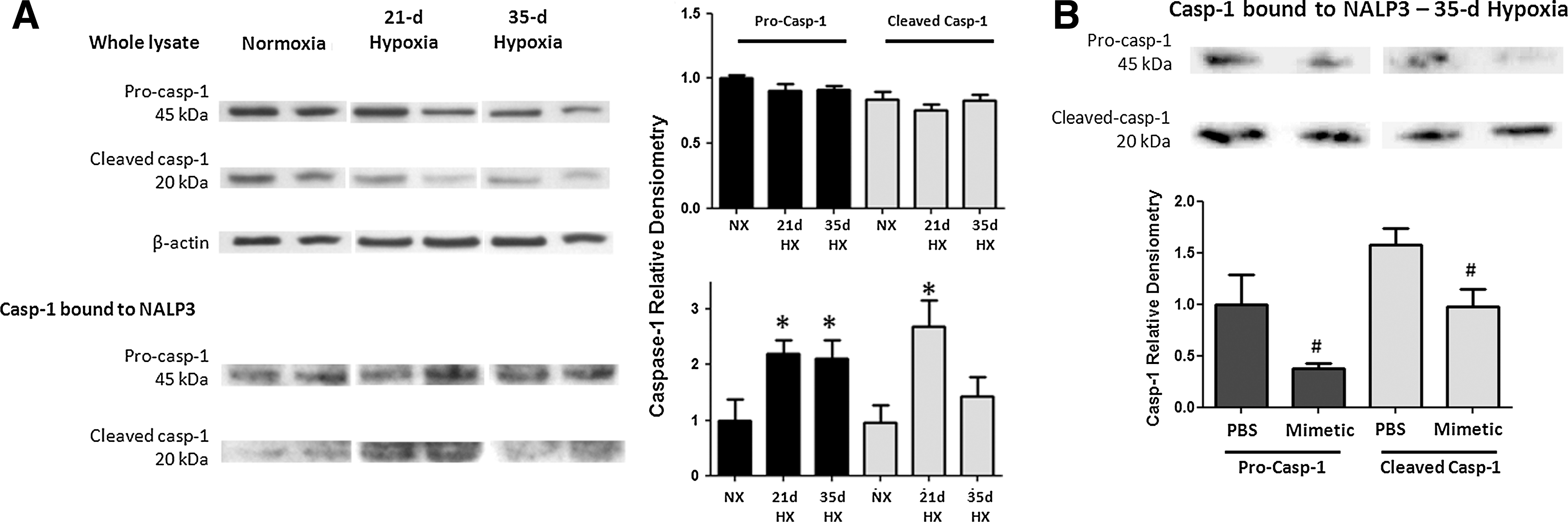
Caspase-1, IL-1β, and IL-18
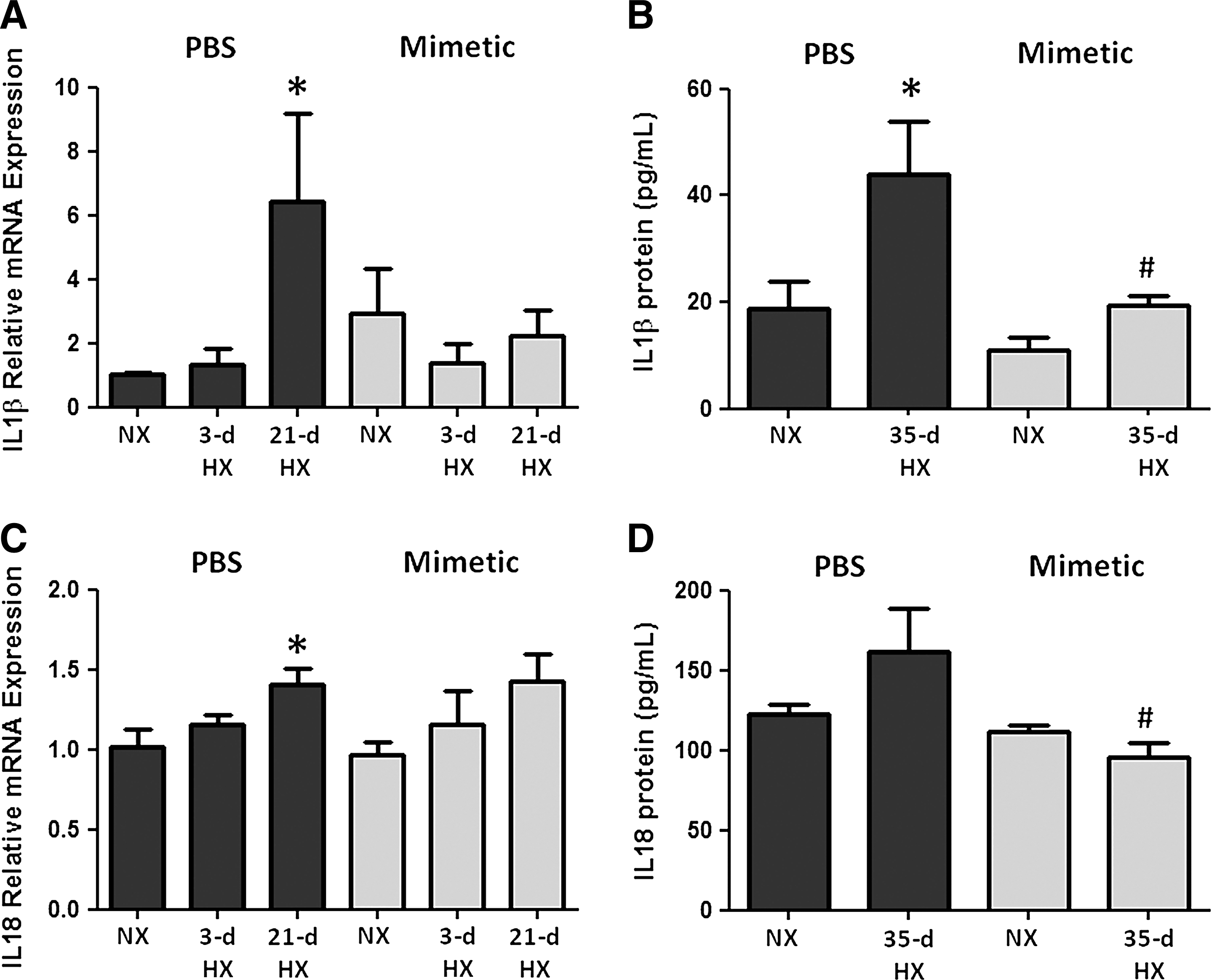
To assess the activation of caspase-1 in this pathway, we quantified pro- and cleaved caspase-1 bound to the immunoprecipitated NALP3. Caspase-1 protein (either pro- or cleaved) was minimally detected in NALP3 immunoprecipitates from normoxic mice. Following 21 and 35 days of chronic hypoxia, pro-caspase-1 and cleaved caspase-1 expression in mouse lung was increased significantly (Fig. 5A). There was an attenuation of both forms of caspase-1 expression in hypoxic mice treated with SOD mimetic compared to PBS controls (Fig. 5B). Western blots of tissue lysates without immunoprecipitation showed no changes upon hypoxic exposure, further supporting the concept that NALP3 activation involves the binding of caspase-1 (Fig. 5A). Hypoxia increased IL-1β and IL-18 transcript expression, whereas SOD mimetic treatments attenuated IL-1β expression but not IL-18 expression (Fig. 6A, B). Caspase-1, IL-1β, and IL-18 protein expression after a 21-day hypoxic exposure did not significantly increase (data not shown). However, after a 35-day chronic hypoxic exposure, there was increased IL-1β and a trend toward increased IL-18 protein. Importantly, treatment with the SOD mimetic in 35-day hypoxia-exposed mice significantly decreased both IL-1β and IL-18 production compared to the 35-day hypoxic PBS-treated group.
Discussion
This study demonstrated that chronic hypoxic PH was attenuated in mice treated with MnTE-2-PyP. This is of importance because MnTE-2-PyP has been shown to target both intracellular and extracellular superoxide (43, 49) in contrast with other tested antioxidant strategies, including tempol and allopurinol (9, 21, 38, 39). We are particularly interested in its role as an extracellular superoxide scavenger because of compounding evidence that support the critical role of extracellular oxidative stress and EC-SOD in oxidative pulmonary injury. EC-SOD is the major antioxidant enzyme in blood vessels, and recent studies have shown that chronic hypoxia causes decreased EC-SOD expression and activity in the lung (16, 32). Loss of EC-SOD contributes to the pathogenesis of PH, which provided the rationale to test antioxidant strategies that restore extracellular SOD activity in the vasculature (47). In addition, increased oxidative stress in mouse lungs is protected either by overexpression of lung EC-SOD (32) or adenoviral administration (6, 24) in chronic hypoxic or monocrotaline induced PH models. SOD mimetics are protective in other models of lung injury that are also associated with reduced EC-SOD expression (3, 11, 15). Our studies demonstrated that the extracellular properties of MnTE-2-PyP contribute to its protective effects in hypoxia-induced oxidative stress in pulmonary arteries, which lead us to further investigate two novel extracellular targets, HA and the NALP3 inflammasome.
Since this mimetic is a positive charged antioxidant similar to EC-SOD that scavenges superoxide and reverses diseases characterized by insufficient EC-SOD, it has been most commonly described as an EC-SOD mimetic. Although the activity has been shown to predominantly be SOD activity, the mimetic also has a low amount of catalase activity, scavenging hydrogen peroxide generated after the dismutase of superoxide. The catalase activity can be attributed to the ability of the Mn moiety to undergo higher oxidation states, such as Mn IV or Mn V, and thus converting hydrogen peroxide into water and oxygen. The advantages of this mimetic are its higher efficacy to dismutate superoxide (log kcat=7.76 as compared to log kcat=8.84–9.3 of SOD enzymes), its high efficacy to reduce peroxynitrite, (log kred=7.53), and also scavenge carbonate radical. The ability to eliminate superoxide, peroxynitrite, and carbonate radical exceeds that of other compounds presently in preclinical and clinical studies such as Mn salen derivatives, Mn cyclic polyamines, nitrones, nitroxides, MitoQ, and similar compounds (36). One of our hypotheses focused on protection of extracellular remodeling that is linked to inflammation, via HA- and CD44-mediated activation of the NALP3 inflammasome.
MnTE-2-PyP protects against both early and late processes indicative of hypoxia-induced vascular remodeling, showing by its attenuation of early vessel wall cell proliferation and late small vessel muscularization. These findings support the premise that the extracellular compartment plays a critical role in vascular remodeling. One key finding demonstrated that HA, an important ECM component in the lung vasculature, was modulated in response to hypoxic exposure, and was suppressed by treatment with MnTE-2-PyP. In normal and healthy conditions, HA exists as intact, large-molecular-weight glycosaminoglycan polymers in the vascular adventitia. Upon lung injury, fragmented HA can be found in other compartments such as the bronchoalveolar lavage fluid and serum of patients with idiopathic pulmonary arterial hypertension (1, 35). In bleomycin lung injury, a model of PH, mice overexpressing EC-SOD had attenuated release of pulmonary HA into the bronchoalveolar fluid (50). HA content is regulated within a more intricate system, involving HA synthases and hyaluronidases. Published studies show that isolated IPAH pulmonary artery smooth muscle cells (PASMC) contained more HA in the culture supernatants compared to control donors and that HA content is associated with increased hyaluronan synthase (Has1) and decreased hyaluronidase (Hyal1) gene expression in the PASMC (1, 35). Fragmented HA can have a number of actions through different macrophage receptors that ultimately increase NALP3 inflammasome activation and interleukin release. Fragmented HA binds to the CD44 receptor to activate NALP3 inflammasome; the Rhamm receptor on macrophages to promote macrophage recruitment; and TLR4 to increase NF-kB and IL gene expression (30). Our data suggest that hypoxia caused initial degradation of intact HA, while HA synthesis may have begun in response to longer hypoxic exposure. In addition, our data show that the HA receptor, CD44, which binds fragmented HA and can activate the NALP3 inflammasome, is also initially upregulated. HA content or CD44 expression in the SOD mimetic-treated groups did not change upon hypoxic exposure, suggesting that antioxidant treatment may stabilize HA modulation. MnTE-2-PyP may impact other extracellular signals or intracellular ROS that regulate NALP3 inflammasome activation independent of HA and we expect that these effects could be modulated by other antioxidants as well. Further studies are needed to elucidate the role of HA production, degradation, and source of HA fragments in chronic hypoxic PH. Regardless, the overall modulation of HA content in the lung is an important contributing factor because it is known to be involved in the activation of inflammatory pathways such as the NALP3-mediated production of IL-1β and IL-18.
MnTE-2-PyP attenuates IL-1β and IL-18 production, important lung cytokines that perpetuate inflammation, oxidative stress, and the development of PH. Studies have shown that IL-1β plays a role within a complex inflammatory environment in the pulmonary vascular remodeling process that involves inflammatory cell recruitment, proliferation of smooth muscle and endothelial cells, and mitochondrial and ion channel dysregulation (4, 18). Although other models show that early inflammation markers (46) precede hypoxia-induced PH, interestingly, our studies show that IL-1β and IL-18 are activated at a later time point. Changes in IL-1β transcript expression and protective effects of MnTE-2-PyP may be attributed to direct activation by extracellular ROS (31), or by a HA-TLR4 receptor signaling pathway, via the HA receptor CD44 and assembly of the NALP3 inflammasome (48). The NALP3 inflammasome is a component of the innate immune system and is responsible for caspase-1 dependent activation and secretion of IL-1β and IL-18 (2, 10, 31, 33, 45). Our data show that hypoxia induces caspase-1-mediated activation of the NALP3 inflammasome and subsequent production of IL-1β and IL-18, and that this response was attenuated by MnTE-2-PyP. Critical mechanisms involved in the pathogenesis of pulmonary vascular remodeling include modulation of the ECM and inflammation, two processes that are linked to oxidative stress in the lung. Because small molecular weight HA can act as endogenous signals to also activate the NALP3 inflammasome, our studies provide a rationale for further investigation into the contribution of extracellular HA as an activator of the NALP3 inflammasome in CHPH models. It may also inhibit other inflammatory pathways. For example, it has been shown that the SOD mimetic alters NF-kB by oxidizing one of the subunits and inhibiting its ability to bind to DNA. It has also been shown that the SOD mimetic inhibits AP-1 and HiF-1alpha, thus inhibiting inflammatory pathways directly. This study has provided a foundation to study pathways by which antioxidant replacement can impact inflammation, in this case, NALP3-mediated inflammation in CHPH, and further studies are needed to fully characterize this pathway.
In summary, our studies demonstrate that the SOD mimetic, MnTE-2-PyP, has a protective role in hypoxia-induced inflammation, pulmonary vascular remodeling, and development of PH. We have identified potential novel targets of oxidative stress in chronic hypoxia in the extracellular compartment that are modulated by MnTE-2-PyP. Future studies will evaluate the mechanisms inducing HA regulation and inflammasome activation, and provide a foundation to consider the therapeutic use of catalytic antioxidants for hypoxia-associated lung diseases.
Materials and Methods
Hypoxic mouse model and SOD mimetic treatment
Studies were performed on 4-week-old, C57/BL6 male mice (Jackson Lab, Bar Harbor, ME) maintained in normobaric normoxia or hypobaric hypoxia for up to 35 days. The hypoxia exposures were performed in hypobaric chambers at a simulated altitude of 18,000 ft above sea level (395 torr), conditions equivalent to 10% atmospheric oxygen. Normobaric conditions were at ∼1500 ft above sea level (Denver, CO). Subcutaneous injections of phosphate buffered saline (PBS) or 5 mg/kg of SOD mimetic (MnTE-2-Pyp) dissolved in PBS were given three times weekly beginning on day 1 of hypoxic exposure or under normoxic conditions. Hemodynamic measurements and lung tissue were taken at the 3-, 21-, and 35-day time points according to the protocols as described below. Animal studies were approved by the Institutional Animal Care and Use Committee.
Assessment of PH
Mice were anesthetized by inhaled isofluorane (2%–4%) mixed with room air (21% oxygen, 79% nitrogen). RVSP was measured by direct RV puncture in a closed chest. A 25-gauge needle attached to a pressure transducer was introduced into the RV and live pressure tracings were measured using the Cardiomax III Cardiac Output program (Columbus Instruments). Pressures were monitored for at least 30 s, and averaged every 10 s to account for beat-to-beat variability. The blood was then drained from the lungs and heart by flushing 5 ml cold PBS into the right atrium and through the left atrium. The hearts were resected, and the right and left ventricle (including the septum) were separated under a dissecting microscope and then weighed. Right ventricular hypertrophy was quantified by comparing the ratio of the right ventricular/left ventricular+septum weights (RV/LV+S).
Measurement of vascular cell proliferation and muscularization
Lungs were flushed through the pulmonary artery with PBS, and then tissue was inflation fixed, embedded, and sectioned for immunohistochemistry according to previous methods (32). Briefly, the left lung was inflation-fixed at 20 cm H2O pressure in 4% paraformaldehyde for 30 min and then dissected from the chest cavity and placed in 4% paraformaldehyde at 4°C for 2 days. Lungs were then transferred to 70% ethanol, paraffin-embedded, and sectioned. Lung sections were immunostained with monoclonal Ki67 antibody (1:200 Lab Vision NeoMarkers) or mouse monoclonal α-smooth muscle actin antibody (1:100, Clone 1A4). The block, secondary antibody, and avidin–biotin complex (ABC) reagent were provided in the Mouse-on-Mouse kit (Vector Laboratories) for the mouse monoclonal antibodies. The slides were developed with ImmPact DAB diluent (Vector Laboratories) and counterstained with hematoxylin. Tissue sections were examined by light microscopy and photographed. The number of proliferating Ki67-positive cells within the vessel wall was measured by counting Ki67-positive cells within the intimal and medial layer of muscularized pulmonary arteries between 50 and 150 μm (20× magnification). Ten vessels were counted for each mouse lung. The number of small vessels (<50 μm) with positive α-SMA staining was counted in 10 fields of view (10× magnification) to evaluate muscularized small pulmonary vessels. The analysis of Ki67 and α-SMA staining was evaluated by an investigator blinded to treatment groups.
Detection of HA content in mouse lung
HA content was measured from flushed and flash-frozen right lung tissue, using a competitive binding assay. This assay measures the competition of HA present in the sample verses HA coated on a 96-well plate for binding to a biotinylated HA-binding protein (bHABP Seikagaku). About 60 μL of sample or Healon standard (Pharmacia) was loaded onto nonfat dry milk-blocked Covalink-NH 96-Microwell plates (Nunc, Fisher Corp.) after overnight protease digestion. After addition of 60 μL bHABP to each well and incubation at 37°C for 1 h, 100 μL of the sample-bHABP incubation solution was transferred to a HA-coated Covalink plate and incubated for 1 h at 37°C to allow to competitive binding (0.2 mg/ml HA, ICN, Inc.). HA-binding was detected by an ABC reagent (Vectastain) and o-phenylenediamine (Sigma). The change in absorbance at 450 nm after a 15-min incubation was measured.
To visualize HA content around pulmonary vessels, inflation fixed, and embedded lungs were sectioned and stained. Lung sections were deparaffinized with citrisolve and rehydrated in graded ethanol. Negative controls were treated with hyaluronidase (Sigma) at a concentration of 1.5 μg/ml in sodium acetate buffer for 2 h at 60°C. Slides were blocked with Avidin/Biotin blocking kit (Vector Laboratories) and 0.1% BSA in PBS. Biotinylated HA-binding protein (Calbiochem) was then incubated on the slides overnight at 4°C at a concentration of 25 μg/ml in PBS. Slides were then washed with PBS, treated with 0.3% hydrogen peroxide, and then developed with Elite ABC reagent, Impact DAB, and hematoxylin QS (Vector Laboratories).
NALP3, IL-1β, and IL-18 transcript expression
RNA was isolated from frozen lungs to quantify NALP3, IL-1β, and IL-18 transcript expression by semi-quantitative RT-PCR. RNA was extracted using RNeasy RNA isolation kit (Qiagen). Semi-quantitative PCR was performed using cDNA generated from 1 μg RNA. Enzymes and reagents used were Maxima First Strand cDNA Synthesis kit and Maxima SYBR Green/Fluorescein qPCR Master Mix (Fermentas). The threshold cycle for each sample in triplicate was measured using the Bio-Rad MyiQ™ Real-Time PCR Detection System. The PCR conditions used were 1 cycle at 95°C for 3 min and 60 cycles at 95°C for 30 s, 56°C for 30 s, and 72°C for 30 s. All transcripts were normalized to the endogenous control transcript, hypoxanthine phosphoribosyltransferase 1 (HPRT). Values are reported as transcript levels relative to the normoxic PBS group. The following primer sets were used: mNALP3 forward 5′-GGTTGGTGAGCTGCTGTCTCACATC-3′, mNALP3 reverse 5′-CTGTGTCTCCAAGGGCATTGCTTC-3′; mIL1β forward 5′-CTCCACCTCAATGGACAGAATATCAACC-3′, mIL1β reverse 5′-GGTGGGTGTGCCGTCTTTCATTAC-3′; mIL18 forward 5′-GGACTGGCTGTGACCCTCTCTGTG-3′, mIL18 reverse 5′-CAAACTCCATCTTGTTGTGTCCTGGAAC-3′; mHPRT forward 5′-TGCTCGAGATGTCATGAAGGAG-3′, mHPRT reverse 5′-TTTAATGTAATCCAGCAGGTCAGC-3′.
NALP3, Caspase-1, IL-1β, and IL-18 protein expression
Protein was isolated from frozen lungs to quantify NALP3, Caspase-1, IL-1β, and IL-18 expression by immunoprecipitation, Western blot, and ELISA. Protein was extracted by homogenizing lung samples in RIPA buffer with protease inhibitors (Sigma) for immunoprecipitation and Western blot or in T-PER buffer with protease inhibitors (Pierce Biotechnology) for ELISA assays. For Western blot analysis, 30 μg total protein was loaded onto the gel. For immunoprecipitation, 200 μg total protein was immunoprecipitated using 2 μg NLRP3/NALP3 mouse IgG2b (Enzo Life Sciences) and goat anti-mouse IgG Magnabind beads (Pierce Biotechnology). Samples were applied to a magnet to separate protein bound to beads, and then washed eight times. After the last wash, protein was eluted using NuPage LDS sample buffer and reducing agent (Invitrogen). Samples were loaded on a 4%–12% Bis-Tris Gel and separated by electrophoresis (NuPAGE MES SDS Running Buffer; Invitrogen) and then transferred to a polyvinylidene difluoride membrane using the semi-dry method (NuPAGE Transfer Buffer; Invitrogen). The membranes were blocked for 1 h at room temperature with 5% milk in TBST buffer (0.05% Tween 20) and then probed with primary antibody, NLRP3/NALP3 mouse IgG2b (1:500 dilution; Enzo Life Sciences), or Anti-Caspase-1/ICE rabbit polyclonal (1:1000 dilution; Invitrogen), overnight at 4°C. Membranes were then incubated in HRP-conjugated secondary antibody (1:10,000 dilution), goat anti-mouse IgG, or goat anti-rabbit IgG (Millipore), for 1 h at room temperature. Blots were developed using SuperSignal West Femto Chemiluminescent Substrate (Thermo Scientific) and exposure to film. Band intensities were quantified by densiometry. For immunoprecipitated samples, band intensities were normalized to total protein of each sample (200 μg). For nonimmunoprecipitated samples, band intensities were normalized to β-actin. Densitometric analysis was reported as values relative to the normoxic or PBS control groups.
For ELISA assays, 100 μg total protein was loaded onto IL-1β- or IL-18-coated 96-well plates (eBioscience) and processed according to manufacturer's protocol. The absorbance was read at 450 nm. The concentration of each sample was extrapolated from mouse IL-1β or IL-18 recombinant protein standard curve.
Statistical analysis
For animal studies, we used n=4 to 6 for each group. Assays were performed at least in triplicates to generate the mean and the standard error of the mean. The data comparing multiple groups were first analyzed by ANOVA, followed by Bonferonni post-test and Student's two-group t-test to evaluate differences between means of each group. p<0.05 defines statistically significant differences.
Footnotes
Acknowledgments
We would like to thank James D. Crapo for helpful discussions and for supplying the SOD mimetic, MnTE-2-PyP. We would also like to thank Hagir B. Suliman and Kurt R. Stenmark for constructive feedback, and Jie Liao for excellent technical assistance. This work was funded by NIH grant R01-HL086680 to E.N.G., as well as R01-HL093535 and a grant from The Children's Medical Center Foundation to R.C.S. L.R.V. was supported by institutional training grant T32-HL007171.
Author Disclosure Statement
No competing financial interests exist.