
Abstract
Introduction
Traditional candidates for EDHF include a number of EDRFs (NO and carbon monoxide [CO]); arachidonic acid metabolites (PGI2, trihydroxyeicosatrienoic acid, and epoxyeicosatrienoic acids [EETs] derived from COX, lipoxygenase, and cytochrome P450 mono-oxygenase pathways, respectively); hydrogen peroxide (H2O2); potassium ion (K+); and C-type natriuretic peptide, etc. (16 –18, 29, 40, 41). The endothelium-dependent hyperpolarization of vascular SMCs can be initiated by an increase in [Ca2+]i in ECs via two basic pathways (16 –18, 29), both involving the activation of endothelial SKCa and IKCa channels (15). In one pathway, the resulted EC hyperpolarization can be transmitted to the underneath SMCs through gap junctions (7, 31). Alternatively, EC hyperpolarization leads to K+ efflux, which then activates Na+/K+-ATPases and/or KIR channels in SMCs to generate SMC hyperpolarization and vasodilatation (15).
Innovation
In this study, we used glass microelectrode technique to impale a single endothelial cell (EC) of intact vascular tissue or reach smooth muscle cell (SMC) after penetrating the endothelium layer to measure membrane hyperpolarization in situ, which is a gold standard to identify hydrogen sulfide (H2S) as an endothelium-derived hyperpolarizing factor. Via this technique, the comprehensive lines of electrophysiological evidence were obtained to demonstrate that H2S produced by endothelium causes the hyperpolarization of EC via the activation of IKCa and SKCa channels. This endothelium hyperpolarization results in adjacent SMC hyperpolarization, which is related to the increased expression of SKCa channel and underlie the molecular mechanism of endothelium-dependent vasorelaxation.
Hydrogen sulfide (H2S) is produced in both vascular ECs and SMCs with L-cysteine as the substrate and cystathionine γ-lyase (CSE) as the catalyzing enzyme (9, 38, 39, 46, 49). Endogenously generated H2S induces vasorelaxation in part through an endothelium-dependent mechanism (9, 46, 49). The blockade of SKCa and IKCa channels by the co-application of charybdotoxin (ChTX) and apamin reduced H2S-induced and endothelium-dependent relaxation of rat aorta and mesenteric artery (MA) (9, 49). Our previous studies have also shown that the vasorelaxing effects of H2S were more potent on small resistance MA than on large conduit aorta (9, 46, 49). Knocking out CSE expression led to membrane depolarization of the whole vascular tissues, detected with a voltage-sensitive fluorescent dye probe (28). Indeed, acetylcholine-induced membrane hyperpolarization of the isolated ECs from wild-type (WT) mice was significantly greater than that from CSE-knockout (KO) mice (28). These reports suggested in many ways that either H2S itself is an EDHF or that H2S releases EDHF from the endothelium (40, 41). However, the key question regarding the nature of H2S being an EDHF remains unsettled. Whether EC-produced H2S hyperpolarizes EC itself and whether the presence of ECs is the prerequisite for H2S-induced SMC hyperpolarization have been unknown. Whether this endothelium-dependent SMC hyperpolarization induced by endothelium-generated H2S depends upon blood vessel types or gender has not been reported. An EDHF role of endogenous H2S produced by ECs cannot be convincingly established without directly recording membrane potential changes of vascular SMCs in the presence of intact ECs. These challenges are successfully addressed in the present study by linking the endothelium-dependent SMC hyperpolarization to the functional and structural changes of SKCa and IKCa channels induced by endothelium-produced H2S.
Results
Differential contribution of CSE to endothelium-dependent relaxation of different vascular tissues
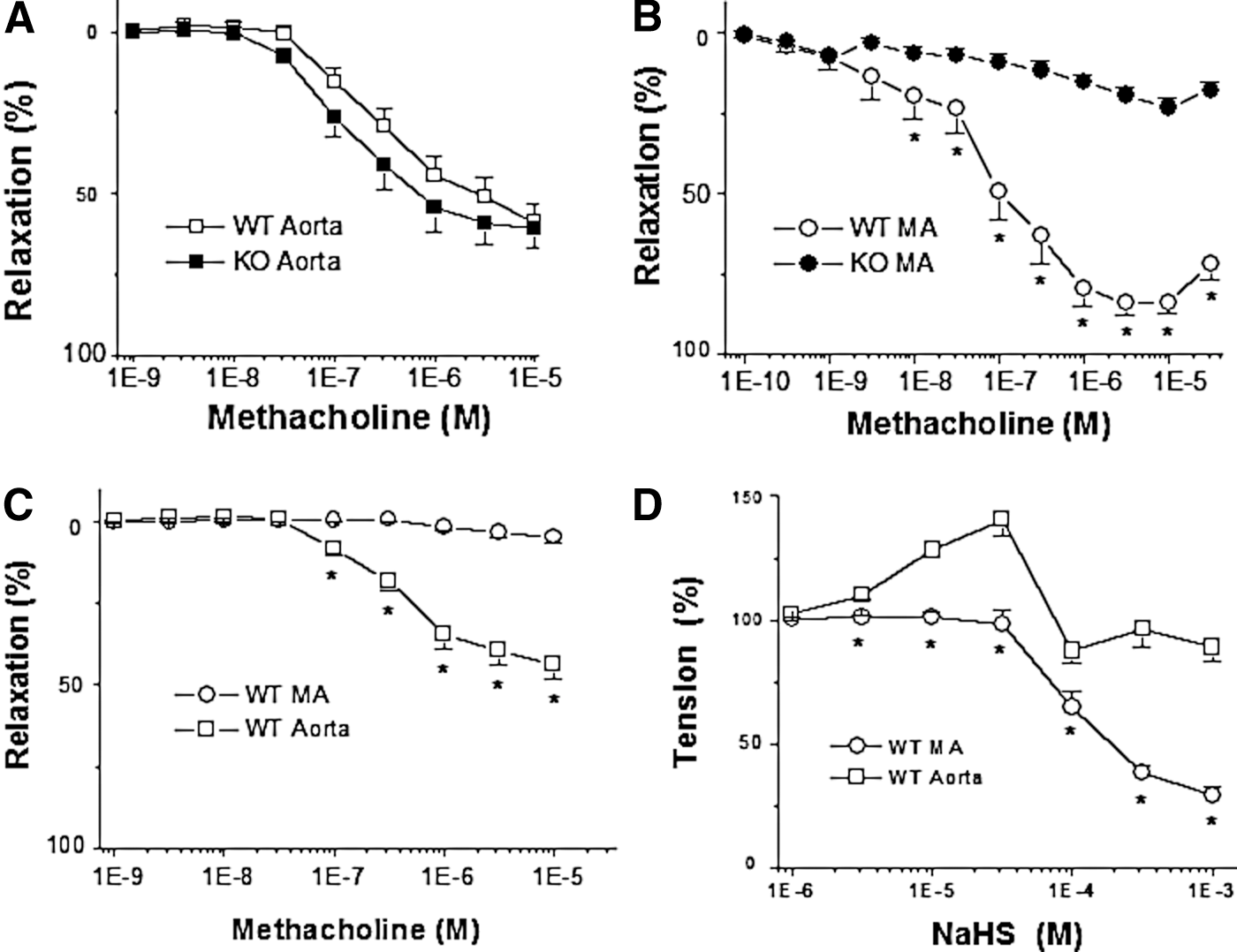
The endothelium-intact aorta rings from WT mice exhibited the same degree of vasorelaxation as the aortic rings from CSE-KO mice in response to methacholine (Mch)-induced cholinergic stimulation (Fig. 1A). In contrast, Mch-induced endothelium-dependent relaxation of resistance MA was significantly decreased in CSE-KO mice in comparison with that of WT mice (Fig. 1B). Pretreatment of vascular tissues with ChTX and apamin had no influence on Mch-induced endothelium-dependent relaxation of aortic tissues, but abolished that of MA from WT mice (Fig. 1C). Exogenously applied NaHS, a H2S donor, mimicked the Mch-induced endothelium-dependent relaxation of WT-MA in a dose-dependent fashion (Fig. 1D). The physiological concentration of H2S in circulation has been reported in the range from high nano-molar to low micro-molar (12). In our study, NaHS at more than 10−4 M tremendously induced MA relaxation, probably due to a toxicological effect of H2S at this concentration on vascular tissues. Interestingly, NaHS caused a biphasic vascular tone change, contraction followed by relaxation, in WT mouse aortic tissues (Fig.1D).
CSE activity and endothelium-dependent hyperpolarization of SMCs in intact vascular tissues
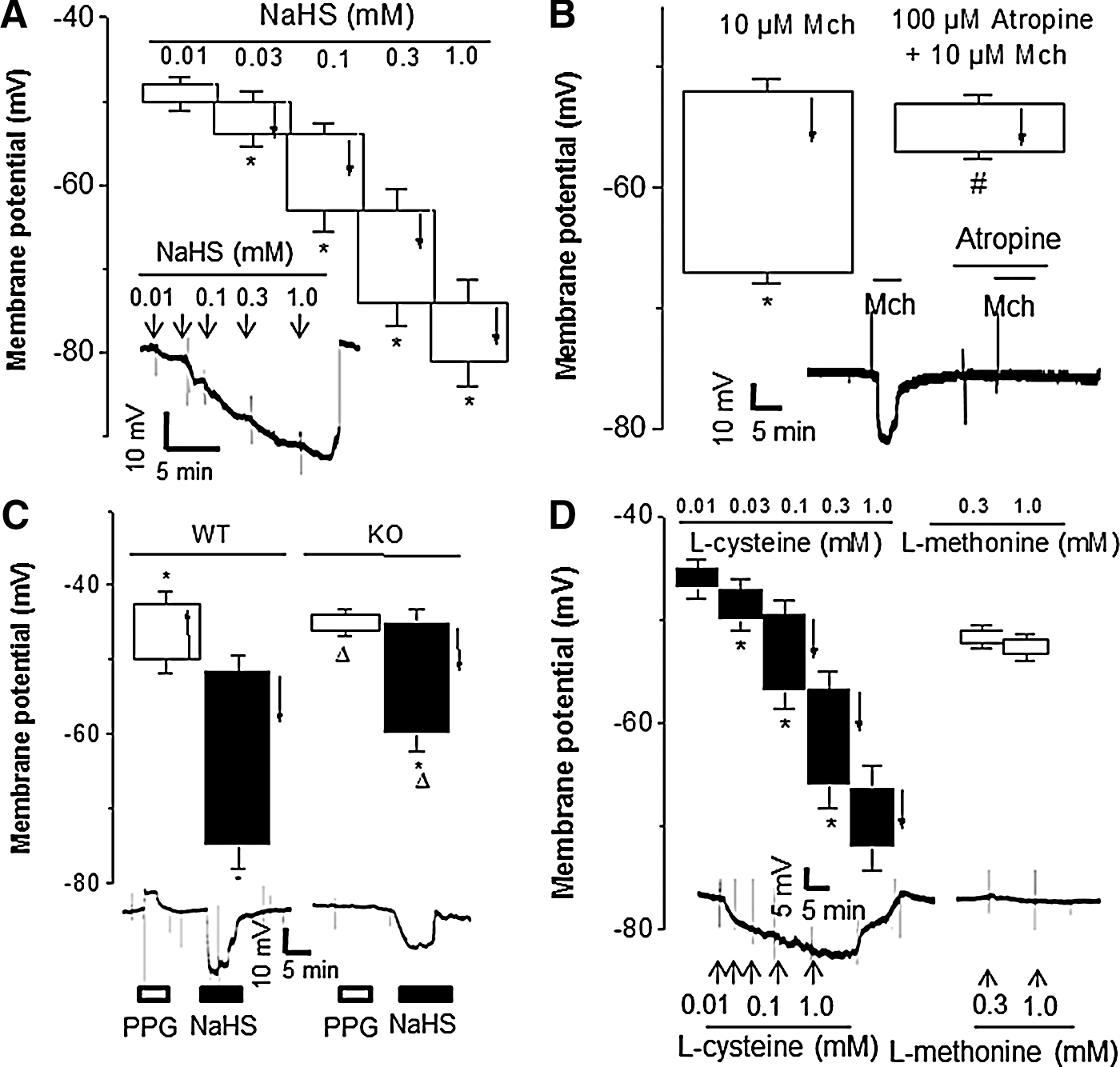
In order to detect endothelium-dependent membrane potential changes of SMCs, we advanced an electrode to impale vascular SMCs of the intact third order branch of MA from the adventitial side with the undamaged ECs on the luminal side. We found that NaHS induced a dose-dependent hyperpolarization of SMCs of WT-MA (Fig. 2A). Mch also hyperpolarized SMCs, an effect that was blocked by atropine (Fig. 2B). Inhibiting CSE enzymatic activity with DL-propargylglycine (PPG, an inhibitor of CSE) depolarized membrane potential of WT-MA SMCs, but this maneuver had no effect on CSE-KO MA (Fig. 2C). Because endogenous production of H2S in ECs and SMCs is virtually eliminated in CSE-KO mice (28, 46), the above data suggest that endogenous H2S contributes to the maintenance of the resting membrane potential of WT mouse MA. L-cysteine is a substrate of CSE and it has been shown to hyperpolarize neurons (24). Indeed, L-cysteine induced a dose-dependent hyperpolarization of WT-MA SMCs (Fig. 2D). As a control, L-methonine (0.3 and 1.0 mM) had no effects on membrane potential of WT-MA SMCs (Fig. 2D).
Compared with that (−54±1.3 mV, n=20) in WT-MA SMCs, the resting membrane potential (−49±1.0 mV, n=25, p<0.05) in CSE-KO SMCs shifted to more negative (Fig. 3A, B). The NaHS-induced hyperpolarization was similar in MA-SMCs from WT mice and CSE-KO mice (Fig. 3A, B). Mch hyperpolarized WT-MA SMCs (p<0.05), but not those from CSE-KO mice (Fig. 3A, B). It appears that Mch-elicited SMC hyperpolarization requires the expression of CSE and production of H2S in vascular endothelium. ChTX and apamin inhibited the hyperpolarizing effect of Mch on WT-MA SMCs as well as the effects of NaHS on SMCs from both WT and CSE-KO MA (Fig. 3A, B). Removal of endothelium abolished SMC hyperpolarization induced by Mch in WT-MA, but not that induced by NaHS in WT and CSE-KO MA tissues (Fig. 3C, D).
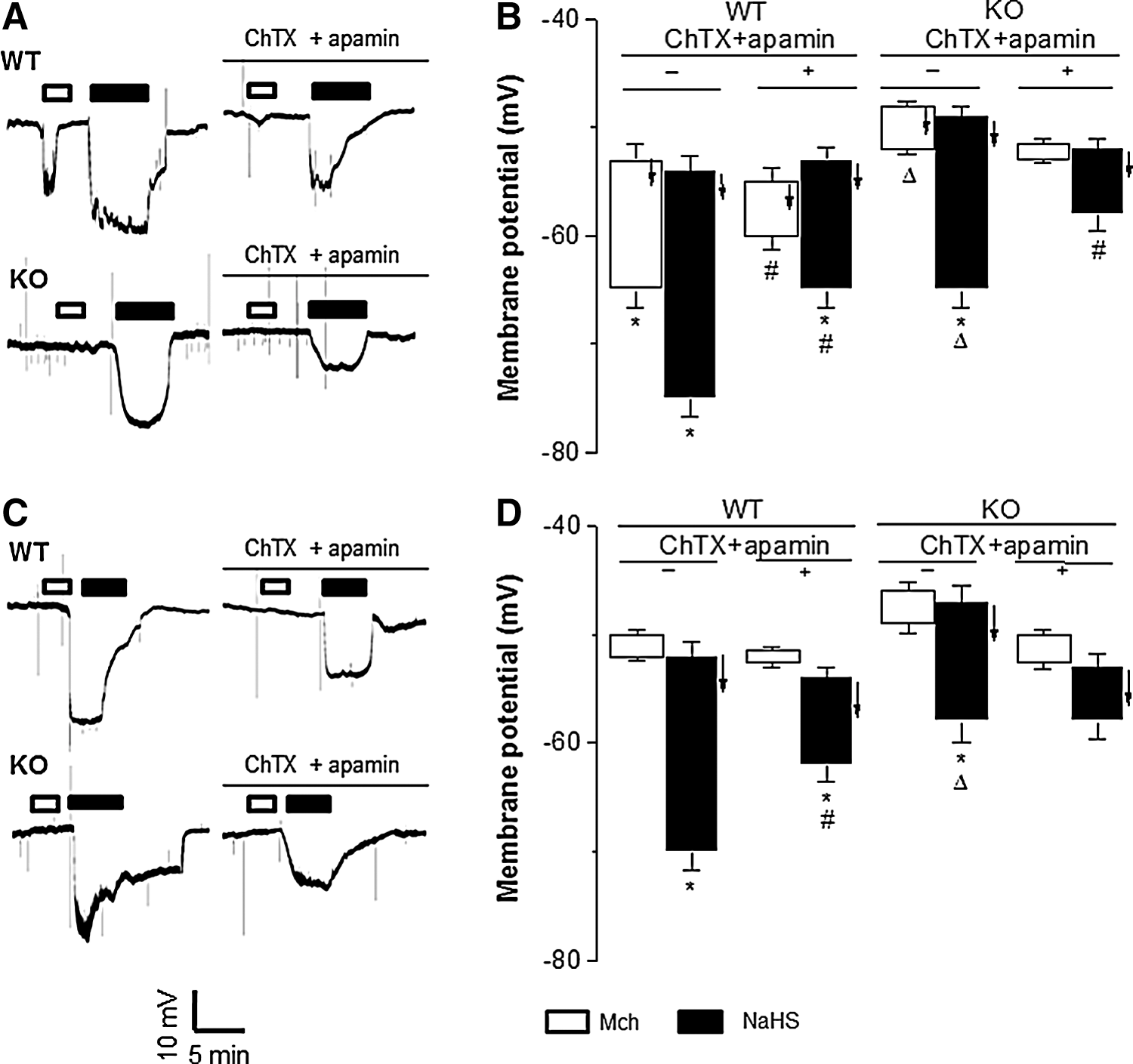
Similar to the differential vasorelaxant effects of Mch on mouse arteries (Fig. 1), Mch did not hyperpolarize aortic SMCs from WT or KO mice (Fig. 4C, D). In contrast, NaHS caused similar hyperpolarization of SMCs from mouse aorta and MA regardless the presence of ChTX/apamin treatment or an intact endothelium (Figs. 3A, B and 4C, D).
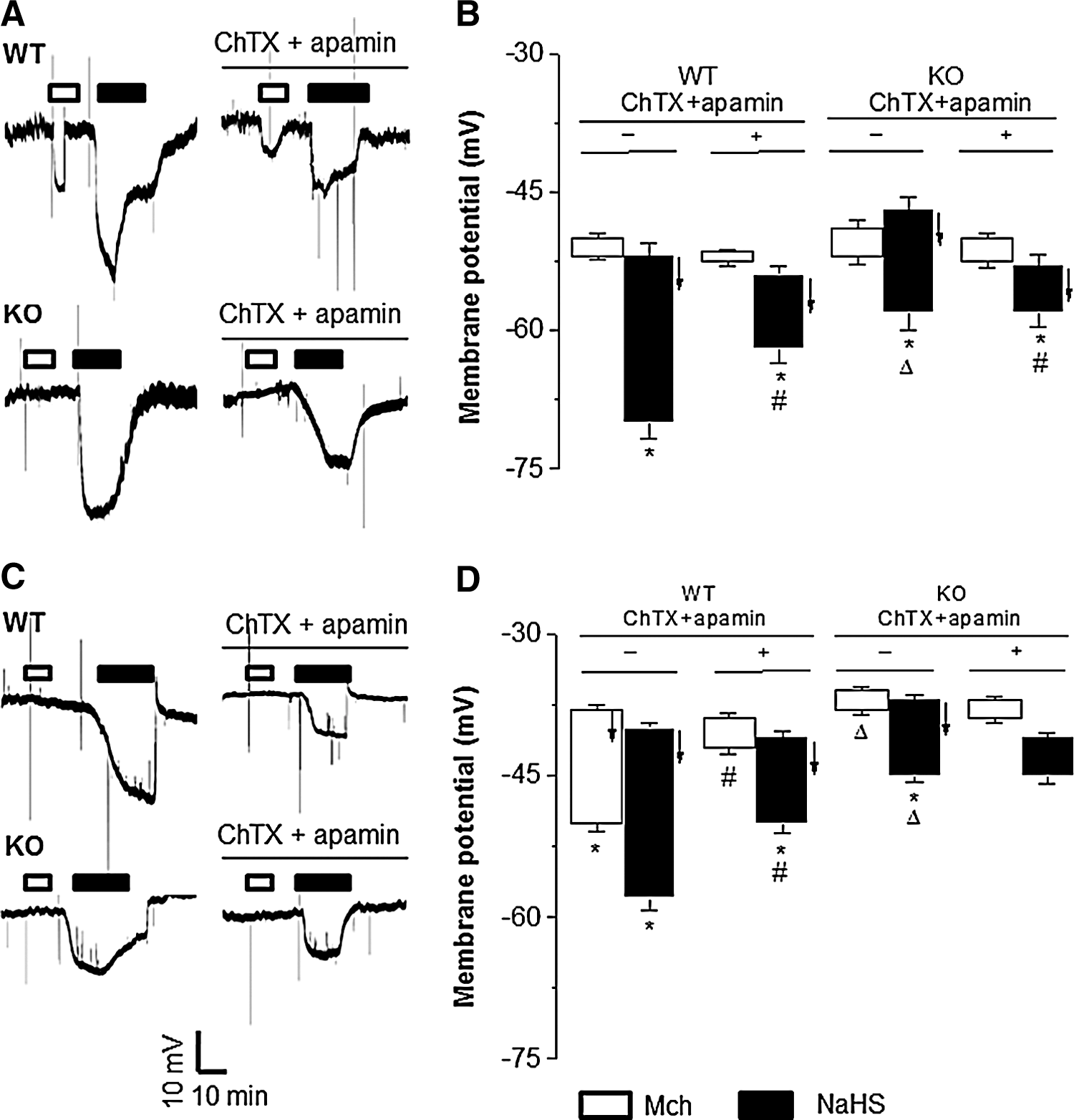
Mch hyperpolarized ECs of the intact WT-MA tissues, but not that of CSE-KO MA, which was inhibited by ChTX/apamin (Fig. 4A, B). NaHS induced similar EC hyperpolarization in intact WT-MA and CSE-KO MA, and this effect was inhibited by ChTX/apamin (Fig. 4A, B). These results indicate that the hyperpolarizing effect of Mch is mediated by endothelium-located CSE that generates H2S, but the exogenously supplied H2S can act on both ECs and SMCs to cause hyperpolarization.
Gender difference of the hyperpolarizing effects of H2S on mouse arteries
Mch and NaHS hyperpolarized SMCs of the endothelium-intact MA from female WT mice, which was blocked by ChTX/apamin (n=12, p<0.05) but not by NG-nitro-L-arginine methyl ester (L-NAME, an inhibitor of NO synthase) or indomethacin (Indo, an inhibitor of cyclooxygenase) (Fig. 5A, B). The ChTX/apamin-sensitive hyperpolarizing effect of Mch and NaHS on SMCs of male WT-MA was weaker than that on female WT-MA (Fig. 5A, B). We also compared membrane potential changes of aortic SMCs from female and male WT mice. Mch and NaHS induced hyperpolarization of female aortic SMCs, which was inhibited by ChTX/apamin but not by L-NAME or Indo (Fig. 5A, C). Mch did not hyperpolarize aortic SMCs in male WT mice. NaHS hyperpolarized male WT mouse aorta SMCs, which was also inhibited by ChTX/apamin but not by L-NAME or Indo (Fig. 5A, C). However, since we only tested the effects of L-NAME and Indo at one given concentration, the effectiveness in blocking NO pathway and prostagladin pathway in our mouse vascular preparation should be cautiously interpreted.

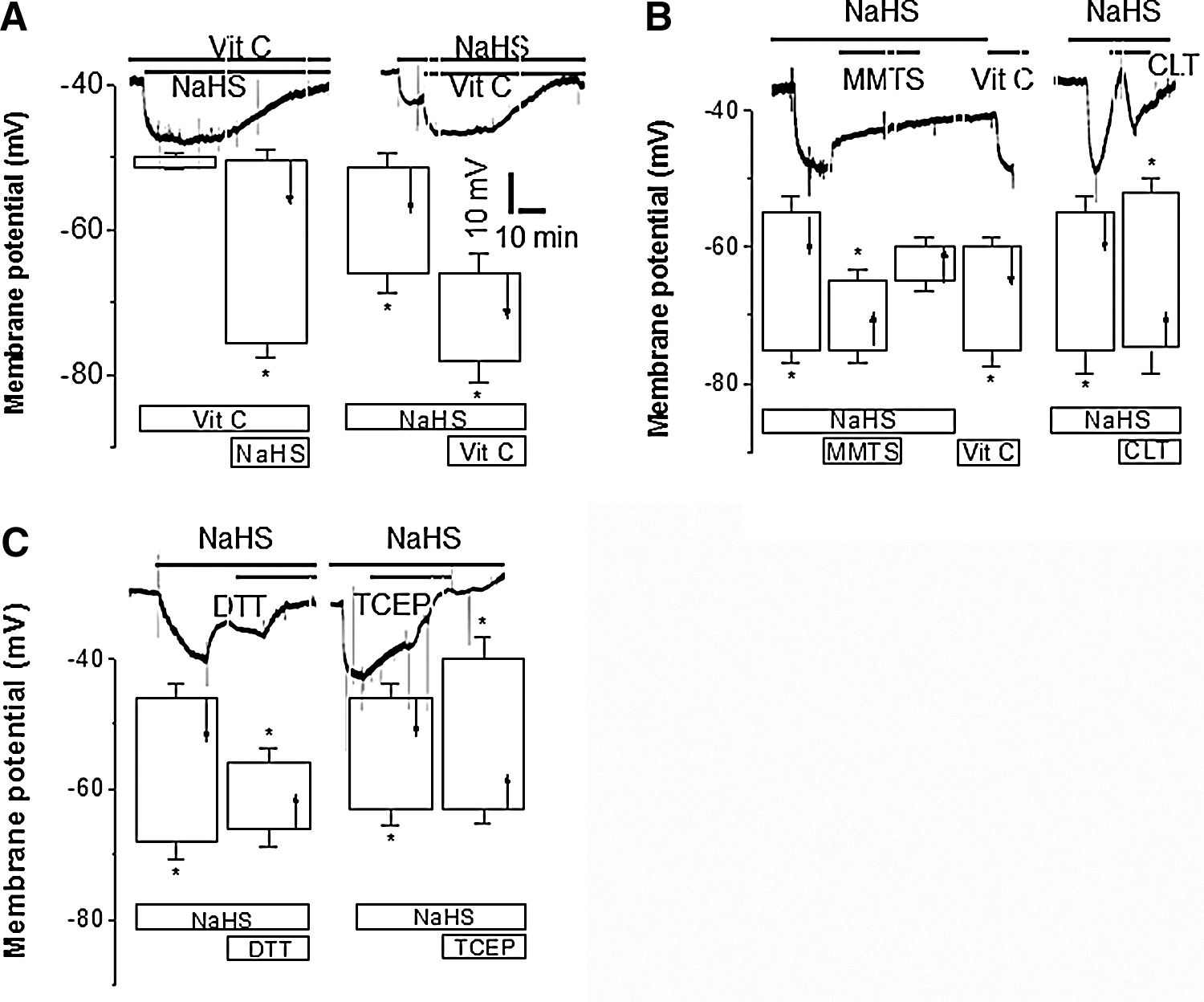
Modulation of H2S-induced hyperpolarization of vascular SMCs by various −SH group-modifying compounds
L-ascorbate (Vit C) can selectively expose free −SH group of protein amino acid residues (26). In the presence of Vit C, NaHS-induced SMC hyperpolarization was potentiated (Fig. 6A) although Vit C itself had no effect on membrane potential of SMCs of WT MA. Vit C following NaHS application further enhanced NaHS-induced SMC hyperpolarization (Fig. 6A). Methyl methanethiosulfonate (MMTS), a selective free −SH reactive compound, abolished NaHS-induced SMC hyperpolarization (Fig. 6B), which was reversed by Vit C. Chloramines T (CLT), an oxidizing agent of free −SH groups of amino acids (27), also blocked reversibly NaHS-induced hyperpolarization (Fig. 6B). Tris (2-carboxyethyle) phosphine (TCEP), the most potent persulfide cleaver (1), significantly abolished NaHS-induced SMC hyperpolarization (Fig. 6C). Furthermore, NaHS-induced hyperpolarization of SMCs was blocked by dithiothreitol (DTT), a reducing agent (Fig. 6C).
H2S-induced activation of whole-cell and single-channel IKCa and SKCa channels in ECs
In primarily cultured ECs from WT mouse mesenteric arteries, outward macroscopic K+ currents were stimulated by NaHS (Fig. 7A, B). NaHS-enhanced K+ currents in ECs were not inhibited by iberiotoxin (IbTX). In the presence of IbTX, NaHS-stimulated K+ currents were inhibited by apamin and further by apamin in conjunction with ChTX (Fig. 7C, D). In outside-out patch configuration, single-channel recordings were performed with asymmetric 5.4/140 mM [K+]. Two types of K channels with unitary conductances of 6.8±0.3 pS and 15.3±0.5 pS (n=5 and 7, respectively) were observed (Fig. 8A, B). The open probability of 6.8 or 15.3 pS channels was 0.27±0.02 or 0.30±0.03 (n=5), which was increased by NaHS to 0.89±0.07 or 0.84±0.06, respectively. NaHS-evoked increase in the open probability was not altered by IbTX, but inhibited by apamin at 100 nM by 48.4%±2.7%, and further inhibited by ChTX by 34.2%±2.4% (Fig. 8C, D).


The expression of IKCa and SKCa proteins
Both SK2.3 and IK3.1 channel proteins were expressed in cultured ECs (Fig. 9A). SK2.3 channel protein expression in ECs was increased by NaHS treatment for 12 h but reduced by PPG (Fig. 9A), but NaHS and PPG seemed to have no effect on IK3.1 channel expression. In addition, SK2.3 protein level in MA and aorta was significantly lower from CSE-KO mice than that from WT mice (Fig. 9B). IK3.1 expression level in MA and aortic tissues was similar between CSE-KO and WT mice (Fig. 9B).

Discussion
Endothelium-dependent relaxation/vasodilation had been generally attributed to the release of NO and PGI2. However, after COX and NO synthase activities are inhibited, endothelium-dependent vasorelaxation can still be observed in specific blood vessels, which has been attributed to the effect of putative EDHF (16 –18, 30). The existence of EDHF was firstly proposed in the early 1980s (11, 37). Numerous putative EDHF candidates have been since tested, including NO, CO, H2O2, C-type natriuretic peptide, PGI2, trihydroxyeicosatrienoic acid, EETs, and K+ ion itself (16 –18). Until now, none of the aforementioned EDHF candidates can fully explain the effects of the proposed EDHF. For example, the relaxant effects of NO (NO*, NO−, ONOO−) on vascular SMCs result primarily from guanylyl cyclase activation and the stimulation of cGMP-dependent protein kinases. Both endogenous and exogenous NO are able to hyperpolarize arteries by activating various subtypes of K+ channels, including BKCa, KATP, KV, and KIR channels (35). Whereas PGI2 and its analogs also activated different K+ channels (BKCa, KATP, and KV channels) via activation of adenylyl cyclase-cAMP-PKA pathway (43). Thus, NO and PGI2 do not hyperpolarize SMCs via endothelial IKCa and SKCa channel activation. Notwithstanding the evidence of EET-induced BKCa activation, it has been speculated that EETs might be EDHF, particularly in coronary vessels. However, there is no evidence that EETs activated IKCa and SKCa channels in ECs (42). C-type natriuretic peptide and other peptides induced relative small hyperpolarizatin of arterial SMCs via stimulation of a G-protein-coupled KIR3.x or glibenclamide-sensitive KATP channels (6). CO also relaxes and hyperpolarizes vascular SMCs via BKCa channel activation (44). The activation of BKCa, KATP, KV, and KIR channels in SMCs has been implicated in the dilator/hyperpolarizing actions of H2O2 and other reactive oxygen species (O2 −, ONOO−, OH−) (13, 25). All these EDHF candidates do not cause the hyperpolarization of ECs, and their effects on SMC hyperpolarization cannot be altered by blocking SKCa and IKCa channels.
Our study provides direct evidence that H2S released from ECs causes endothelial hyperpolarization as well as SMC hyperpolarization, thus fully fulfilling the role of an EDHF. More specifically, we demonstrated that (i) cholinergic stimulation-induced hyperpolarization of ECs and SMCs was ChTX/apamin sensitive. Mch, an analog to acetylcholine, can also release NO from endothelium to hyperpolarize vascular SMCs via cGMP-PKG-activated BKCa channels located in SMCs (5). However, the elimination of Mch-induced SMC hyperpolarization after the inhibition of endothelial CSE expression and H2S production indicates a mediating role of endogenous H2S as an EDHF, rather than NO. (ii) Exogenous H2S mimics ChTX/apamin-sensitive hyperpolarizing effect of cholinergic stimulation on both ECs and SMCs. A well-recognized fingerprint of EDHF is the blockade of its effect by the combined application of apamin and ChTX, which block SKCa and IKCa channels (15 –18, 29). These ion channels mediate the passage of EDHF signal from ECs to the targeted underlying SMCs (15, 30, 32). (iii) Inhibition of CSE activity with PPG depolarizes, and supplementation of CSE substrate hyperpolarizes, SMCs. These observations emphasize that the endogenously generated H2S is key for the regulation of membrane potentials in vascular SMCs. (iv) Endothelium-dependent SMC hyperpolarizing effects of cholinergic stimulation and H2S are greater in resistance artery than conduit vessel. The relative contributions of EDRF/EDHF to endothelium-dependent vasorelaxation are related to the types of blood vessels (33). Our previous observation that Mch and H2S induce more profound endothelium-dependent relaxation of peripheral resistance MA than big conduit aortae suggests that CSE-generated H2S from endothelium could be a ChTX/apamin-sensitive EDHF. A greater hyperpolarization induced by H2S in resistance artery MA than in conduit arterial aorta may be because the number of myoendothelial gap junction increases as the sizes of arteries decrease (31). (v) Mch- and H2S-induced endothelium-dependent hyperpolarization of SMCs is greater in female mice than in male mice. This is consistent with the reported EDHF-mediated vasorelaxation that is more pronounced in female mice than male ones (31). The estrogen-related endothelium-dependent hyperpolarization and vasorelaxation are likely due to increasing the expression of SK3 or Cx40/Cx43 (19, 23). It is likely that Mch activates G-protein-coupled muscarinic receptor in ECs and elevates [Ca2+]i level. The rise in [Ca2+]i activates CSE via calmodiulin and increase H2S production (38, 46). H2S activates ChTX/apamin-sensitive SKCa and IKCa channels in ECs as well as SMCs and causes K+ efflux, leading to hyperpolarization. This EC hyperpolarization may also be directly conducted to underlying SMCs via myoendothelial gap junctions. The present study showed that NaHS caused a biphasic change in vascular tension of the isolated WT mouse aortic tissues (Fig. 1D). This may be explained by different molecular targets being affected by NaHS at different concentrations. Although we do not have evidence for the identities of these NaHS targets, it is clear that the vascular effects of NaHS are different between mouse aortic tissues and mesenteric arteries. It has been reported previously that H2S induces a dual vascular effect in rat mesenteric arterial bed. H2S at lower concentration stimulates vasoconstrictioon but induces relaxation in higher concentration. NaHS-induced arachidonic acid release was suggested (14).
S-sulfhydration is the covalent modification of cysteine residue(s) of a protein, yielding a persulfide moiety (–SSH) (27). H2S has been demonstrated to sulfhydrate Kir6.1 subunits of KATP channels complex heterologously expressed in HEK-293 cells (27, 28) and IKCa channels in primary human aortic ECs (28). Redox modification of the dynamic balance between free sulfhydryl groups (–SH) and disulfide bond (–S-S–) is an important mechanism for controlling ion channel function. In particular, redox modification of −SH groups has been reported to modulate the gating of IKCa channels (4). Our Western blot results demonstrated that SK2.3, but not IK3.1, channel expression was enhanced by NaHS and reduced by PPG or CSE gene-KO although IK3.1, channel protein is expressed in vascular tissues. We had previously demonstrated that H2S concentration in unstirred and covered culture medium dropped about 55% within 30 min of the addition of 100 μM NaHS (45). Deleon et al. reported that in a stirred and uncovered Hepes buffer solution the 50% of added H2S (100 μM) was lost in about 5 min (12). H2S as a gasotransmitter may instantly activate the downstream molecules to generate biological responses. Once the signaling is cascaded, it may not be necessary to continuously maintain higher level of H2S in the milieu. Moreover, the concentrations of free H2S in vascular tissues were reported to be 100 times greater than that of the other tissues (22, 24). For these considerations, we treated the ECs with 200 μM NaHS for 12 h to study channel protein expression.
It is quite unknown how H2S changes the protein expression of SK2.3 but not IK3.1. We speculate that H2S may first S-sulfhydrate some upstream molecules, such as transcription factors or kinases, which afterward induce SK2.3 transcription or translation. Of three characterized SKCa channel α-subunit isoforms (SK2.1, SK2.2, SK2.3) in ECs, only SK2.3 protein is expressed on the plasma membrane (2). A homotetramer IKCa α-subunit (IK3.1) is also identified in ECs (3). The differences in expression of SK2.3 and IK3.1 observed in the present study might be also due to the following reasons: (i) SK2.3 channels are diffusely distributed in caveolae throughout the plasmalemma of ECs. In contrast, IK3.1 channels are clustered within endothelial projection microdomains that form myoendothelial gap junctions with the adjacent SMCs (23). (ii) The activation of SKCa channels (especially SK2.3 channels) is responsible for acetylcholine-induced EC hyperpolarization (34), whereas IK3.1 channel activation mediates SMC hyperpolarization and vessel relaxation (10).
H2S, as a reducing agent, can donate proton (H+) and −SH group. The latter in the presence of protons is easy to covalently modify the cysteine residues of K+ channels via formation of persulfide and then causes K+ channel protein configuration change and K+ channel opening, leading to membrane hyperpolarizaton. This process is potentiated by −SH reducing agent (Vit C), blocked by −SH oxidizing agent (CLT, MMTS), and abolished by most potent persulfide cleaver (TCEP) and the disulfide bond breaker (DTT). Vit C maintains the reduced micro-environment of −SH groups and favor formation of −SSH. MMTS covalently modifies free −SH groups and prevent the formation of −SSH by NaHS. CLT oxidizes free −SH groups and decomposed of −SSH, which is consistent to our recent observation that CLT abolished the stimulatory effects of H2S via oxidizing extracellular cysteine residues of expressed KATP currents (rvKir6.1/rvSUR1) (20). TCEP selectively binds to −SSH and reduces its formation. DDT breaks the disulfide bond of −SSH and reduced the formation of −SSH, which is in line with the reported effect of DTT that reverses H2S-induced sulfhydration of IKCa channels (28). It has also been reported that H2S inhibits L-type calcium channels only in the presence of free sulfhydryl groups in the channel proteins, pointing to a critical role of sulfhydryl groups in regulating the biological effects of H2S (48).
In summary, our study provides solid evidence that H2S is an EDHF via using microelectrode technique to impale a single SMC or EC of intact vascular tissue to measure membrane hyperpolarization in situ. Although both microelectrode technique and voltage-sensitive fluorescent probe (28) can detect membrane potential change in arterial tissues in response to H2S and Ach/Mch, microelectrode technique overcomes the shortcomings of voltage-sensitive fluorescent probe that cannot differentiate measured membrane hyperpolarization from SMCs or ECs (8, 28). Using electrophysiological microelectrode technique in our study clearly has demonstrated the unique EC dependence of H2S-induced hyperpolarization and linked it to increased SKCa channel expression. We further demonstrated that the EDHF role of H2S is greater in peripheral resistance arteries than in conduit arteries, and more important in arteries from female animals than in male ones. The identification of H2S as an EDHF will not only help better understand the mechanisms underlying endothelium-dependent vasorelaxation of different types of vascular tissues, but also shed light on devising novel therapeutic agents to deal with specific cardiovascular diseases.
Materials and Methods
Animals
CSE-KO mice were generated as previously described (46) and in-house bred. The second and third generation of 10–16-week-old CSE-KO offspring and age-matched wild-type littermates on C57BL/6J×129SvEv background were used. PCR-genotyping of CSE-KO mice was performed using a three-primer assay in two reactions (46). All animal experiments were conducted in accordance with approved protocols by the Animal Care Committee of Lakehead University, Canada. All animals were maintained on standard rodent chow, and had free access to food and water.
Reagents
NaHS, Mch, DTT, L-cysteine, L-methionine, homo-cysteine, glutathione, atropine, KCl, tetraethylammonium, apamin, ChTX, IbTX, Vit C, MMTS, PPG, L-NAME, Indo, TCEP, and CLT were purchased from Sigma Chemical.
Preparation of MA and aortic tissues
Mice (WT and KO, male and female) were killed by decapitation following CO2 asphyxiation. This process lasted for 3 min, not causing respiratory and circulatory collapse. MA or aorta of mice were removed and immediately immersed into ice-cold Kreb’ solution, which comprised of (in mM): 118.7 NaCl, 4.7 KCl, 1.18 KH2PO4, 1.17 MgSO4, 2.5 CaCl2, 24.8 NaHCO3, and 10.1 glucose (pH 7.4), as described in our previous publications (10, 14). Arterial tissues were carefully dissected out and cleaned from the surrounding adipose or adherent connective tissues under a dissection microscope (Wild M3B). Aorta and MA were cut into 2–3-mm-long aortic rings or MA strips (1.5–2.0 mm long, 100–180 μm diameter) for myographic tension assay and the measurement of membrane potentials. Great care was taken to ensure that the endothelial layer was not damaged during processing of the tissue preparation.
Myograph recordings of the tension development of MA and aorta
Blood vessel aortic rings or MA strip segments were mounted in a four-channel chamber of a wire Multi Myogragh (Model 610M) (35) and the Kreb’ solution was aerated with 95% O2 and 5% CO2 at 37°C. Two tungsten wires (25 μm in diameter) were guided through the lumen of each ring or strip segment. One wire was connected to a force transducer and the other to an adjustable micrometer. All segments were normalized with a 30-min equilibration for optimal force development. Thirty minutes after normalization, the viability of segments was tested by alternatively stimulating tissues with 10 μM noradrenaline plus 140 mM K+ and 10 μM noradrenaline alone. Each stimulation lasted for 2–5 min with a 5-min interval, during which the tissues were washed four times with Kreb. Only those vessel segments that generated a minimum tension of 1.0 mN/mm in response to stimulation were used in the following studies. Vessel segments were equilibrated for another 30 min before starting the experimentation. Myodaq (2.01 version; DMT) and Myodata (2.02 version; DMT) software were used for collecting and analyzing data. The vessel segments were preconstricted with phenylephrine at the concentration that induces 60%–70% of maximal contraction to evaluate the concentration-dependent relaxant response to the tested compounds. Endothelium was kept functionally undamaged unless otherwise specified, which was confirmed by 10 μM Mch-induced vascular tissue relaxation. When the concentration of KCl in Kreb solution was changed to 25 or 100 mM in some experiments, the osmolality of solution was adjusted by equimolarly removing NaCl. Agents were added to the 10 ml bath under static conditions.
Recording of the membrane potentials of SMCs and ECs in intact vascular tissues using glass microelectrodes
Blood vessel preparations were maintained in Kreb’ solution and gassed with 95% O2 and 5% CO2. ECs were removed from the vessels in selective experiments by gently rubbing the intimal surface of the vessel with a moistened cotton ball for longitudinally opened aorta or by exposing the lumen to distilled water for 30 s for MA branch. The loss of vascular ECs was confirmed by the lack of response to 10 μM acetylcholine. The isolated blood vessels were transferred to the customer-made transparent gel-filled dish. To record the membrane potential of aortic ECs, the adventitia side of longitudinal opening aorta strip was downward (endothelium side up) and pinned down into the gel by a pair of tiny stainless steel pins. A single microelectrode filled with 3 M KCl (resistance 70–100 MΩ) punctured cell membrane of ECs and entered the intracellular space to record the changes in the resting membrane potentials of ECs (16). Criteria for a successful impalement include an abrupt negative drop in voltage on impalement, followed by a stable negative potential for at least 3 min, and a rapid return to previous potential upon withdrawal of the microelectrode. To record the membrane potential of aortic SMCs, the single microelectrode was further advanced until new sudden negative drop in voltage appeared. For membrane potential recording of MA, all side branches from superior MA were cut off and connective tissues surrounding the vessel were removed under a microscope. The tiny tissue without longitudinal opening was cut in a helical direction to give a strip with a width of about 1 mm and a length of about 10 mm. A glass microelectrode was inserted into vascular SMCs from the adventitial side of strip, while ECs were impaled from the luminal side via a hole in the main MA wall produced by removal of a side branch to access the intimal surface. The changes in the membrane potential were measured through a high-impedance amplifier (Duo 773 Electrometer; World Precision Instruments). Electrical signals were continuously recorded with gap-free acquisition protocol at zero voltage for 1-h duration, and controlled by a Digidata 1440A interface and a pCLAMP software (Version 6.02; Molecular Device) (36, 47). Following a stable recording of membrane potential for at least 5 min, NaHS, Mch, or other chemicals were applied to the preparations through the superfusing fluid, whereas toxins (ChTX/apamin) were directly added to the chamber. When L-NAME (100 μM) or Indo (10 μM) was used, these agents were applied 15 min before the addition of NaHS and Mch. Only the results of the experiments that a single impalement was successfully maintained are used for data analysis. All experiments were conducted at room temperature (20°C–22°C). The osmolarity of recording solutions was always adjusted to 300 mOsms.
Patch-clamp experiments for recordings of macroscopic and unitary K+ currents
The whole-cell and single-channel patch-clamp techniques (20, 21, 38, 44) were used to record K+ currents in primarily cultured ECs. In brief, ECs were mechanically isolated by placing the endothelial surface of an opened artery in contact with a Matrigel1 pretreated cover slip and applying gentle pressure to the back of the artery with a brush, and cultured for overnight by Dulbecco's modified Eagle's medium containing 10% fetal bovine serum and 1% penicillin (100 IU/ml). K+ currents were measured at room temperature using whole-cell mode and excised outside-out patch configuration of the patch-clamp technique. The external solution contained (mM): NaCl 140, CaCl2 1.8, MgCl2 1, KCl 5.4, and Na-HEPES 10 (pH 7.4). Recording pipettes were filled with a solution containing (mM): KCl 130, NaCl 5, MgCl2 1, Mg-ATP 3, EGTA 10, and K-HEPES 10 (pH 7.4). The above recording solutions were used for both whole-cell and single-channel K+ currents recordings. Drugs were added cumulatively to the recording chamber in bolus doses. Patch-clamp data were recorded at 5 kHz using an Axopatch 200B amplifier (Molecular Devices) with compensation for series resistance and cell capacitance. Data were filtered at 1 kHz and digitized using an Axon interface, and data analysis was performed using pClamp-10 software (Molecular Devices). Variance analysis and histogram distribution were used to determine the unitary conductance of single K+ channels. Single channel current amplitudes were calculated by fitting amplitude histograms to a Gaussian distribution. Channel open probability was expressed as P open =NPo/n where NPo=[(to )/(to +tc )], Po=open probability for one channel, to= sum of open times, tc=sum of closed times, N=total number of all single channels in the patch, and n=number of different types of single channels in the same patch.
Western immunoblotting
Mouse MA and aorta tissues or cultured ECs were harvested and lysed in a lysis buffer (EDTA 0.5
Immunoblots were probed with rabbit polyclonal anti-SK2.3 or anti-IK3.1 (1:250 dilution; Santa Cruz Biotechnology) and horseradish peroxidase (HRP)-conjugated goat anti-rabbit secondary antibody (1:10,000 dilution, New England Biolabs). Anti-β-actin primary antibody (1:11,000 dilution; Santa Cruz Biotechnology) and HRP-conjugated rabbit anti-mouse secondary antibody (1:10,000 dilution; Sigma) were employed to normalize SK2.3 and IK3.1 expression. The blots were developed using chemiluminescence (ECL Western Blotting Detection Reagents; Amersham Biosciences).
Statistical analysis
Experiments were repeated at least three times and the mean, as well as the standard error of the mean (SEM) were calculated. All data were expressed as means±SEM. Statistical analyses were conducted using Student's t-test for unpaired samples between the control and the tested group. Multiple comparisons were made with one-way analysis of variance followed by a post hoc analysis (SPSS 10.0 for Windows). Statistical significance was set at p<0.05.
Footnotes
Acknowledgments
This study was supported by a research fellowship from the Heart and Stroke Foundation of Canada to G.T. and operating grants from Canadian Institutes of Health Research to R.W. and L.W.
Author Disclosure Statement
There is no conflict of interest.