
Abstract
Introduction
Silent mating-type information regulation 2 homolog (sirtuin 1 or SIRT1) is an NAD+-dependent class III histone deacetylase that is upregulated under caloric restriction (CR), extending the lifespan of many organisms (60,128,147). Activating via deacetylation of a myriad of stress-responsive transcription factors (TFs), coregulators, and enzymes, SIRT1 plays a key role in metabolic control at cellular, tissue, and whole-body levels (133). Because of its profound antioxidative and anti-inflammatory effects, a growing body of work focuses on SIRT1 in EC biology. In general, elevation of SIRT1 expression and/or activation exerts beneficial effects on ECs. In contrast, stress conditions such as excessive ROS and aging decrease SIRT1 expression/activity, which leads to endothelial dysfunction (114).
MicroRNAs (miRNAs), ranging from 18 to 24 nucleotides in length, belong to a class of noncoding small RNAs. The precursors of miRNAs, that is, primary transcripts (pri-miR), are induced transcriptionally in response to environmental stimuli in a manner similar to mRNA. miRNAs generally downregulate gene expression by binding to the complementary sequences in their mRNA targets at the 3′-untranslated region (3′UTR). As of yet, over 1500 human miRNAs have been reported (
As summarized in Table 1, alterations in miRNAs, SIRT1, and its regulators have been demonstrated in multiple conditions that involve oxidative stress and predispose to cardiovascular diseases. This review focuses on the interplay between miRNAs and SIRT1 in the context of oxidative stress-induced endothelial dysfunction, the pertinent approaches for experimental validation, and the translational implications of this supposition.
These miRNAs have been identified as H2O2-inducible miRNAs in vitro.
↑: upregulation;↓: downregulated; (+): activation; and (−): inactivation; SIRT1, sirtuin 1; miRNAs, microRNAs; AGEs, advanced glycation endproducts; PKC, protein kinase C; NOX, NAD(P)H oxidase; PARP, poly-ADP ribose polymerase; AMPK, AMP-activated protein kinase; STZ, streptozotocin; GK, Goto-Kakizaki; LDL, low-density lipoprotein; HFD, high-fat diet; AT1R, angiotensin II type 1 receptor; NAMPT, nicotinamide phosphoribosyl transferase; H2O2, hydrogen peroxide.
Oxidative Stress and SIRT1 in ECs
SIRT1 is highly sensitive to the cellular redox states (14). In ECs, SIRT1 activation can alleviate oxidative stress, prevent endothelial senescence, increase eNOS-derived NO bioavailability, and promote mitochondrial biogenesis (3,21,97,176). Transgenic mice with EC-specific SIRT1 overexpression on an ApoE −/− background ameliorates atherosclerosis, demonstrating the atheroprotective capacity of SIRT1 (170). Many of the effects of SIRT1 are due, in part, to its deacetylation of multiple targets, including eNOS, peroxisome proliferator-activated receptor-γ coactivator 1-α (PGC1α), p53, Forkhead Box O family (FoxO), and NF-κB (15,91,97,127,154,167) (Fig. 1). Through the deacetylation of eNOS at Lys-496 and Lys-506, SIRT1 increases eNOS activity and hence contributes to endothelial homeostasis (97). The atheroprotective shear stress induces SIRT1 in ECs. In concert with AMP-activated protein kinase (AMPK) phosphorylation of eNOS, SIRT1 deacetylation of eNOS further enhances NO bioavailability (21). SIRT1 expression is upregulated in vivo in response to CR or resveratrol, an SIRT1 activator, and this is correlated with enhanced eNOS activity and reduced oxidative stress (103,125).
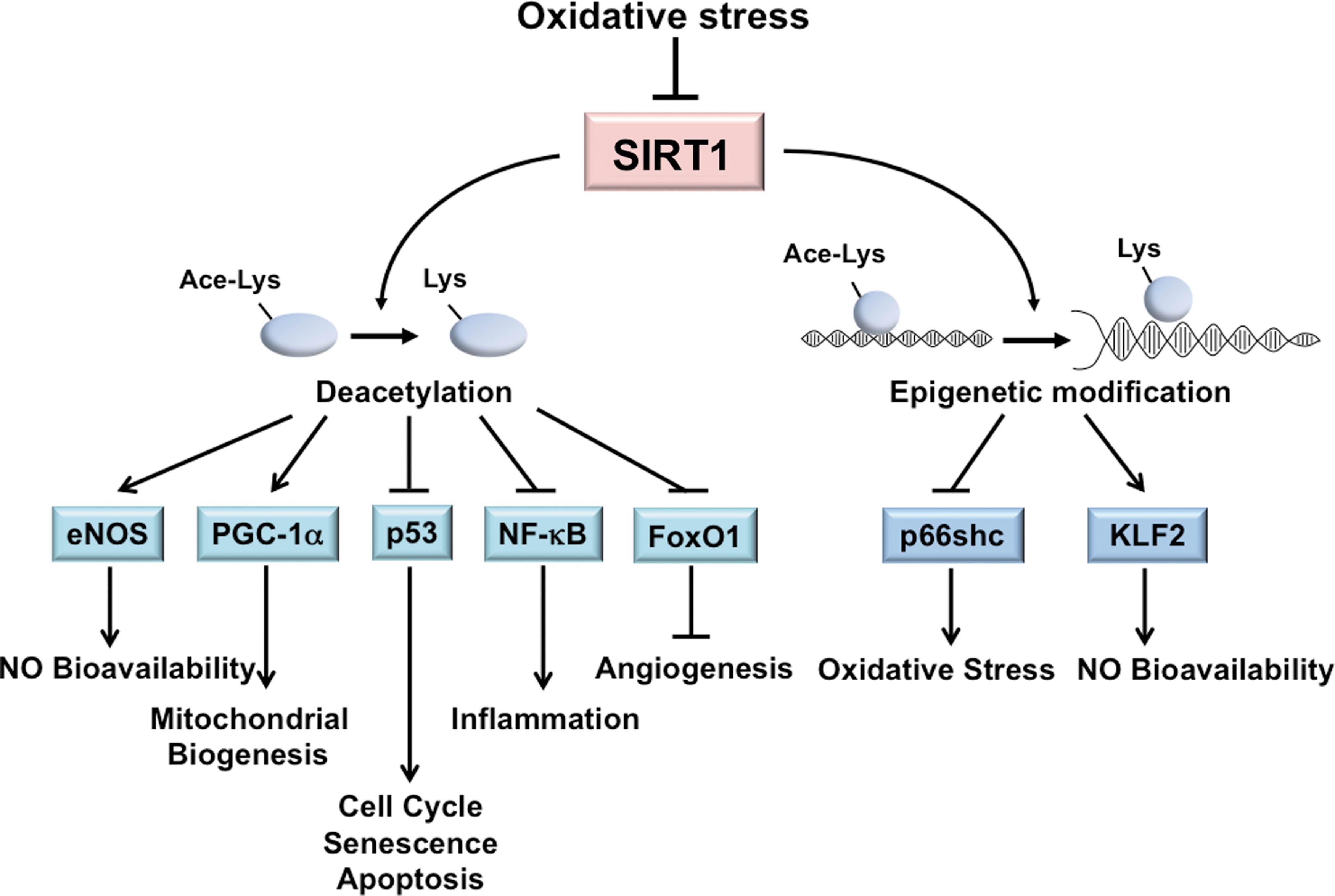
PGC1α is a peroxisome proliferator-activated receptor-γ transcriptional coactivator and functions as a metabolic regulator. SIRT1 deacetylation of PGC1α is linked to mitochondrial biogenesis, which is essential for the maintenance of cellular redox homeostasis (99). PGC1α increases the level of mtDNA and upregulates many antioxidant enzymes, including manganese superoxide dismutase (MnSOD) and glutathione peroxidase 1, through PGC1α modulation of nuclear respiratory factors and mitochondrial transcription factor A (53,161). In ECs, shear stress-induced SIRT1 causes PGC1α induction and activation, and a concomitant increase in mitochondrial biogenesis (21). Additionally, SIRT1 induction by resveratrol or sera from CR rats significantly reduces mitochondrial ROS levels in cultured ECs (26,150).
SIRT1 can also regulate the cellular redox state through deacetylation of nuclear factor erythroid 2-related factor 2 (Nrf2), a master TF in the cellular response to oxidative stress. Under oxidative stress, Nrf2 binds to the antioxidant-responsive elements (AREs) in the promoter regions of many antioxidative genes, thus transactivating their expression (49,57). Resveratrol activates Nrf2 in cultured human coronary arterial ECs, which is associated with the upregulation of several Nrf2 target genes such as NAD(P)H:quinone oxidoreductase 1, γ-glutamylcysteine synthetase, and heme oxygenase-1 (149). However, SIRT1 probably deacetylates Nrf2 at Lys-588 and Lys-591 to promote its translocation from the nucleus to the cytoplasm in HepG2 cells. Such modification results in decreased binding of Nrf2 to AREs and therefore the inhibition of the expression of antioxidative genes. This regulation is to counteract the acetylation of Nrf2 by cAMP response element-binding protein (CREB)-binding protein (CBP), which results in nuclear retention of Nrf2 and its transactivation of the ARE-containing genes (63). Clearly, differential regulation of Nrf2 by SIRT1 in ECs warrants further examination.
SIRT1 activity also antagonizes the oxidative stress-induced senescence of ECs, largely through its negative modulation of p53 by deacetylation of Lys-373, Lys-382, and Lys-320. Such a response counteracts the effects of p300 and p300/CBP-associated factor (PCAF), which stabilizes p53 to promote cell cycle progression, senescence, and apoptosis (91,110). In line with this negative regulation of p53, the level of SIRT1 is related to premature senescence of human umbilical vein endothelial cells (110). The protective effects of SIRT1 on ECs can also be conferred by its deacetylation of NF-κB and FoxOs. SIRT1 deacetylates RelA/p65-NF-κB at Lys-310 and suppresses the NF-κB-mediated inflammatory and apoptotic processes (167). By deacetylating FoxO1 or FoxO3, SIRT1 increases the cell resistance to oxidative stress and promotes angiogenesis (15).
Because hypoxia affects the cellular redox state, SIRT1 is also likely to be modulated by changes in the NAD+/NADH ratio. Dioum et al. (31) demonstrated that SIRT1 activated by hypoxia deacetylates hypoxia-inducible factor (HIF)-2α to transactivate erythropoietin, an HIF-2α target gene. This is preceded by acetylation of HIF-2α by CBP, both of which are required for the maximal HIF-2 signaling under hypoxia (20). Conversely, HIF-1α has been shown to be inhibited by SIRT1 deacetylation (81). Because SIRT1 is downregulated under hypoxia (169), HIF-1α acetylation by PCAF should be enhanced and thus augmenting the transactivation of the HIF-1α target genes. These findings suggest a negative role of SIRT1 in tumor growth and angiogenesis. Although SIRT1 is an essential player in hypoxic response, the molecular basis for SIRT1 regulation of HIF-1α or HIF-2α remains to be determined.
Excessive oxidative stress may also cause the endothelial-to-mesenchymal transformation (EnMT), a process involved in cardiovascular development as well as in pathophysiological conditions such as pulmonary hypertension (4). Transforming growth factor-β (TGF-β) is a potent inducer of EnMT and can convert ECs to spindle-shaped α-SM actin-expressing cells. SIRT1 deacetylates Smad7 to causes its proteosomal degradation and thereby inhibit TGF-β signaling (72). Thus, SIRT1 may maintain the endothelial lineage and prevent the TGF-β-induced EnMT.
In addition to post-translational modification, SIRT1 can regulate several key molecules modulating endothelial homeostasis through transcriptional regulation. SIRT1 suppresses the transcription of the prooxidant molecule p66shc, which protects ECs from hyperglycemia-imposed oxidative damage (173). Moreover, resveratrol increases Krüppel-like factor 2 (KLF2), a critical transcriptional factor controlling eNOS expression in a SIRT1-dependent manner (44). Together, the critical role of SIRT1 in the regulation of endothelial homeostasis is manifested by its comprehensive regulation of molecules involved in the redox and inflammatory state and the endothelial phenotype. Given that SIRT1 can be regulated by miRNAs under oxidative stress, these oxidative stress-responsive miRNAs are likely to constitute the crosstalk between SIRT1 and its downstream target genes as well as between the deacetylation targets of SIRT1. For example, hypoxia-induced HIF-1 attenuates Nrf2-dependent transactivation in ECs (88). It is possible that oxidative stress-responsive miRNAs may regulate Nrf2 through SIRT1 and HIF-1.
Oxidative Stress Regulates SIRT1 Through miRNAs
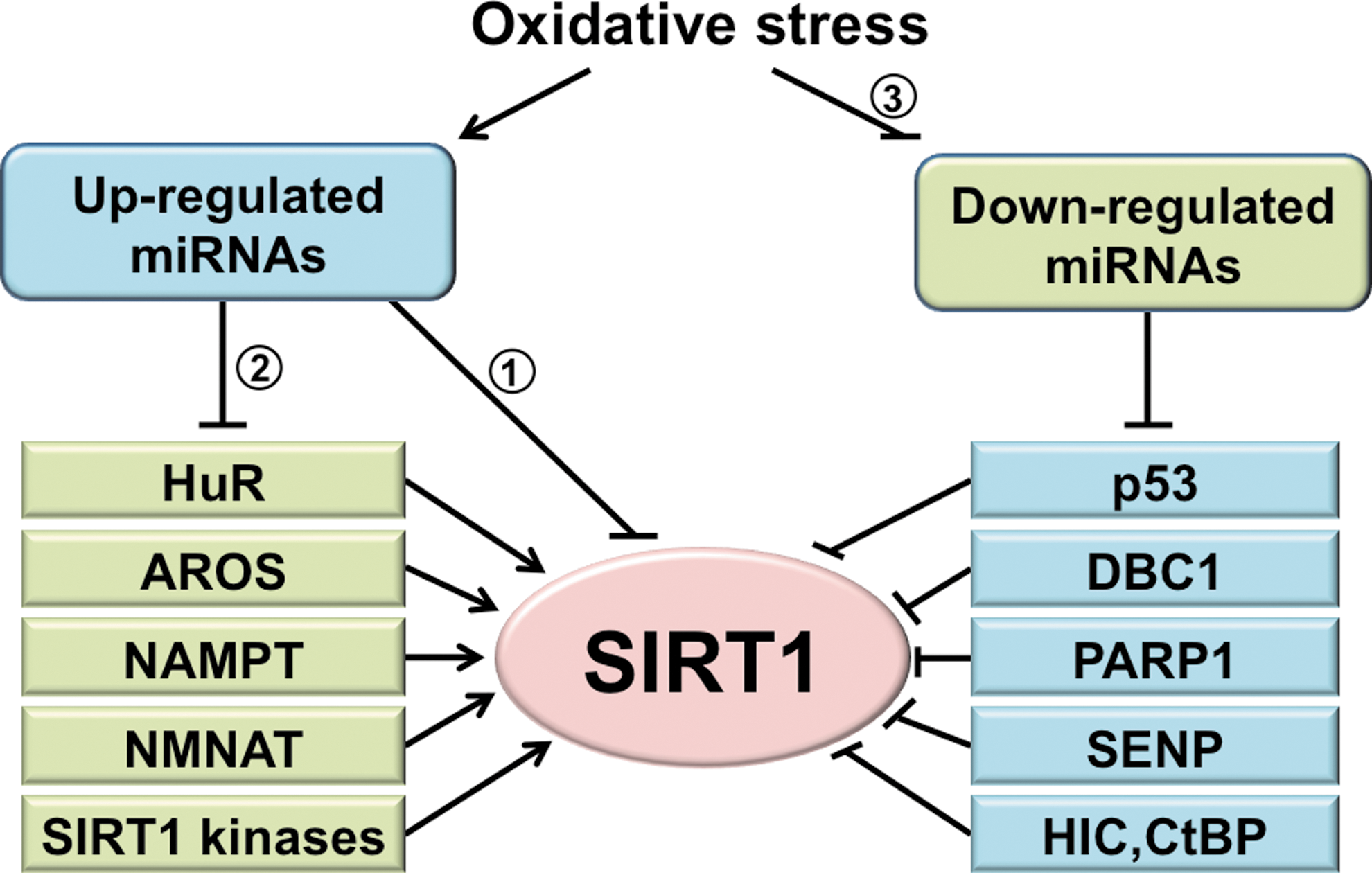
Oxidative stress regulates a panel of miRNAs (95,136). Some of these miRNAs have been predicted in silico (computer-assisted bioinformatics analysis) and/or experimentally determined to target the 3′UTR of SIRT1 mRNA. In parallel to the direct regulation, miRNAs may suppress SIRT1 indirectly through the actions on SIRT1 regulators (Fig. 2). This section first focuses on miRNAs that directly target SIRT1 and then on those that indirectly regulate it. Given the unique characteristics of miRNAs, a research strategy that integrates bioinformatics predictions and experimental validation is discussed as well. Finally, because SIRT1 can regulate miRNAs through the modulation of their upstream TFs, the mechanisms by which SIRT1 regulates miRNAs under oxidative stress are discussed.
Direct targeting of SIRT1 by oxidative stress-induced miRNAs
Based on the complementary sequences of SIRT1 3′UTR, a panel of miRNAs that target SIRT1 under various physiological and pathophysiological conditions was predicted and experimentally validated. The first reported miRNA to downregulate SIRT1 during the senescence of human cell lines was miR-34a (165). The targeting SIRT1 by miR-34a was also observed in ECs and endothelial progenitor cells (EPCs) (58,171). Similarly, miR-217 was identified as an endogenous SIRT1 inhibitor, which promotes endothelial senescence (99). Several other miRNAs, including miR-181, miR-138, and miR-199, downregulate SIRT1 in various cell/tissue types. MicroRNA-181 is upregulated by a high-fat diet in the liver, resulting in decreased SIRT1 and impaired insulin sensitivity (172). Both miR-181 and miR-138 levels increase in senescent keratinocytes, and their targeting SIRT1 promotes senescence (126). In contrast, miR-199 is downregulated in hypoxic preconditioning, leading to SIRT1 desuppression and decreased apoptosis in cardiomyocytes (119). Table 2 summarizes the experimentally validated miRNAs that negatively regulate SIRT1 expression via 3′UTR targeting as well as experimental approaches employed in their validation. Although there are numerous studies on miRNA regulation of SIRT1, few studies correlated the oxidative stress-responsive miRNAs and their regulation of SIRT1 with functional consequences. Because oxidative stress is a primary cause of EC dysfunction, it is conceivable that the oxidative stress-induced miRNAs downregulate SIRT1 with dysregulated EC functions and phenotypes.
↑: miR upregulated and↓: miR downregulated by the stimuli/model; R, reporter assay; O, overexpression; I, inhibition; TF, transcription factor; ESC, embryonic stem cell; mESC, murine ESC; EC, endothelial cell; EPC, endothelial progenitor cell; p53, tumor protein 53; FXR, Farnesoid X receptor; p63, transformation-related protein 63; ROS, reactive oxygen species; MSC, mesenchymal stem cell.
Identification of SIRT1-targeting miRNAs under oxidative stress
Bioinformatics algorithms have emerged as powerful tools to predict miRNAs that target SIRT1. For example, miR-92, a key regulator in neovascularization, was predicted to target SIRT1 mRNA (12). Such information is available in online databases, including

GEO, Gene Expression Omnibus; NGS, next-generation sequencing; TFBS, transcription factor-binding site; LNA, locked nucleic acid.
The discovery of stimulus-regulated miRNAs can start with miRNA profiles from a high-throughput screening, that is, microarrays or next-generation sequencing. This information can be obtained from published or experimentally derived datasets. Because hydrogen peroxide (H2O2), senescence, high glucose, oscillatory shear stress, and angiotensin II all trigger oxidative stress, miRNAs that are commonly induced by these stimuli are likely to be oxidative stress-responsive miRNAs. Importantly, because H2O2 is the direct agent that elevates cellular oxidative stress, H2O2 treatment offers arguably the most definitive stimuli for any screening approach. For example, miR-200c, miR-21, and miR-146a were identified from H2O2-treated cells and were also elevated in animal models associated with higher oxidative stress (11,82,95). The subsequent prediction of SIRT1 targeting by these miRNAs can be achieved by using the databases mentioned above. Additional considerations can be used to increase the validity of these predictions. These include, but are not limited to, (i) whether the induction of these miRNAs and suppression of SIRT1 are concurrent with a given cellular event, for example, the increased acetylation of SIRT1 targets, cellular senescence, impaired mitochondrial biogenesis, NO bioavailability, and angiogenesis; (ii) the conservation of miRNA-targeted sequences across species; and (iii) whether the TFs that putatively induce these miRNAs also downregulate SIRT1, which is facilitated by the TF-binding site (TFBS) analysis of the miRNA promoters.
Putative miRNA promoter regions can initially be obtained from miRStart (22) and then scanned for TFBSs. TFs such as NF-κB, activator protein 1, CREB1, signal transducer and activator of transcription 3, MYC, and specificity protein 1 are well-characterized TFs that are activated by oxidative stress. Their TFBS in the miRNA promoter regions can be predicted using position weight matrices available in databases such as TRANSFAC (158) and JASPAR (16). The results can be further subjected to University of California, Santa Cruz (UCSC) genome browser (
In vitro and in vivo validation
A multitude of studies demonstrate in vitro and in vivo methods to validate the mechanisms by which miRNAs regulate their mRNA targets and hence produce functional changes. The methods employed to validate SIRT1 targeting by oxidative-responsive miRNAs are listed in Table 3. Several in vitro assays are routinely performed to examine whether SIRT1 is indeed a bona fide target of a given miRNA. Luciferase reporters, in which luciferase cDNA is linked to the SIRT1 3′UTR, can be cotransfected with putative pre-miR (miRNA mimetics) or anti-miR (miRNA inhibitors or antagomirs) to assess the functionality. The direct targeting of miRNA on the complimentary sequences of SIRT1 mRNAs can thus be tested. To confirm the direct targeting through the predicted binding sites, 3′UTR mutation experiments should be performed as a negative control.
To manipulate the levels of miRNAs, gain-of-function approaches, in which miRNAs are elevated by either pre-miR miRNA mimics or viral vectors, can be utilized. Additionally, loss-of-function assays where miRNAs are inhibited by locked nucleic acids (LNAs) or other types of antagomirs can be performed to examine whether the SIRT1 mRNA and/or protein levels change in response to perturbations in the miRNA levels. These experiments should be accompanied by the examination of the functional readouts of SIRT1, for example, the deacetylation of its targets. These gain- and loss-of-function approaches can also be used in vivo with additional considerations of the delivery efficiency and tissue specificity. In parallel to these functional studies, a biochemical approach is frequently used to provide more direct evidence in active miRNA–mRNA targeting. Instead of assessing the overall miRNA or mRNA changes in cells, one can immunoprecipitate miRNA-induced silencing complex components, typically, Ago proteins, and quantify the associated miRNA and mRNA species by using quantitative polymerase chain reaction or a microarray (9,160). With cross-linking immunoprecipitation–sequencing, RNAs can be crosslinked with RNA-binding proteins, and the Ago-bound miRNA–mRNA complexes can be comprehensively identified by subsequent deep sequencing. In addition to the approaches listed above, Thomson et al. have reviewed other useful methods with relevant examples (145).
Indirect inhibition of SIRT1 by oxidative stress-responsive miRNAs
Some of the oxidative stress-responsive miRNAs can modulate SIRT1 through the regulation of SIRT1 upstream regulators without directly targeting SIRT1. Recent advances in systems biology offer promising approaches to identify indirect regulators of SIRT1. For example, the miRNA targets predicted in silico can be categorized and stratified based on the existing information from the literature and on a network-based analysis. The network-based analysis tools include gene annotations (gene ontology) (5), Database for Annotation, Visualization and Integrated Discovery (54), Kyoto Encyclopedia of Genes and Genomes (107), and so on. The analyses of the protein–protein interactions (129,138), kinase–substrate interactions (51), and metabolic interactions (73) can be further applied to generate an interactome map of the target genes. Based on the visualized interactome and its relation to SIRT1, miRNAs and their putative targets can be selected for validation. Notably, miRNAs can target multiple genes within a network. Therefore, miRNAs predicted to target multiple genes that coordinately regulate SIRT1 (including those targeting SIRT1 mRNA) should be candidates for further study. An overview of SIRT1 regulators and their potential regulation by miRNAs is presented in Table 4. Such indirect regulation of SIRT1 by miRNAs is a part of oxidative stress-regulated SIRT1. Finally, although endothelial SIRT1 is most likely regulated by endogenous miRNAs produced by ECs, miRNAs released into the circulation from other tissues/organs; that is, circulating miRNAs might also regulate the SIRT1 network.
AROS, active regulator of SIRT1; NMNAT, nicotinamide mononucleotide adenylyl transferase; HuR, human antigen R; HIC1, hypermethylated in cancer 1; CtBP, carboxyl-terminal binding protein; SENP, sentrin-specific protease; JNK, c-Jun N-terminal kinase; CK2, casein kinase 2; PKA, protein kinase A; DYRK, dual-specificity tyrosine-phosphorylation regulated kinase; CDK1, cyclin-dependent kinase 1.
miRNA regulated by SIRT1 under oxidative stress
As discussed above, SIRT1 modulates many TFs and coactivator/repressors and hence regulates the transcription of their target genes. Thus, SIRT1 may also control the expression of an array of miRNAs that affects the EC biology. This aspect should be integrated into the network of oxidative stress-regulated SIRT1, and is exemplified by a recent study (41). Seeking miRNAs mediating SIRT1 downregulation of CREB in the brain, miRNAs in the mouse brain lacking SIRT1 activity were profiled and demonstrated that SIRT1 modulates synaptic plasticity and memory formation via its suppression of miR-134 by cooperating with YY1, a corepressor of miR-134 transcription. Conversely, loss of SIRT1 activity resulted in miR-134 induction, which in turn downregulated miR-134 targeting CREB and brain-derived neurotrophic factor to impair synaptic plasticity (41). Although there is as yet no supportive evidence, it is possible that miR-134 is oxidative stress-responsive in ECs and hence plays a functional role in oxidative stress-altered endothelial biology. The similar approach can be used to identify the oxidative stress-responsive miRNAs, in which transcription is regulated by SIRT1.
Oxidative Stress-miRNA-SIRT1 Pathway Is Implicated in Vascular Diseases
Oxidative stress and the associated endothelial dysfunction are common features of vascular diseases, including atherosclerosis, hypertension, aneurysm, stroke, and restenosis. Numerous animal and human studies support the hypothesis that oxidative stress is a major cause leading to inflammation and impaired NO bioavailability (112). In the dysfunctional ECs, the major sources of ROS/RNS are NAD(P)H oxidase (NOX), mitochondrial electron leakage, and uncoupled eNOS, which are opposed by antioxidant defense enzymes (29,101). Modulations of these molecules in vivo can alter vascular outcomes. Genetic ablation of p47phox, a key component of NOX, results in lower levels of superoxide with decreased atherosclerotic lesions in ApoE−/− mice (7). Similarly, NO induction decreases atherosclerosis in hypercholesterolemic animals (25). In contrast, ApoE −/− mice deficient in MnSOD, the antioxidant enzyme expressed in the mitochondria, show accelerated atherogenesis at the arterial branch points (6).
The extensive crosstalk between SIRT1 and these ROS-generating molecules reveals that SIRT1 plays a crucial role in maintaining endothelial homeostasis (126). Although their regulation appears to be fine-tuning, miRNAs could be an essential mechanism in SIRT1 regulation, as explored above. A role for the oxidative stress-miRNA-SIRT1 pathways in vascular diseases, particularly, atherosclerosis and abdominal aortic aneurysm (AAA), has been described in recent studies. It is likely that the miRNA regulation of SIRT1 is not limited to the pathobiology of these two diseases. For comprehensive review of miRNAs and cardiovascular diseases, readers are referred to Small et al. (137) and Ono et al. (108).
Oxidative stress is tightly associated with the initiation and progression of atherosclerosis, as well as plaque rupture (92). Many risk factors for atherosclerosis such as hyperglycemia, hypercholesterolemia, and atheroprone flow patterns are associated with enhanced oxidative stress and a decreased SIRT1 level in ECs and/or in the arterial wall (21,170,173). Thus, miRNAs capable of targeting SIRT1 are likely to be involved in the development of atherosclerosis. Among these, miR-34a and miR-217 were found to be upregulated in human atherosclerotic plaques (99,117). Other SIRT1-targeting miRNAs may also play a role in atherogenesis. For example, miR-520c and miR-373 increase matrix metallopeptidase 9 (MMP9) partially through targeting SIRT1 in human fibroscarcoma cells (86). Because MMP9 contributes to atherosclerosis and destabilization of the plaques (89), it is possible that the miR520c/miR373-SIRT1-MMP9 pathway is engaged in atherosclerosis.
AAA is caused by progressive weakening of the aortic wall, creating ballooning of the vessel. Supraphysiological oxidative stress, in concert with inflammatory processes and protease-mediated extracellular matrix (ECM) degradation, contributes to the pathogenesis of AAA (98,163). It is likely that SIRT1 is inhibited during the pathogenesis of AAA, and its impairment exacerbates the formation of aneurysms. Indeed, resveratrol, an SIRT1 activator, prevents AAA in mice, with a preserved ECM structure as well as attenuated inflammation, oxidative stress, and neovascularization (61). Data from a limited number of reports support the role of oxidative stress-responsive miRNA in AAA via SIRT1 targeting. Microarray profiling reveals that 10 miRNAs, including miR-92a and miR-132 (2 miRNAs potentially target SIRT1 in ECs), are upregulated in the aortic tissues of rat AAA (84). Because MMP activation, elastin breakdown, and collagen remodeling are common features of AAA, miRNAs involved in regulating MMP should be crucial in the pathogenesis of AAA. Maegdefessel et al. reported that miR-29b is downregulated in AAA development both in mouse and human tissue along with increased collagen levels (94). The inhibition of miR-29b using LNA reduces AAA progression in mice. In contrast, the overexpression of miR-29b leads to enlarged AAA (94). MiR-21 is also upregulated in AAA. By targeting phosphatase and tensin homolog protein, miR-21 overexpression increases proliferation and decreases apoptosis in the aorta, which are protective against aneurysm expansion. In contrast, systemic administration of LNAs antagonizing miR-21 worsened AAA (93). Due to the close correlation between MMP, vascular remodeling, and SIRT1 (102), the oxidative stress-miRNA-SIRT1 pathway may be involved in AAA pathogenesis. Considering that oxidative stress is a detrimental factor common to vascular dysfunction, it is reasonable to propose that miRNA regulation discussed above may be relevant for the onset of a wide spectrum of vascular diseases.
miRNA Therapeutics for Vascular Diseases
The reduced level of SIRT1 associated with various diseases and the beneficial effects of SIRT1 in alleviating endothelial dysfunction point to a therapeutic potential of SIRT1 activation for vascular diseases (52). However, the canonical SIRT1 activators (polyphenols) are plagued with pharmacokinetic properties (poor absorption and rapid metabolism) that limit their clinical utility (23). The discovery of miRNA opens a new window for the development of novel expression regulators in the form of oligonucleotides that either inhibit or mimic the actions of native miRNAs (14,68). Many reviews have summarized the potential use of miRNA in cardiovascular therapy (96,146,151,152). Here, we would like to make a few comments on these strategies in relation to miRNA in treating vascular disorders with SIRT1 aberration.
Conceptually, miRNAs can be used for intervention or palliation of vascular damage that involve oxidative stress-impaired SIRT1. An advantage of the miRNA strategy is that because miRNAs appear to act on functionally related targets in a coordinated fashion, their regulation is likely to produce pleotropic benefits with a minimum of side effects. Unfortunately, the downside of such large target spaces of miRNAs, that is, the lack of an absolute requirement for complementary target interaction, may enable the off-target effects (manipulating one miRNA may alter thousands of mRNA transcripts). Particularly, targeting some miRNAs may be tumorigenic.
There are additional challenges in the use of oligonucleotides as therapeutic agents. First of all, the size and polyanionic character of potential nucleotide agents limit their cell uptake, and they do not fit the drug-likeness rules of Lipinski (83). Their inherent metabolic instability imposes another challenge. However, these problems appear manageable through chemical modification, combination with lipid particles, and/or association with virus-based delivery systems. The recently developed antagomirs such as LNAs show great promise as therapeutic agents (35,71). They can be readily uptaken by cells due to their relatively small size and cholesterol conjugation. In addition, they are resistant to exo- and endonuclease, rendering them highly stable. Ideally, vascular tissue-specific delivering and targeting systems would greatly minimize untoward effects.
With this in mind and within the context of miRNA, SIRT1, and vascular diseases, there are a variety of strategies that in theory could utilize oligonucleotides to enhance SIRT1 activity and/or promote its downstream actions. For example, antisense oligonucleotides (antagomirs such as LNAs) could be administered to antagonize miRNAs that suppress SIRT1 expression and in turn increase its level. Of the currently known miRNAs that suppress SIRT1 expression (Table 2), all, but four, appear to be tumor suppressors. Therefore, if true in vivo, their antagomirs may be potential tumor promoters and of little therapeutic value. Antagomirs of the four miRNAs that suppress SIRT1 expression without tumor suppressor activity (i.e., miR-92, miR-93, miR-132, and miR-181) may be appropriate candidates to examine for their therapeutic potential for vascular diseases. Particularly, miR-92 may suppress the expression of both SIRT1 and KLF2 (12,132). Targeting miR-92 may hence exert pleotropic benefits to the vasculature.
miRNAs that modulate SIRT1 upstream regulators may also be considered as potential therapeutic targets. Notably, the activation or upregulation of most SIRT1 kinases appears to be associated with tumors and thus may not offer a safe approach to SIRT1 activation. Alternatively, increased expression of SIRT1 can be achieved by using pre-miR-targeting proteins that negatively regulate SIRT. Future studies elucidating the associated mechanism will reveal the possibility of such approaches.
Additional strategies enhancing the levels/activities of various kinases that act in concert with SIRT1 in imposing antioxidative and anti-inflammatory effects should be considered as well. One good example of such is AMPK. Because miR-33 may target AMPK (28), antagonizing miR-33 may enhance AMPK and consequentially enhance SIRT1 activity. Further, among the downstream effects of SIRT1 activation is the enhanced expression of the cholesterol transporter ATP-binding cassette, subfamily A, member 1 (ABCA1), which is also post-transcriptionally targeted by miR-33 (79,121). Therefore, antagomirs to miR-33, such as SIRT1, would increase the expression of ABCA1 leading to lower cellular cholesterol and higher high-density lipoprotein (HDL) levels, which is antiatherosclerotic. Indeed, the inhibition of miR33a/b has been proved to increase HDL and lower very low-density lipoprotein triglyceride, which is atheroprotective (120). However, because the miR-33a mimetic has antitumor effects (56), the oncogenic possibility of using miR-33 inhibitors warrants caution.
Conclusion and Perspectives
Clearly, increased SIRT1 expression and/or activity are indispensable in maintaining the endothelium in health. Oxidative stress, commonly exerted by pathophysiological conditions, on the other hand, leads to endothelial dysfunction. Given these opposing outcomes resulting from augmented SIRT1 versus oxidative stress, one should expect that an imbalanced redox state will negatively regulate SIRT1. Information provided in the review indicates that the oxidative stress-responsive miRNAs may play a role linking the increased oxidative stress and impaired SIRT1. It appears that miRNAs can both directly and indirectly regulate SIRT1 in ECs. Through coordinated regulation, such a network suppresses SIRT1 or desuppresses SIRT1 inhibitors, so that the aggravated inflammatory and oxidative statuses affect the endothelial function. This new paradigm of miRNA-involved EC biology not only is regulated at multiple levels (transcription, post-transcription, post-translation, etc.) but is also coordinated temporally and spatially. Because of this complexity, high-throughput screening, bioinformatics, and system biology should be integrated with experimental approaches in future studies. The usefulness of these state-of-the-art methods also include the identification of novel miRNAs and previous unidentified targets of miRNA involved in either pro- or antioxidative stress in ECs. The accuracy of this new approach heavily depends upon experimental validation in vitro and in vivo, which can be achieved by conventional methods. Because of the fine-tuning nature of miRNA regulation, the relative abundance of miRNA should be prudently considered. To avoid false-positive results obtained from miRNA overexpression experiments, genetic or siRNA deletion of the miRNA with ensuing gain of function should be considered. Notwithstanding, overexpression of miRNAs exceeding the physiological amounts may still have its therapeutic potential.
The approach outlined here can be applied to explore miRNA networks elicited by other environmental stimuli, which affect endothelial homeostasis either positively or negatively. To increase the translational potential, disease-based rather than cell culture-based screening (e.g., atherosclerotic tissues vs. H2O2-treated ECs) is more relevant to investigate the role of the miRNA-SIRT1 pathways in the pathophysiology of cardiovascular diseases. Last but not least, our understanding of miRNA in relation to the potential use in therapy is in its infancy. The above comments will hopefully spur future research on its therapeutic applications.
Footnotes
Acknowledgments
This work was supported in part by National Institutes of Health Research Grants HL106519 and HL105318 (to J.S.) and the American Heart Association Postdoctoral Fellowship Grant 11POST7590128 (to Z.C.). The authors wish to thank Dr. Wei Sun for his critical comments on the article.