
Abstract
Protein action in nature is largely controlled by the level of expression and by post-translational modifications. Post-translational modifications result in a proteome that is at least two orders of magnitude more diverse than the genome. There are three basic types of post-translational modifications: covalent modification of an amino acid side chain, hydrolytic cleavage or isomerization of a peptide bond, and reductive cleavage of a disulfide bond. This review addresses the modification of disulfide bonds. Protein disulfide bonds perform either a structural or a functional role, and there are two types of functional disulfide: the catalytic and allosteric bonds. The allosteric disulfide bonds control the function of the mature protein in which they reside by triggering a change when they are cleaved. The change can be in ligand binding, substrate hydrolysis, proteolysis, or oligomer formation. The allosteric disulfides are cleaved by oxidoreductases or by thiol/disulfide exchange, and the configurations of the disulfides and the secondary structures that they link share some recurring features. How these bonds are being identified using bioinformatics and experimental screens and what the future holds for this field of research are also discussed. Antioxid. Redox Signal. 18, 1987–2015.
I. Introduction
The best characterized post-translational modifications of proteins are covalent change of amino acid side chains and hydrolytic cleavage of peptide bonds (200). This review focuses on an emerging post-translational modification of proteins involving reductive cleavage of disulfide bonds. Protein disulfide bonds are the links between the sulfur atoms of two cysteine amino acids. This motif is also called a cystine residue. Some of these bonds control the function of the mature protein in which they reside by being cleaved in a regulated way.
II. Post-Translational Modifications
A. Covalent modification of amino acid side chains
The most common type of post-translational modification is covalent modification of side chains (Fig. 1). This means of post-translational protein control is operative in nearly every cellular process. The side chains of 15 of the 20 common amino acids of proteins can be covalently modified by a variety of small (e.g., oxygen) and large (e.g., ubiquitin) molecules (Table 1). There are currently no known modifications of Leu, Ile, Val, Ala, and Phe side chains. In most cases, enzymes catalyze the transfer of a cosubstrate molecule onto the side chain, although some events do not require a catalyst. More than 40 types of covalent modifications of side chains have been identified (200) (Table 1). The most common post-translational modification in eukaryotes is phosphorylation. The human genome encodes 518 protein kinases (135) and 148 protein phosphatases (86), which catalyze covalent phosphorylation and dephosphorylation of proteins.

The side chains of 15 different protein amino acids can be covalently modified (200). This is catalyzed by enzymes and requires cosubstrates in most cases. We have attempted to capture the more common modifications. Some rarer modifications have not been listed, such as the residues involved in autocleavage of peptide bonds, autocyclization of peptides, and modifications of N-terminal residues of polypeptides.
B. Hydrolytic cleavage or isomerization of peptide bonds
The second most common type of post-translational modification is hydrolytic cleavage of specific peptide bonds by proteases (Fig. 1). Proteolytic cleavage of peptide bonds controls the half-life and turnover of proteins. In contrast to most covalent modifications of side chains, hydrolytic cleavage of peptide bonds is irreversible under physiological conditions. As a consequence, proteases are tightly regulated, both temporally and spatially. Temporal control is often achieved by making zymogen forms of proteases, which are activated when needed by other proteases. Spatial control in the intracellular environment can be achieved by compartmentalization such that the protease cannot access its substrate until required. In the extracellular environment, proteases are usually controlled by protease inhibitors. An example is conversion of prothrombin to thrombin by the prothrombinase complex in the blood, and then inhibition of thrombin by antithrombin. The human genome encodes 569 different proteases (112).
The other peptide bond modification is cis-trans isomerization of the X-Pro peptide bond. Proline is unique among amino acids as the side chain forms a cyclic structure with the peptide bond nitrogen, allowing the X-Pro peptide bond to adopt both cis- and trans-conformations. The other peptide bonds exist almost exclusively in the trans-configuration. Prolyl isomer interconversion plays a role in protein function (114) and is catalyzed by peptidyl prolyl cis-trans isomerases.
C. Reductive cleavage of disulfide bonds
A third type of post-translational control of proteins is reductive cleavage of disulfide bonds, which is the focus of this review.
It is important to note that disulfide bond cleavage is separate and distinct from modifications of unpaired cysteine residues. A number of modifications of the thiolate anion of free cysteine residues have been identified, such as oxidation, nitrosylation, and prenylation, which are examples of covalent modification of a side chain (see Table 1). For instance, oxidation of a cysteine thiolate leads to formation of a disulfide bond in some proteins, and these are examples of type 1 modifications (Fig. 1).
D. Distribution of post-translational modifications in the cell
The chemistry of post-translational modifications restricts to a large degree where the modifications happen in the organism. Enzymes that modify substrate side chains require a cosubstrate, so they are nearly always restricted to specific intracellular environments where all three components are available. In contrast, cleavage of peptide or disulfide bonds usually does not require a cofactor, so these modifications occur in both intracellular and secreted proteins. The enzymes or factors that mediate these cleavage events are found inside and outside the cell and only need to find their substrate to function.
III. Where Disulfide Bonds Form and Are Found
Disulfide bonds are formed in proteins as they mature in the cell (39). This maturation occurs in the ER, Golgi complex, post-Golgi complex vesicles, and mitochondrial intermembrane space in eukaryotic cells (21) and in the periplasmic space in bacteria (145). The formation of disulfide bonds is assisted by the oxidoreductases. Oxidoreductases have a reactive dithiol/disulfide in their active sites that undergoes cycles of oxidation/reduction with disulfides or dithiols in the protein substrate resulting in reduction, formation, or interchange of disulfide bonds in the maturing protein (16).
There were 15,662 disulfide bonds in 3,758 human proteins in UniProt as of January, 2013. A large number of disulfide bonds (8,183) are in proteins (1,204) that are secreted or function in the topologically extracellular environments of the endoplasmic reticulum, Golgi and endosome. Plasma and organelle membrane proteins (1,989) also contain a high proportion of the disulfide bonds (7,097). Notably, 368 proteins that function in the cytoplasm and/or nucleus contain 143 disulfide bonds, a cellular environment that is generally thought not to support disulfide bond formation (J. Wong and P. Hogg, unpublished observations).
IV. Evolution of Disulfide Bonds
The cysteine content of proteins has increased as proteins have evolved, and the expansion of this amino acid continues today (22, 82). Not surprisingly, considering their role in protein structure and function, cysteines involved in disulfide bonds are significantly more conserved than cysteines not involved in disulfide bonds (212). In fact, disulfide bond cysteines are the most conserved amino acid. The vast majority (>95%) of disulfide bonds in eukaryotes are conserved once acquired, and there is a positive correlation between the rate of disulfide bond acquisition and organismal complexity (212). The faster rate of accrual of disulfide bonds in complex organisms follows the greater diversity and sophistication of protein function in these species.
V. Allosteric Disulfide Bonds
Disulfide bonds perform either a structural or functional role (8). Most disulfide bonds are structural and assist protein folding and stabilize the tertiary and quaternary structure. The disulfides in the active sites of oxidoreductases are functional bonds that catalyze redox changes in dithiols/disulfides in other proteins.
The disulfide bonds that are cleaved for post-translational control of protein function have been called allosteric disulfides (174, 175). The definition of allosteric control is a change in one site, the allosteric site, which influences another site by exploiting the protein's flexibility (133). A classic example of allosteric regulation is binding of the allosteric effector, 2,3-bisphosphoglyceric acid, to hemoglobin, which reduces its affinity for oxygen. By definition, therefore, cleavage of an allosteric disulfide bond influences another site in the protein.
VI. How Allosteric Disulfide Bonds Are Cleaved
The allosteric disulfides are cleaved either by oxidoreductases or by thiol/disulfide exchange and sometimes both forms of cleavage occur in a protein (Fig. 2). There are three examples (plasminogen, CD4, and von Willebrand factor) where an allosteric disulfide is cleaved by an oxidoreductase, and then one of the resulting cysteine thiolates cleaves a second allosteric bond nearby the first. It is possible that other forms of disulfide bond reduction/oxidation will be identified in time, such as acid–base-assisted hydrolysis (69).

The redox potentials of four allosteric disulfide bonds have been determined (see specific examples below) and fall within the redox potentials required for cleavage by an oxidoreductase. The potentials range from −184 (transglutaminase 2 [TG2]) to −255 [adenosine 5′-phosphosulfate (APS) kinase] mV. The catalytic bonds have redox potentials ranging from −120 to −270 mV (74), while structural disulfides can have redox potentials as low as −470 mV (46). A fundamental question that remains to be answered is how the oxidoreductases function in the extracellular environment. That is, do the reduced enzymes function as single-turnover catalysts or is the reduced form regenerated, so that one enzyme can cleave more than one allosteric bond. For example, oxidized thioredoxin is reduced by thioredoxin reductase and NADPH inside the cell. Electrons are shuttled from NADPH via thioredoxin reductase to the active-site disulfide of thioredoxin, reducing the bond, so that the enzyme can reduce another substrate disulfide. Thioredoxin reductase is present in extracellular environments such as blood (197), but it is not clear where the reducing equivalents would derive from.
Reductive cleavage of disulfide bonds occurs via a bimolecular nucleophilic substitution (SN2) reaction where the three sulfur atoms involved (the sulfur ion nucleophile and the two sulfur atoms of the disulfide bond) must form a ∼180° angle (47, 210) (Fig. 2). Considering this strict stereochemistry, it is not surprising that stretching, twisting, pulling, and shear forces influence the stability of disulfide bonds (10, 209, 210). These factors can enhance or inhibit cleavage of a disulfide bond by changing the alignment of the three sulfurs involved (10). Mechanical forces, therefore, may play a significant role in regulating cleavage of some allosteric disulfide bonds. An example is the adhesive activity of von Willebrand factor (VWF), which is altered by thiol/disulfide exchange that is influenced by the shear forces generated in flowing blood (31).
Cleavage of allosteric disulfides by oxidoreductases or by thiol/disulfide exchange will also be influenced by the electrostatic milieu of the cysteines involved. The proximity to charged amino acid side chains influences the pKa of the attacking cysteine thiol group. This is important, as it is the cysteine thiol anion that is the reactive species (see Fig. 2). For example, Asp26 functions as an acid/base in the oxidation/reduction reactions catalyzed by Escherichia coli thioredoxin by protonating or deprotonating the Cys35 active-site thiol (30). Also, introduction of positively charged amino acids in the vicinity of a redox-sensing disulfide bond engineered in yellow fluorescent protein resulted in an up to 13-fold increase in reactivity of the disulfide. This is in part due to a decrease in the pKa of one of the disulfide cysteine thiols (64). The electrostatic environment of allosteric disulfides, therefore, will likely prove to be an important determinant of susceptibility to cleavage. There are no studies of this property of allosteric disulfides at this stage.
It is becoming apparent that there are some recurring features of allosteric bonds in terms of the configuration of the disulfide and the secondary structures that they link. How disulfide bonds are characterized is summarized in the following section, and we use this convention when discussing specific examples of allosteric bonds.
VII. Classification of Disulfide Bonds
The geometry of a disulfide bond is defined by the six atoms linking the two α-carbons (Cα) of the cysteine residues named Cα-Cβ-Sγ-Sγ′-Cβ′-Cα′ (Fig. 3). These six atoms plus the amide nitrogen attached to Cα and Cα′ are used to define the five χ-angles of a disulfide bond, which are calculated by the rotation around the bonds linking the atoms (174). The first χ 1 angle is defined by the atoms N-Cα-Cβ-Sγ; the second χ 2 angle is defined by atoms Cα-Cβ-Sγ-Sγ′; and so on. The χ angles can be positive or negative, enabling 20 different possible combinations of disulfide bond based on the sign of each χ angle. The three main types of bond classification are the spirals, hooks, and staples. Each disulfide bond can then be further classified as right- or left-handed (RH or LH) depending on if the sign of the χ 3 angle is positive or negative, respectively.
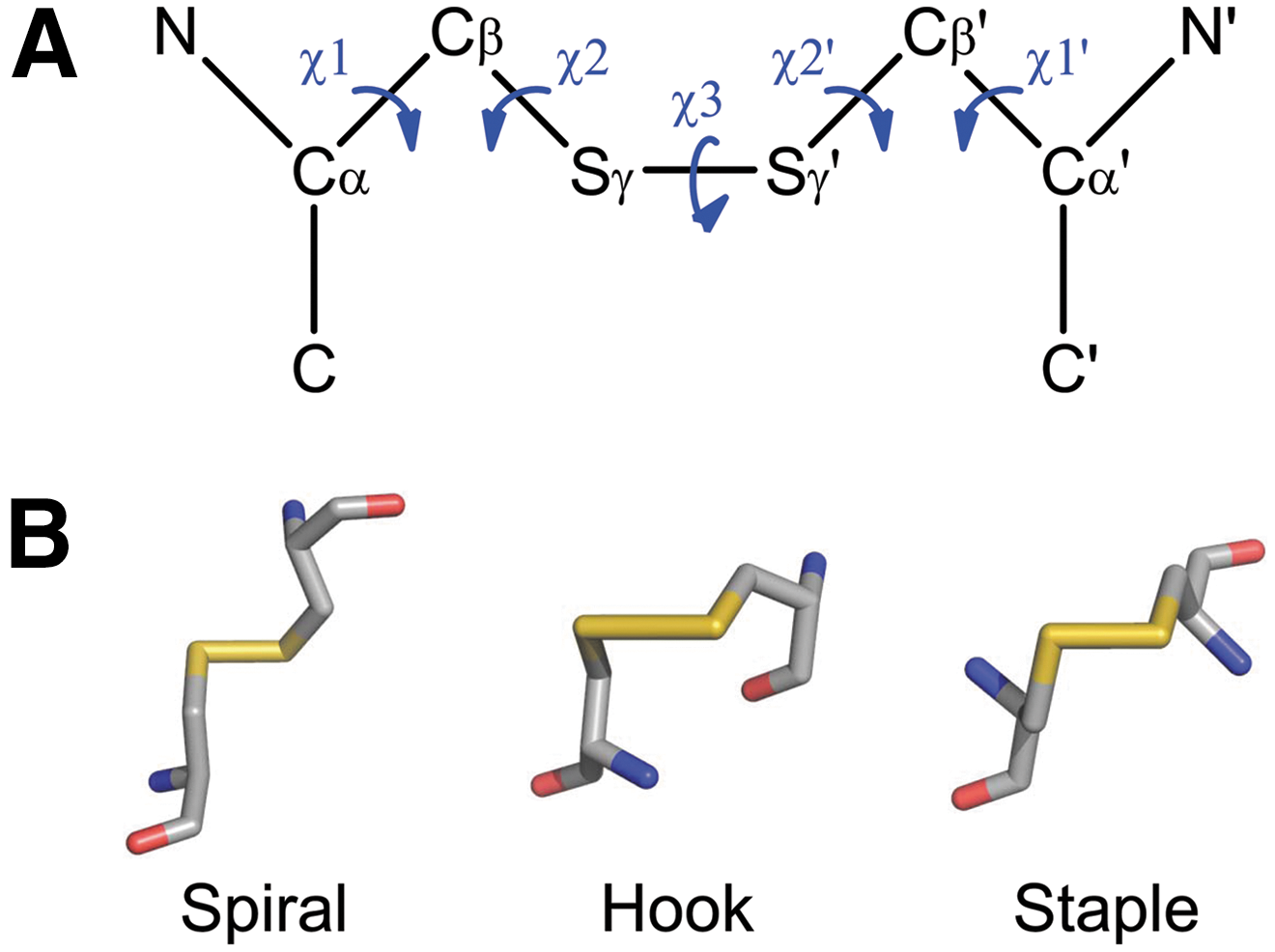
The most prevalent type of disulfide bond is –LHSpiral (∼25% of all disulfides), which is considered to be the primary structural disulfide. This configuration also has the lowest mean dihedral strain energy (DSE) at ∼10 kJ·mol−1 in X-ray structures (174, 175). Oxidoreductase active-site disulfide bonds are nearly all±RH Hooks, as they occur in a conserved Cys-X-X-Cys motif at the end of an α-helix in a thioredoxin fold. The catalytic disulfide bonds cycle between a reduced and oxidized state that coordinates with the oxidized and reduced state of a substrate protein, respectively.
VIII. Grouping of Allosteric Disulfides According to the Type of Change in Protein Function Triggered by Their Cleavage
Cleavage of an allosteric disulfide bond in a protein can result in change in ligand binding, change in substrate hydrolysis, change in proteolysis, or oligomer formation of the protein (Fig. 4). We have gathered examples of allosteric disulfides into these four groups and will discuss specific bonds in some detail. The bonds we have chosen to highlight are those that we feel have been sufficiently characterized to qualify as an allosteric disulfide. The allosteric disulfide cysteines have been identified in all cases; there is at least one crystal structure of the bond, and how the disulfide is cleaved and the functional consequences of this cleavage are known for most examples. There are many other examples at a more preliminary stage of characterization that we will not discuss. Some examples, such as the CD4, gp120, MICA, and β2-glycoprotein I allosteric disulfides, will be addressed only briefly as they have been discussed in detail elsewhere (8). Key features of the bonds that we highlight are summarized in Table 2. We have also chosen to discuss only those examples involving cleavage of an allosteric disulfide. There are some examples, such as the Cys186-Cys209–RHStaple disulfide in tissue factor (8), which appear to regulate the function of the protein in which they reside when they form, rather than when they are cleaved.
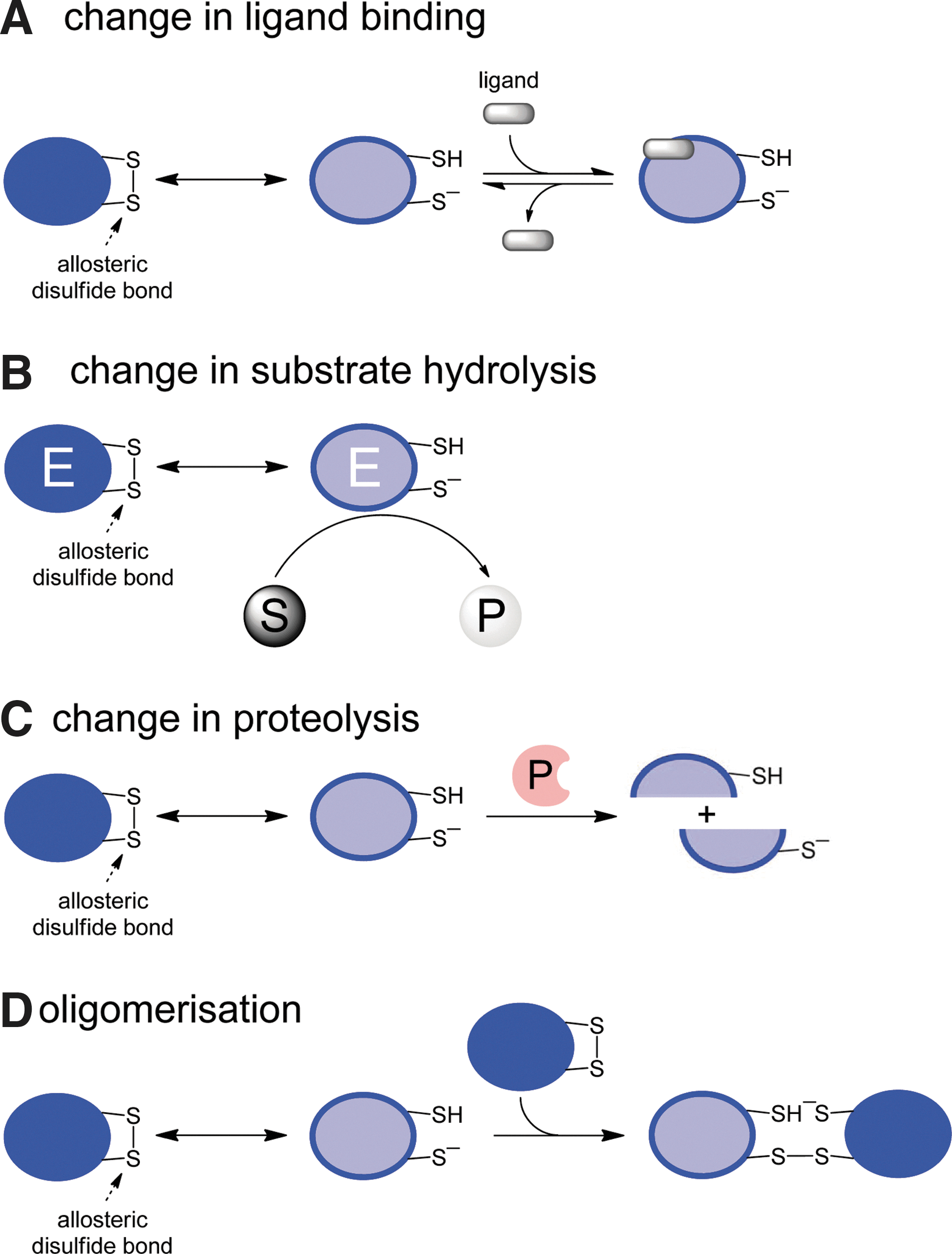
The configuration of the allosteric disulfide bond, the secondary structures that the bond links, and the mechanism of cleavage are indicated, where known.
A number of prokaryote and eukaryote proteins contain one or more cysteine thiolates that can be oxidized by compounds such as H2O2, which leads to formation of new intra- or intermolecular disulfide bonds. The cysteine sulfur anion is oxidized to the sulfenic acid, which is unstable and reacts with a neighboring cysteine thiolate, releasing water and leading to a new disulfide bond. Examples are prokaryote (OcyR, Spx, CprK, etc.) and eukaryote (Yap1p, Nrf2, etc.) transcription factors (5), and human peroxiredoxin IV (26), NMDA receptor (194), Ca2+/calmodulin-dependent protein kinase phosphatase (9), guanosine 3′,5′-monophosphate-dependent protein kinase Iα (23), and apoptosis signal-regulated kinase-1, Ask1 (142, 143). In many systems, it has been proposed that the new disulfide bond can also be reduced. This aspect has not been clearly demonstrated in any system, so we have chosen not to include these disulfides, which could be considered allosteric if they are indeed cleaved in a regulated way. Thioredoxin has been implicated in reducing the Ask-1 intermolecular disulfide bond (142, 143), but how and why this occurs is not clear, and the specific cysteines involved are not known.
A. Change in ligand binding
1. Interleukin receptor subunit-gamma
Interleukin (IL) receptor subunit-gamma (CD132) is the common gamma-chain of the transmembrane cytokine receptors for IL-2, 4, 7, 9, 15, and 21 on the surface of lymphocytes. CD132 binds to additional cell surface proteins to form a particular cytokine receptor. For example, the IL-2 receptor is made up by three subunits: an α, β, and γ (CD132) chain, while IL-7 is composed of only an α-chain and CD132. CD132 forms a complex for the different cytokine receptors that activate cell-signaling pathways upon binding their specific ligand (199).
The CD132 Cys183-Cys232 disulfide is an allosteric bond. It is reduced on the surface of cultured T cells by small-molecule and protein reductants and on the surface of activated thymocytes in mice after an inflammatory challenge (126). Cleavage of the disulfide inhibits IL-2 binding to the receptor complex and, therefore, receptor signaling and cell proliferation. The specificity of this effect was established using IL-2-dependent and independent T-cell clones. Thioredoxin, protein disulfide isomerase (PDI), and gamma interferon-inducible lysosomal thiolreductase (GILT), all oxidoreductases secreted by immune cells during immune activation, were able to reduce the Cys183–Cys232 disulfide bond in cell culture. In accordance with these observations, mutation of Cys183 and Cys232 to serine or alanine severely disrupts binding of IL-2 to the receptor complex (154).
Loss of the disulfide bond through reduction or mutation likely alters the structure of the ligand-binding region. This is relevant to human disease, as mutations in these particular cysteines are among those identified in patients with X-linked severe combined immunodeficiency (X-SCID) (148, 161). X-SCID results from a mutation in the gene encoding CD132 (IL2RG) and is characterized by very low numbers of lymphocytes and susceptibility to various infections from a lack of functional CD132, which would normally direct proper lymphocyte development.
In the crystal structure of CD132, the Cys183–Cys232 disulfide bond is located at the protein surface close to the IL-2-binding site (208) (Fig. 5). The disulfide bond forms a −LHHook conformation linking two β-loops, with a relatively short Cα-Cα′ distance of 5.3 Å, as compared to the average length (5.6 Å) of other disulfide bonds present in the α- and β-chains of the IL-2 receptor complex (PDB: 2ERJ) (96, 188, 208). Interestingly, the structure of CD132 contains a –RHStaple disulfide between Cys63 and Cys73, and Cys63 mutations have also been found in patients with X-SCID (161). The significance of this disulfide for CD132 function has yet to be tested experimentally.

The finding that the Cys183–Cys232 disulfide bond can regulate CD132 function during inflammatory processes may represent a new mechanism of immune regulation. It's likely that these findings have implications for the regulation of cytokine receptors in general, as well as other cell surface proteins during immune activation.
2. HIV gp120
The human immunodeficiency virus (HIV) envelope glycoprotein (env) is a trimer of gp41/gp120 heterodimers on the viral surface that is activated by binding to the immune coreceptor, CD4, and a chemokine receptor, CXCR4 or CCR5, on cells expressing these receptors. gp120 contains nine disulfide bonds, and at least one of them is cleaved during fusion of the viral and target cell membranes (7, 11, 49, 119, 169). It has been proposed that cleavage of the gp120 disulfide bonds aids the elaboration of the gp41 fusion peptide and its insertion into the target cell membrane. Five of the seven disulfide bonds resolved in crystal structures of gp120 can exist in either –RHStaple or –LHStaple configurations (175).
gp120 can be reduced by PDI, thioredoxin, or glutaredoxin-1 (6, 7, 11, 49, 155, 164). Six of the nine gp120 disulfides bonds (Cys54–Cys57, Cys126–Cys196, Cys131–Cys157, Cys296–Cys331, Cys378–Cys445, and Cys385-Cys418) can be substrates for one or more of these reductants. Moreover, cleavage of at least one of the bonds (Cys296–Cys331 bond) is enhanced when gp120 is bound to CD4 (7), which implies that ligand binding influences cleavage of the gp120 allosteric disulfides.
Notably, the disulfide pairing in the V1/V2 domains of a recombinant gp120 trimer was found to differ from the pairing in a gp120 monomer (55). The disulfide pairings were Cys119–Cys157, Cys126–Cys131, and Cys196–Cys205 in the trimer, and Cys119–Cys205, Cys126–Cys196, and Cys131–Cys157 in the monomer. This can arise in one of two ways: either the gp120 bonds pair differently during maturation of the protein, or the bonds can exchange in the mature protein. The later explanation is intriguing, and if correct may be triggered or regulated by CD4 or chemokine receptor binding. In either case, this finding may have significant implications for the immunogenicity of gp120 and HIV vaccine design.
In addition, the V1/V2 domain Cys126–Cys196 disulfide bond has been targeted by CD4 mimetics that contain a cysteine sulfur ion nucleophile (27, 120). The mimetic thiolate attacks the Cys126-Cys196 disulfide forming a stable mixed disulfide linking the two molecules. This approach to targeting allosteric disulfide bonds could be employed in other proteins in other systems.
3. β2-glycoprotein I
β2-glycoprotein I (β2GPI) is a blood protein involved in thrombosis through interactions with serine proteases, anionic phospholipid, and cell surface receptors (131). It is an autoantigen in the antiphospholipid syndrome, which is associated with vascular thrombosis, accelerated atherosclerosis, and recurrent miscarriages (54). The β2GPI–antibody complex is thought to prime the arterial and venous vasculature for the development of thrombosis through effects on blood cells and coagulation and fibrinolysis proteins.
β2GPI consists of four complement modules each with two conserved disulfides and a fifth domain that contains three disulfide bonds (19, 178). The Cys288–Cys326 disulfide bond in the fifth domain, which has a±RHHook configuration, is reduced in blood (76, 156). Reduced β2GPI protects endothelial cells from oxidative stress and forms a mixed disulfide complex with von Willebrand factor (76, 156). There is less-reduced β2GPI in the blood of patients with antiphospholipid syndrome, which is associated with increased immunogenicity of the protein and increased thrombosis (77).
4. C-reactive protein
C-reactive protein (CRP) is a plasma protein of the innate immune system that is involved in the response to inflammation. CRP recognizes microbes and dying/dead cells, which activates the complement system. CRP is a well-known predictor of cardiovascular disease, with increased levels of plasma CRP associated with an increased risk of acute cardiovascular events. A number of studies now show that CRP is also an active participant in cardiovascular disease. However, the exact role of CRP in the pathogenic process is difficult to characterize, as CRP can have both proinflammatory and anti-inflammatory properties [reviewed in (42)].
This may be explained in part by the evidence for at least two conformational forms of CRP that appear to have distinct and overlapping activities. One form of CRP is monomeric, and the other is a pentamer composed of five identical CRP subunits. Upon binding to apoptotic cell membranes or activated platelets, native pentameric CRP dissociates into monomeric units (41, 79, 87, 88, 132). The monomer form is further regulated by an allosteric disulfide bond (Cys36–Cys97), which defines a third form of the protein. The disulfide bond is not cleaved in the CRP pentamer, likely due to restricted access to the disulfide bond in each of the subunits of the complex (Fig. 6B) (206). Thioredoxin can reduce the Cys36-Cys97 disulfide bond in the monomer, markedly altering CRP activity (Fig. 6C). This reductant colocalizes with CRP in the extracellular space of human atherosclerotic plaques, suggesting that it may regulate the CRP disulfide bond in vivo (206).
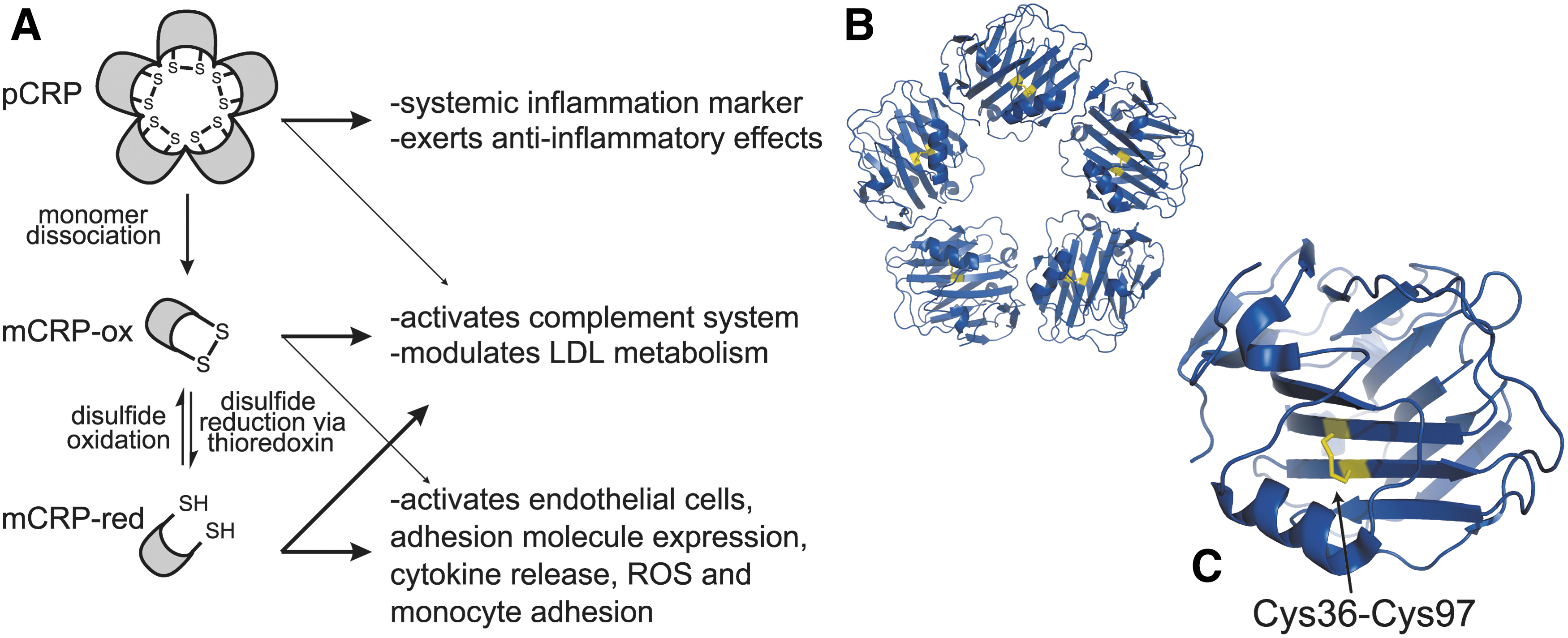
Reduced monomeric CRP has potent proinflammatory effects compared to oxidized CRP. The reduced monomer induces adhesion molecule expression in human coronary artery endothelial cells, resulting in enhanced monocyte attachment and production of IL-8, MCP-1, and reactive oxygen species (206). Reduced monomeric CRP also has higher affinity for C1q, making it more effective at activating complement than the oxidized monomer (206). On the other hand, reduction of the Cys36–Cys97 disulfide bond does not have any apparent effect on monomer binding to low-density lipoprotein (LDL).
Based on these findings, a two-step model for the regulation of CRP has been proposed (206). First, the pentamer form of CRP undergoes conformational changes causing dissociation of the pentamer into monomers, which have distinct effects on cellular processes. Second, the CRP monomer exists in oxidized and reduced forms defined by the redox state of the Cys36–Cys97 disulfide bond, which also have different physiological effects. The reduced monomer form has multiple proinflammatory effects, likely due to structural changes in the cholesterol-binding region of CRP where Cys36 is located. In pentameric CRP, the cholesterol-binding region is mostly buried within the intersubunit contact areas. Dissociation into monomers and reduction of the Cys36–Cys97 disulfide bond would expose this region, enhancing cholesterol binding and enabling tight binding of CRP to the endothelial cell membrane where it mediates proinflammatory responses (78, 206). Both the oxidized and reduced monomer can activate complement and influence LDL metabolism, which may provide atheroprotective effects (Fig. 6A) (179).
The CRP Cys36–Cys97 disulfide bond is an –RHStaple with a short Cα-Cα′ distance of ∼4.2 Å. The cysteines in CRP are evolutionarily conserved, indicating that redox regulation is likely to be operative in other species. Indeed, reduction of the disulfide bond in rabbit monomeric CRP was required for induction of IL-8 and MCP-1 release from human coronary artery endothelial cells (206). Redox regulation of CRP is also likely to be relevant to other inflammatory diseases. For instance, increased levels of both thioredoxin and CRP are observed in the serum of patients with rheumatoid arthritis (80).
Further studies are needed to determine if the reduced form of monomeric CRP exists in vivo and to understand the relevance to cardiovascular and inflammatory diseases, but the initial studies provide intriguing insight into the regulation of the conflicting physiological activities of CRP (206).
5. Interleukin-4
IL-4 is a cytokine involved the differentiation of naïve T-helper cells to lymphocytes. IL-4 signaling has been associated with autoimmune diseases and allergic reactions (36).
IL-4 contains six cysteines that form three intramolecular disulfide bonds (Cys3–Cys127, Cys46–Cys99, and Cys24–Cys65) (Fig. 7). Cleavage of IL-4 disulfide bonds occurred when recombinant IL-4 was incubated with lipopolysaccharide (LPS)-activated monocytes and HeLa cells. Reduction of disulfides was not observed using unstimulated monocytes (36). IL-4 reduction could be prevented by preincubating the HeLa cells with the thiol alkylator N-ethylmaleimide or a thioredoxin reductase inhibitor, which implicates HeLa-derived thioredoxin as the IL-4 reductant. Reduction of one or more of the three IL-4 disulfides by thioredoxin or PDI inhibits IL-4 binding to its receptor, IL-4Rα (36).

There are a number of crystal and solution structures of IL-4, though only of the disulfide bonded forms. In three of the solved crystal structures, the Cys3–Cys127 disulfide forms a –LHSpiral. The Cys46–Cys99 disulfide shows more structural flexibility, with a different conformation in each structure, appearing as a +LHSpiral,±LHSpiral, and a±RHHook. The Cys24–Cys65 disulfide is present as a –RHHook in all three structures (89, 159, 160, 163, 184, 202, 211). The Cys3–Cys127 is significantly more solvent exposed than the other two disulfides, and it has been suggested that this bond is the thioredoxin target (126).
These findings imply a redox regulation of IL-4 function in the extracellular space surrounding both immune and cancer cells (36). Remarkably, the common gamma-chain of the IL-4 receptor (CD132) has also been found to be redox-regulated during immune activation. How the reduction of disulfides in IL-4 and the common gamma chain of its receptor might be related has not been explored, though it will be interesting to see if there is a relationship.
6. β3 integrin
Integrins are transmembrane cell adhesion receptors that mediate interactions between the cytoskeleton of cells and the extracellular matrix, and also play a signal transduction role (75). The receptors are a heterodimeric complex composed of an α- and a β-subunit. They bind to a wide range of diverse ligands which is partially achieved through the different combination of the different α- and β-receptors. Crystal structures of αvβ3 and αIIbβ3 have shown that integrins alternate between an inactive resting conformation and an active ligand-binding conformation.
Activation of integrins has been associated with reduction of one or more disulfide bonds in the receptors (44, 45, 93 –95, 97, 147, 150, 151, 192, 201, 220, 221, 226). This is based on different types of evidence, including activation of cell surface and purified integrins with reducing agents, the appearance of unpaired cysteine thiols in activated integrins, and the demonstration of endogenous thiol isomerase activity of purified integrins. Cell surface PDI has been implicated in these events.
While there are 18 α-subunits and 8 β-subunits in mammals, the majority of redox studies have been conducted on the β3-subunit, particularly within the αIIbβ3 (fibrinogen receptor) and αvβ3 (vitronectin receptor) complexes for which the crystal structures of the extracellular domain have been solved (217, 228). The β3-subunit (also known as GPIIIa and CD61) consists of a large extracellular domain, a single membrane-spanning region and a 47-amino-acid cytoplasmic tail. Within the extracellular region, there are four epidermal growth factor (EGF)-like domains. Each EGF-like domain contains 8 cysteines that form disulfide bonds in a 1–5, 2–4, 3–6, and 7–8 pattern, with the exception of the EGF-1 domain, which lacks the 2–4 disulfide (228). The crystal structure of αIIbβ3 indicated that all 56 cysteines in β3-form 28 disulfide bonds (228). In the αvβ3 crystal structure, 27 disulfide bonds and two unpaired cysteines were identified in β3 (217). Cys473 and Cys503 in the EGF-2 domain were unpaired in αvβ3 and disulfide bonded in αIIbβ3.
A number of studies have been conducted on the role of the individual disulfide bonds within the β3-subunit for expression and function. The disulfides in the EGF domains are generally important for maintaining the inactive state of β3, as mutation of a single cysteine for most of the disulfide bonds within the EGF domains results in a constitutively active β3 (29, 84, 136, 137). Mutation of disulfides outside the EGF domains usually does not affect the activation state or disrupt function (29, 84, 207). An exception is the C-terminal tail Cys663–Cys687 disulfide, which is important for maintaining the inactive state (25, 84). The long-range Cys5–Cys435 bond has also been found to be important for maintaining the low activity state (84, 190).
A recent detailed study of a disulfide bond only found in β3 EGF domains, the 1–5 position bond, has contributed to our understanding of redox control of integrin function (136). The function of this bond in each EGF domain in both αIIbβ3 and αvβ3 was studied by cysteine to serine substitutions. An allosteric role for the EGF-4 Cys560–Cys583 disulfide in both αIIbβ3 and αvβ3 and for the EGF-3 Cys523–Cys544 bond only in αvβ3 was determined (Fig. 8). Interestingly, ablation of the Cys523–Cys544 disulfide bond resulted in an altered stable conformation in αIIbβ3, but not in αvβ3, using molecular dynamic simulations.

The relative significance of these redox events for integrin activation in mammals is still uncertain. Some evidence for a role in humans is found in αIIbβ3 mutations in the inherited bleeding disorder, Glanzmann's thrombasthenia, and the effect of blood homocysteine on the activation state of αIIbβ3. Cys549Arg, Cys560Phe, Cys560Arg, and Cys598Tyr mutations results in constitutively active αIIbβ3, while Cys374Tyr, Cys457Tyr, Cys506Tyr, and Cys542Arg mutations are associated with reduced surface expression of the integrin (2, 29, 58, 129, 138, 144, 146, 166, 168). Patients with elevated plasma levels of homocysteine have a higher level of platelet αIIbβ3 activation than patients with normal levels of homocysteine (124). As homocysteine is a small redox-active thiol, it is possibly influencing thiol/disulfide events in αIIbβ3.
Compelling evidence for a role for allosteric disulfides in integrin function comes from a recent in vivo screen for functional disulfides (127). Proteins on the surface of mouse splenocytes containing unpaired cysteine residues were identified before and after a strong systemic immunological stimulus. The thiol content of integrins α2, αX, and β5 increased >10-fold after stimulation, whereas no thiols were detected in integrins α3 and β7 until after stimulation. These studies indicate that disulfide bonds are cleaved in integrins during activation in vivo, and it seems likely that this chemical event is functionally relevant in these receptors. The challenge is to decipher what that role is.
7. XRCC1
XRCC1 is a DNA repair protein that forms a complex with DNA polymerase β (Pol β) that enables DNA base-excision repair. Recent crystal structures have shown that a labile disulfide bond forms between Cys12 and Cys20 in human XRCC1 (35). Structures of the oxidized (PDB ID 3LQC) and reduced (PDB ID 3K75) XRCC1 have been solved. The disulfide bond is –LHHook.
The oxidized structure has a slightly altered folding surface, creating an enhanced binding interface for Pol β and a higher affinity interaction (Fig. 9) (35). Because the Cys12–Cys20 disulfide bond is distal to the Pol β-binding interface, reduction or formation of the disulfide bond regulates binding affinity in an allosteric manner through long-range conformational changes. It has not been shown that the redox state of Cys12–Cys20 disulfide bond is regulated in the cell, so the structural studies may prove to be misleading.

8. Transient receptor potential cation channel 5
In humans, the family of transient receptor potential canonical (TRPC) cation channels are thought to act as cellular sensors for one or more chemical factors. The TRPC channels closely resemble the transient receptor potential homologs found in Drosophila melanogaster [reviewed in (149)]. There are seven TRPC channels in humans (TRPC1 to TRPC7), which can form homo- and heteromultimeric channels. TRPC channels are highly expressed in the central nervous system and to a lesser extent in peripheral tissues. The protein has six putative transmembrane domains with a pore region between the fifth and sixth transmembrane domains that is nonselectively permeable to cations. The activation of individual TRPC channels is not fully understood, and there are many conflicting reports. It appears that most channels are activated in response to phospholipase C activation, and some may be activated by Ca2+ store depletion, diacylglycerol, or inositol-1,4,5-triphosphate [reviewed in (219)].
TRPC5 on the surface of HEK-293 cells and fibroblast-like synoviocytes is activated by small-molecule (dithiothreitol [DTT]) and protein (thioredoxin) reductants, whereas thiol alkylators such as nitric oxide have no effect (218). TRPC5 does not form disulfide-linked dimers, so the reductants are affecting an intramolecular disulfide bond. These results imply that the cation channel is regulated by an allosteric disulfide. While the structure of TRPC5 has not been solved, there is a predicted disulfide bond in the third extracellular loop of TRPC5 between residues Cys553 and Cys558. The loop is adjacent to the ion selectivity filter of TRPC5, and the disulfide would likely constrain the channel from opening, restricting cation entry and activation (Fig. 10). Single and double TRPC5 Cys553/Cys558 mutants are constitutively active, and DTT treatment does not result in a further increase in activity (218), which supports the proposal that the Cys553–Cys558 disulfide is an allosteric bond. Moreover, DTT does not activate other TRPC channels that lack Cys553 and Cys558 (218).

Half-maximal activation of TRPC5 was achieved with a thioredoxin concentration of 0.2 μg·ml−1, a concentration well within the extracellular levels of this reductant found in the human body. For instance, a thioredoxin concentration of ∼1.2 μg·ml−1 is found in the serum and synovial fluid of patients with rheumatoid arthritis, and thioredoxin reductase levels in human joints have been found to correlate with disease severity (24, 80, 102, 103, 123, 223). Considering that TRPC5 is expressed by fibroblast-like synoviocytes, which secrete synovial fluid, and both TRPC5 and TRPC1 are expressed in synovial joints of patients with rheumatoid arthritis, it is feasible that thioredoxin could regulate TRPC5 activity in this disease.
9. Antimicrobial peptides
Defensin molecules are effector molecules of innate immunity that protect the host from a wide variety of infectious microorganisms [reviewed in (67)]. Human β-defensin 1 (hBD-1) is one of the most widely expressed peptides within the defensin class of molecules, and has been found in the epithelia of skin, respiratory tract, oral cavity, mammary glands, and the urogenital tract. It has also been detected in breast milk, urine, and plasma. However, until recently, hBD-1 was thought to possess only weak antimicrobial activity as compared to other β-defensins (67).
hBD-1 contains six cysteines that form three disulfide bonds (Cys5–Cys34, Cys12–Cys27, and Cys17–Cys35) (13, 72) (Fig. 11A). It was discovered recently that reduction of the three disulfide bonds in hBD-1 releases potent antimicrobial activity against pathogenic Candida albicans and against anaerobic, gram-positive commensals of Bifidobacterium and Lactobacillus species (177). In addition, the reduced form of hBD-1 is found in human colonic mucosa, intestinal crypts, and the skin epidermis, indicating that reduction of hBD-1 disulfide bonds occurs in vivo (177). Thioredoxin reduces the hBD-1 disulfide bonds in vitro, and both proteins colocalize in human epithelia, suggesting that thioredoxin is the physiological reductant (177). Notably, the colonic mucosa has a low redox potential of −200 to −300 mV due to microbial metabolism and a reduced oxygen partial pressure. This environment is predicted to facilitate cleavage of the hBD-1 disulfide bonds by thioredoxin.

The hBD-1 Cys12–Cys27 disulfide bond links adjacent β-strands, a feature in common with other allosteric disulfides (Table 2). Cys12 is part of a β-bulge motif where the β-strand is deformed to allow formation of the Cys12–Cys27 disulfide bond (Fig. 11A) (72). The β-bulge motif, Gly-X-Cys, is conserved in all known mammalian defensins (72). Reduction of the disulfide bonds results in an unstructured, highly flexible polypeptide chain. Functional analysis of hBD-1 cysteine mutants concluded that the C-terminal cysteines and overall hydrophobicity are important for antimicrobial activity (177).
Defensins are only one of the many different classes of antimicrobial peptides produced in a wide range of organisms. Granulysin is another major antibacterial peptide expressed in the granules of human natural killer (NK) and activated cytotoxic T-cells. This peptide is cytolytic to both microbes and tumor cells. Granulysin contains two –LHSpiral disulfide bonds (3) (Fig. 11B), and cleavage of these bonds by thioredoxin increases cytolytic activity (17). In contrast, thioredoxin cleavage of the disulfide bonds in NK-lysin, an antimicrobial peptide closely related to granulysin, reduces the cytolytic activity of the peptide (4). The disulfide bonds in the related antibacterial peptides, protegrin-1 and α-defensin 1, are also reduced by thioredoxin, although the consequence of this cleavage for the activity of the peptides has not been measured.
It is interesting that reduction of disulfide bonds in some antimicrobial peptides increases activity, whereas in others, it decreases activity. Considering that most cell types express multiple peptides, it may be that this is an adaptation to protect cells in any redox environment: a shift from oxidizing to reducing or vice versa might inactivate some peptides, but activate others, ensuring that the cell always has adequate immune defenses. It is also possible that the antimicrobial activity of an oxidized or reduced peptide depends on the pathogenic target. For example, both oxidized and reduced hBD-1 are active against gram-negative Pseudomonas aeruginosa (181), and only the reduced form is active against Candida albicans and anaerobic, gram-positive commensals of Bifidobacterium and Lactobacillus species (177), and neither the oxidized nor reduced forms are active against gram-negative anaerobe Bacteroides vulgates (177).
B. Change in substrate hydrolysis
1. Transglutaminase 2
TG2 is an ubiquitously expressed enzyme with a wide range of biological roles. Extracellularly, TG2 modifies the substrate glutamine residues by transamidation or by deamidation, converting glutamine residues to glutamate. Intracellularly, TG2 functions as a GTPase and participates in cell signaling. It also has protein kinase, PDI, and isopeptidase activities [reviewed in (62)]. TG2 is associated with several neurodegenerative diseases (62) and is well known as an autoantigen in celiac disease (40). In addition to acting as the target autoantigen, it is thought that TG2 propagates celiac disease by deamidating gluten peptides, increasing their immunostimulating properties.
Human TG2 consists of four domains: an N-terminal β-sandwich, a catalytic domain, and two C-terminal fibronectin type III β-barrels (Fig. 12) (110). As TG2 is a widely expressed protein, it is tightly regulated through three identified allosteric modifiers: Ca2+ ions, guanine nucleotides, and an allosteric disulfide bond. Calcium activates TG2 transamidase activity, while guanine nucleotides (GTP and GDP) inhibit activity. In addition, it has been known for some time that oxidative formation of a disulfide bond in TG2 inactivates the enzyme (18, 32, 33).
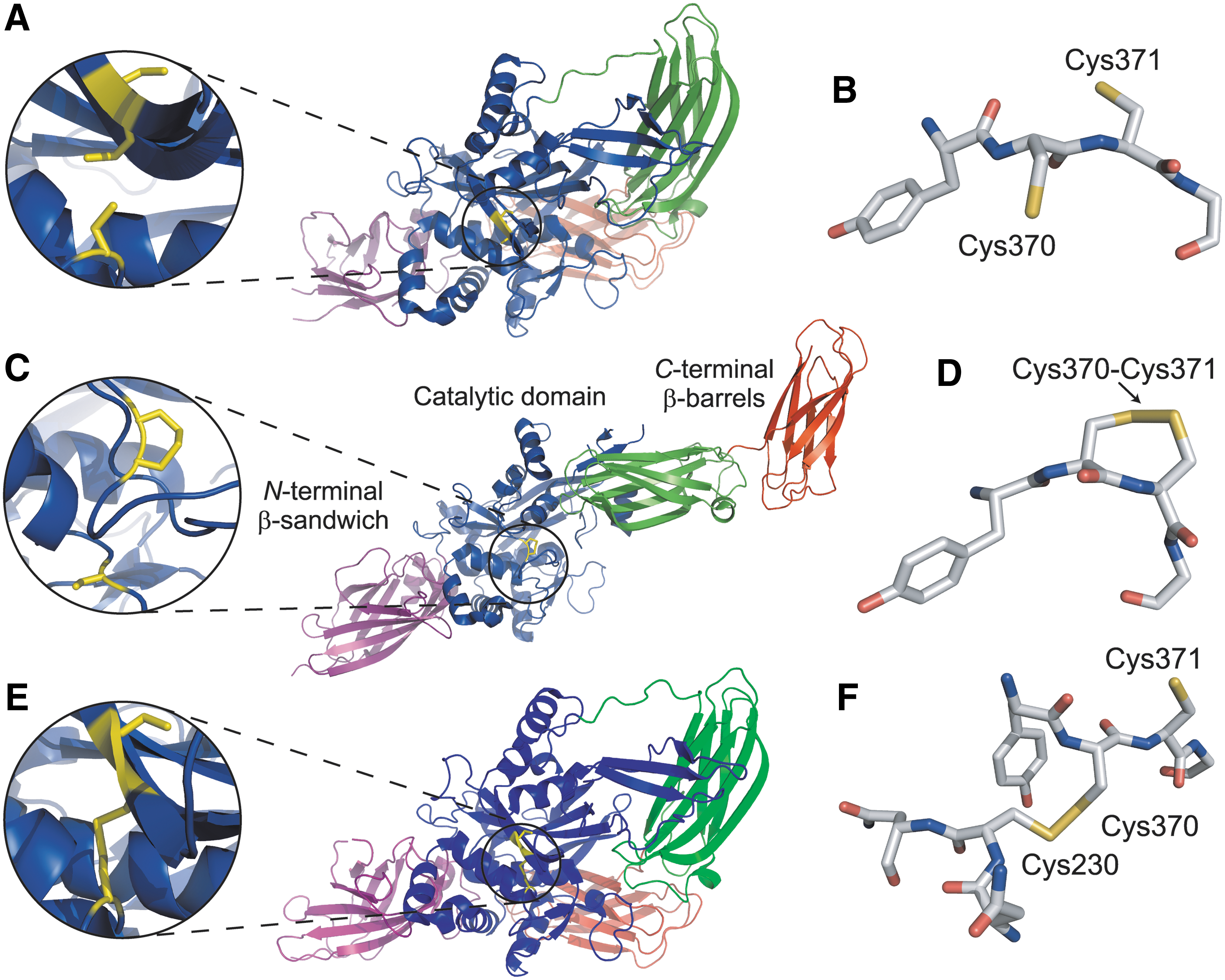
Three crystal structures solved by different laboratories have provided a glimpse into how TG2 is regulated by an allosteric disulfide bond. The first structure shows that in the presence of GDP, TG2 assumes a closed and catalytically inactive conformation with Cys230, Cys370, and Cys371 all in a reduced thiol form (Fig. 12A, B) (PDB: 1KV3) (14, 110). In the presence of calcium and the absence of guanine nucleotides, TG2 forms an open and extended conformation, with a disulfide bond formed between Cys370 and Cys371 (Fig. 12C, D) (PDB: 2Q3Z) (158). The open conformation is believed to be the active form of TG2, though an active-site inhibitor was used to stabilize the active form for crystallization. The role of the Cys370–Cys371 disulfide bond in the open conformation for TG2 activity is less clear.
In addition to possessing GTPase activity, TG2 also hydrolyzes ATP. The crystal structure of ATP-bound TG2 is a closed conformation similar to the GDP-bound structure (Fig. 12E, F) (PDB: 3LY6) (63). However, in the ATP-bound structure, a disulfide bond is formed between Cys230 and Cys370, while in the GDP-bound structure, these cysteine residues were reduced (Fig. 12A, B). The authors proposed that the Cys230–Cys370 disulfide stabilizes the closed and inactive form of TG2 (63). It is possible that the thiol/disulfide exchange might lead to conversion of the closed form of TG2 to the open form (63). The Cys371 sulfur atom is only 7 Å from the Cys370 sulfur atom of the Cys230–Cys370 disulfide bond in the closed ATP-bound structure (Fig. 12E), and the Cys230 sulfur atom is only 11 Å from the Cys370 sulfur atom of the Cys370–Cys371 disulfide bond in the open structure (Fig. 12C). This spacing suggests that the intramolecular disulfide bond could switch between the two forms (see Fig. 2B) depending on the environment.
Mass spectrometry studies of TG2 showed that formation of the Cys370–Cys371 disulfide is progressively favored over the Cys230–Cys370 bond under increasingly oxidizing conditions (185). TG2 deamidase and transamidase activity was found to negatively correlate with the fraction of enzyme containing the Cys370–Cys371 disulfide bond (185). This finding implies that the open conformation of TG2 with a bound inhibitor and a Cys370–Cys371 disulfide bond (158) does not represent the active enzyme and that formation of either of the identified disulfide bonds inactivates the enzyme.
Extracellular TG2 is largely inactive under normal physiological conditions, but can be activated by inflammation and cell injury (81, 183). It is thought that TG2 is released into the extracellular space in the active, reduced form, and is rapidly and reversibly inactivated, possibly through formation of one of the disulfide bonds identified in crystal structures (183, 185). Reduction of the disulfide bond would then reactivate TG2, which is supported by experiments showing that both recombinant thioredoxin and thioredoxin secreted by cocultured monocytes reactivates TG2 on the surface of fibroblasts (81). The small-molecule reducing agent, DTT, also activates TG2.
It will be interesting to determine how allosteric disulfide bond regulation of TG2 might be influenced by the other allosteric regulators of TG2. Calcium and GDP/GTP regulate TG2 activity in different physiological environments in a concentration-dependent manner. For example, inside the cell, the concentration of calcium (a TG2 activator) is low, whereas concentrations of GDP and GTP (TG2 inhibitors) are high, preventing TG2 activation. In contrast, outside the cell, calcium is high, while GDP and GTP are low, conditions that should theoretically activate TG2. TG2 may be kept in an inactive state on the cell surface until reduction of the allosteric disulfide bond activates the enzyme.
The Cys370–Cys371 disulfide in TG2 is a –RHStaple (8) with a standard redox potential of −184 mV (81), so is readily cleaved by the catalytic disulfide of thioredoxin that has a redox potential of −270 mV (90). The Cys230–Cys370 disulfide bond forms a +RHStaple with a calculated DSE of 31 kJ·mol−1, which is significantly higher than the strain on the Cys370–Cys371 bond (14 kJ·mol−1). This is consistent with the hypothesis that the Cys230–Cys370 disulfide bond is a transient intermediate on the way to formation of the more-stable Cys370–Cys371 disulfide.
Given the role of TG2 in pathological processes such as celiac disease, understanding how the disulfide bonds regulate TG2 activity will be essential for understanding TG2 function and developing new treatments that impact the enzyme. Targeting the allosteric disulfide bonds in TG2 and/or the redox processes involved in their regulation may prove to be an attractive strategy for inhibiting unwanted activities of TG2.
2. APS kinase
Sulfur is an essential nutrient for all organisms. In plants, most sulfur is taken up as inorganic sulfate where it can be processed through two metabolic pathways. In both pathways, sulfate is initially activated through adenylation by ATP sulfurylase to form APS. APS kinase then catalyzes the phosphorylation of APS to 3′-phospho-APS [reviewed in (140)]. In Arabidopsis thaliana, adenosine 5′-phosphosulfate kinase (AtAPSK) is essential for reproduction and viability; however, the biochemical regulation of AtAPSK is poorly understood.
The crystal structure of AtAPSK revealed the presence of an intersubunit disulfide bond between Cys86 and Cys119 of two monomers, creating a disulfide-linked dimer (162). Enzyme kinetic studies of AtAPSK showed that reduction of the Cys86–Cys119 disulfide bond increased the catalytic efficiency, but reduced substrate inhibition by APS (162). A mutant enzyme where Cys86 and Cys119 was replaced with alanines had a comparable activity to reduced wild-type AtAPSK.
The Cys86–Cys119 disulfide has a low redox potential of −255 mV at pH 7, approaching that of the catalytic disulfide of thioredoxin (Fig. 13). Formation or cleavage of the Cys86–Cys119 disulfide bond in AtAPSK would likely alter the movements of the N-terminal loop, affecting the catalytic efficiency (162). This allosteric disulfide could determine selectivity for the primary and secondary branches of sulfur assimilation in Arabidopsis thaliana.

3. Ecto-ADP-ribosyltransferase 2.1
ADP-ribosylation is a post-translational protein modification catalyzed by the ecto-ADP-ribosyltransferase (ART) membrane proteins, where an ADP-ribose moiety is transferred from NAD+ to target membrane proteins [reviewed in (230)]. ADP-ribosylation can regulate the activity and function of target proteins. ARTs are expressed as glycosylphosphatidylinositol-anchored membrane proteins or secreted ectoenzymes in mammalian cells. Among the different subtypes of the ART family, the ART2 enzymes have been particularly well characterized in rodent T-cells. In mice, there are two ART2 enzymes, ART2.1 and ART2.2, whereas in rats, the corresponding enzymes are sometimes referred to as Rt6.1 and Rt6.2. They are encoded by two separate, but tandem, genes and share 80% sequence similarity.
Both native and recombinant ART2.1 are inactive until treatment with reducing agents such as DTT and small thiols such as cysteine or glutathione. The subsequent activity is inhibited by alkylation of unpaired cysteine thiols (66). ART2.1 has two more cysteines, Cys80 and Cys201, than ART2.2. Mutation of these cysteines in ART2.1 led to constitutive activity. Conversely, when residues 80 and 201 in ART2.2 were replaced with cysteines, the enzyme was inactive until treatment with reducing agents (66).
The structure of ART2.1 has not yet been solved, but a crystal structure of rat ART2.2 is available. The residues in rat ART2.2 that correspond to the cysteines in ART2.1 (Ser60 and Phe181) are nearby one another, suggesting that a disulfide bond could form between the corresponding two cysteine residues in ART2.1 (139, 165) (Fig. 14). Formation of a disulfide bond between Cys80 and Cys201 in ART2.1 would likely fasten a loop around the NAD-active site and inhibit catalytic activity (139). It has been suggested that ART2.1, but not ART2.2, activity may be regulated by inflammation, where oxidoreductases such as thioredoxin or PDI are secreted (43, 71, 167, 193, 198).

4. Aryl sulfotransferase
Aryl sulfotransferases are bacterial periplasm enzymes that catalyze the transfer of sulfuryl groups between phenolic compounds. They have been implicated in host–pathogen interactions. E. coli aryl sulfotransferase contains a–RHStaple disulfide bond, Cys418–Cys424, which is 12 residues N-terminal of the catalytic His436 (59, 116). The disulfide bond is essential for activity. The DsbL/Dsbl redox couple appears to have coevolved with aryl sulfotransferase to control the redox state of the allosteric bond in the bacterial periplasm (59).
5. Peptidyl-prolyl cis-trans isomerase, AtFKBP13
Photosynthesis and oxygen evolution occur in the thylakoids of chloroplasts in plants and eukaryotic algae. There are several immunophilins within the thylakoid lumen, which are peptidyl-prolyl cis-trans isomerases. AtFKBP13 is a representative of the FKBP immunophilin group. AtFKBP13 contains a–RHStaple disulfide bond, Cys106–Cys111, which is not found in FKBPs in yeast or animals (8, 57). Cleavage of this allosteric bond by chloroplast m-type thioredoxin results in movement of the β-loop motif that it links. This leads to inhibition of peptidyl-prolyl isomerase activity.
6. Carboxyl-terminal Src kinase
Src family kinases are nonreceptor tyrosine kinases involved in many cellular functions. They are negatively regulated by C-terminal Src kinase (Csk) (153). Csk contains an unusual disulfide bond, Cys122–Cys164, in the SH2 domain. Cleavage of the bond changes the structure of the enzyme resulting in a 10-fold increase in kinase activity (130). The disulfide bond exists in either +LHHook or±RHSpiral configurations in the crystal structure of the oxidized enzyme (8). The Csk reductant is not known.
7. Botulinum neurotoxins
Neurotoxins from Clostridium botulinum block synaptic exocytosis in peripheral synapses causing flaccid paralysis [reviewed in (134)]. The toxin is activated by bacterial or host proteases, forming into a heteromer consisting of a 100-kDa heavy chain and a 50-kDa light chain linked by a –RHStaple disulfide bond (Cys429–Cys453 in type A neurotoxins and Cys436–Cys445 in type B).
The neurotoxins enter susceptible cells by receptor-mediated endocytosis where the two-chain molecule integrates into the endosomal membrane and the –RHStaple is cleaved, releasing the catalytic domain of the neurotoxin into the cytosol (48). The released enzyme is an endopeptidase that cleaves the synaptic vesicle fusion complex required for membrane fusion. The –RHStaple disulfide linkage is crucial for the function of the toxin, although the mechanism of cleavage of the allosteric bond is not known.
C. Change in proteolysis
1. Angiotensinogen
Angiotensinogen is a plasma protein and a noninhibitory member of the serpin family of protease inhibitors. It is cleaved by renin and angiotensin-converting enzyme to produce the angiotensin peptides that control blood pressure, fluid homeostasis, and vasoconstriction (34). The crystal structure of angiotensinogen shows that the renin cleavage site is buried in the N-terminal tail and, therefore, inaccessible (225) (Fig. 15). The structure of angiotensinogen bound to renin, though, revealed that the cleavage site is accessible in the complex due to the formation of a disulfide bond between Cys18 and Cys138 in angiotensinogen (225). The Cys18–Cys138 disulfide bond forms a±RHHook with a redox potential of −230 mV (225). Both oxidized and reduced forms of angiotensinogen are found in human plasma.
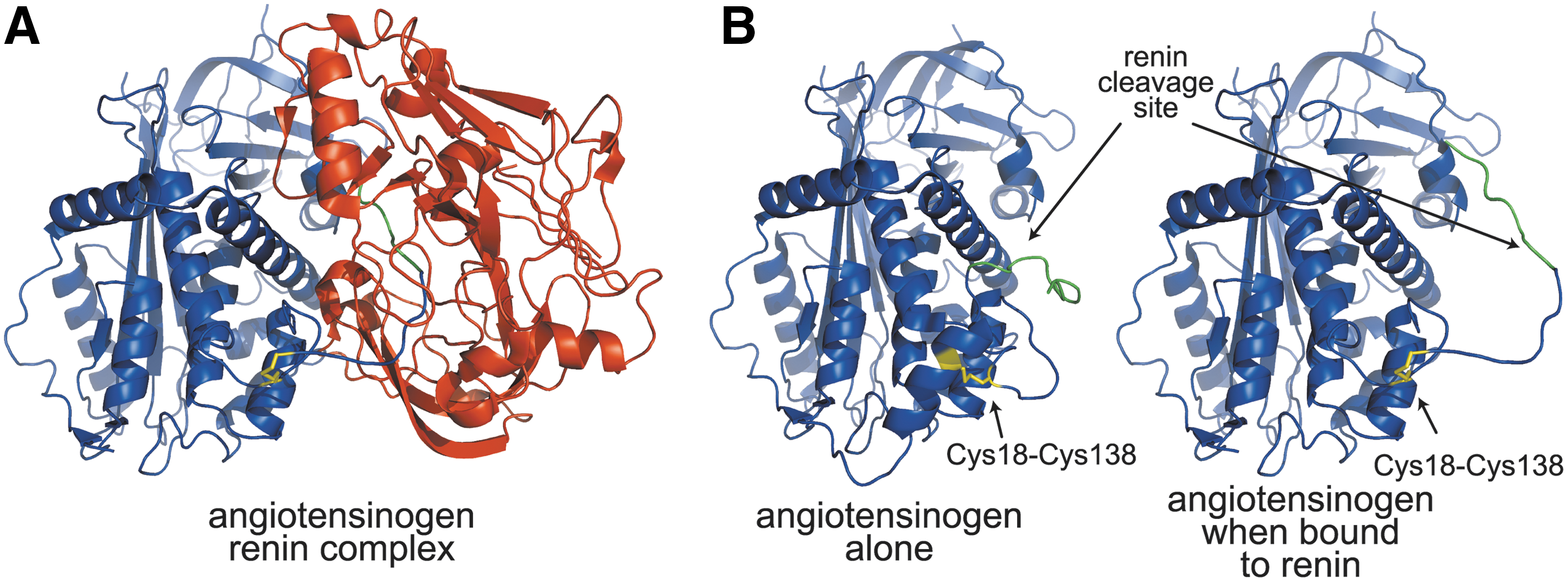
In healthy humans, the ratio of oxidized to reduced angiotensinogen is ∼60:40, which is independent of age or gender. In humans, an increase in the level of oxidized angiotensinogen in the blood leads to increased cleavage by renin and subsequently a rise in blood pressure. This is important, as increased levels of oxidized angiotensinogen have been found to correlate with pre-eclampsia in pregnant women (225). It has been proposed that exposure of maternal angiotensinogen to reactive oxygen species present in the placenta increases the level of oxidized angiotensinogen, contributing to the development of pre-eclampsia (225).
The placental tissue from pre-eclamptic women has been found to have significantly lower levels of antioxidant proteins (such as thioredoxin, thioredoxin reductase, glutathione peroxidase, and others) and decreased enzymatic antioxidant capacity compared to the placental tissue from healthy pregnant women (197). The combination of placental reactive oxygen species and decreased antioxidant capacity in the blood may contribute to the oxidation of angiotensinogen and ultimately the pathogenesis of pre-eclampsia. Because of the essential role of the renin–angiotensinogen system in regulating blood pressure, the allosteric disulfide bond in angiotensinogen may be a useful target for pharmaceuticals for the treatment of pre-eclampsia, estimated to cause 50,000 maternal and 500,000 infant deaths each year globally (225).
2. Plasminogen
The blood protein plasminogen is the zymogen form of the serine protease, plasmin. Plasminogen consists of a Pan-apple domain at the N-terminus, followed by five kringle domains and a serine protease domain at the C-terminus. In the circulation, urokinase or tissue plasminogen activator activates plasminogen to plasmin by cleaving the Arg561–Val562 peptide bond in the serine protease domain. The Pan-apple domain is cleaved from plasmin by autoproteolysis during plasminogen activation. Plasmin primarily cleaves fibrin, which facilitates thrombus dissolution, and it also activates other zymogens and cleaves components of the extracellular matrix (109).
Plasmin is the precursor of the angiogenesis inhibitor, angiostatin (152). Angiostatin is an N-terminal fragment of plasmin and consists of kringles 1–4 and a part of kringle 5. Formation of angiostatin involves cleavage of both disulfide and peptide bonds in plasmin. The evidence implies that disulfide bond cleavage precedes and makes possible the peptide bond cleavage. Reduction of both the Cys462–Cys541 and Cys512–Cys536 disulfide bonds in the kringle 5 domain, followed by proteolysis at the Arg530-Lys531 peptide bond, is the molecular event that results in full-length angiostatin (186). Both the disulfide bonds must be reduced to release angiostatin from plasmin. Additional proteolysis at the Arg474–Val475 and/or Lys468–Gly469 peptide bonds results in smaller angiostatin molecules. A kringle 1–3 fragment can also be produced from these precursors. Both serine- and metalloproteases have been implicated in the peptide bond cleavage events (69, 186, 187).
The Cys462–Cys541 disulfide bond is cleaved in a fraction of plasma plasminogen. We observed that plasminogen can be labeled with a biotin-linked maleimide, and using mass spectrometry identified the Cys462-Cys541 disulfide as being cleaved in a proportion of the protein (D. Butera and P. J. Hogg, unpublished observations). The Cys462–Cys541 disulfide bond is presumably reduced by an oxidoreductase in blood. The recent crystal structure of plasminogen supports the labile nature of the Cys462–Cys541 disulfide. Plasma plasminogen crystallized as a dimer and the structure of the kringle 5 domain differed between the two molecules. Notably, the Cys462–Cys541 disulfide bond is reduced in one molecule and oxidized in the other (Fig. 16A). Cleavage of the Cys462–Cys541 bond changed the structure of kringle 5, as a number of regions of the reduced kringle were not resolved. Law et al. (98) suggested that the bond may have been cleaved by the synchrotron radiation used to reveal the structure. Our findings indicate that the bond is naturally cleaved in a fraction of human plasminogen, what we term reduced plasminogen, and it is the reduced plasminogen that is the substrate for angiostatin formation.
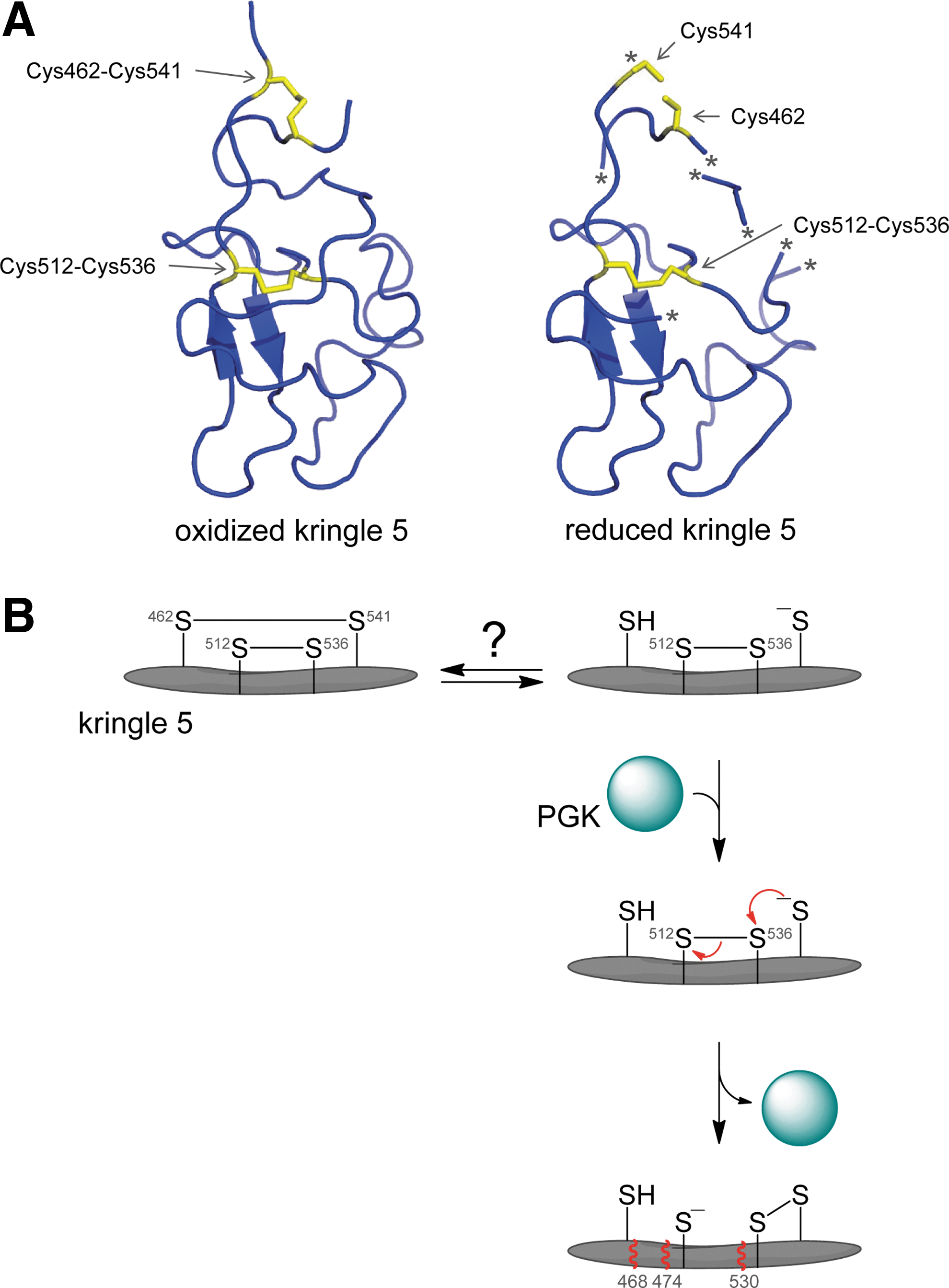
The Cys512–Cys536 disulfide bond appears to be cleaved by intramolecular thiol–disulfide exchange. Stathakis et al. (186) observed that human tumor cells secreted a protein that facilitated angiostatin formation from plasmin. The protein was purified from a fibrosarcoma-conditioned medium and shown to be the glycolytic enzyme, phosphoglycerate kinase (PGK) (100). PGK is the sixth enzyme of the glycolytic pathway where it catalyzes phosphoryl transfer from 1,3-bisphosphoglycerate to ADP to form 3-phosphoglycerate and ATP. PGK is secreted by normoxic tumor cells (37) and influences tumor angiogenesis and tumor growth (204, 205, 229). Human PGK contains seven cysteine residues, and none of them were found to be directly involved in generation of angiostatin from plasmin (101). Alkylation of the PGK cysteine residues, though, or binding of the enzymes substrates, 3-phosphoglycerate and ATP, markedly inhibited angiostatin-forming activity (101). The alkylation and ligand binding presumably change the structure of PGK, which perturbs the interaction with plasmin. We suggest that binding of PGK to reduced plasmin induces a conformational change in kringle 5 that leads to attack by the Cys541 thiolate anion on the Cys512–Cys536 disulfide bond, cleaving the bond (Fig. 16B). This intramolecular bond cleavage results in conformational change and exposure of the peptide backbone to proteolysis C-terminal of residues 468, 474, and 530. This is the simplest sequence of events that can explain all the available data.
Notably, other molecules that interact with plasmin can apparently facilitate the same thiol/disulfide exchange in kringle 5. Binding of the annexin A3-S100A10 heterotetramer (91), plasma-membrane-associated β-actin (203), or a truncated porcine plasminogen activator inhibitor 1 (residues 80–265) (141) to plasmin results in generation of kringle-containing angiostatin fragments. In addition, plasmin undergoes autoproteolysis at alkaline pH, producing kringle fragments and a catalytically active microplasmin fragment with an Lys531 N-terminus (85, 214, 215). It appears that plasmin reduction and proteolysis at alkaline pH are mediated by the same chemical events facilitated by PGK at neutral pH (187). It may be that alkaline pH facilitates angiostatin formation by simply increasing the fraction of Cys541 in the thiolate anion state, which is the state that attacks the Cys512–Cys536 disulfide bond leading to angiostatin release (Fig. 16B).
3. MICA
NKG2D is an activating immune receptor that is expressed by most NK cells, some CD8+ T-cells, and macrophages (56). MICA [a major histocompatibility complex class I (MHCI) homolog] is an NKG2D ligand that is activated by cellular stresses and is present in intestinal epithelium and epithelium-derived tumors (56, 60, 61). NKG2D-bearing cells can mediate tumor rejection, but tumors can evade this immune response by expressing and shedding MICA (56, 83). Shed MICA induces internalization and degradation of NKG2D.
MICA contains three disulfide bonds: Cys96–Cys164, Cys202–Cys259, and Cys36–Cys41. Cleavage of the Cys202–Cys259 disulfide by tumor cell surface endoplasmic reticulum protein 5 (ERp5) triggers proteolytic shedding of MICA (83). The Cys202–Cys259 bond is inaccessible in the crystal structure, which implies that a major conformational change is necessary for ERp5 to react with the disulfide (83).
4. Factor XI
Factor XI is a disulfide-linked homodimer that circulates in plasma. Proteolytic activation of factor XI to XIa initiates the intrinsic/consolidation phase of coagulation. The Cys362–Cys482 and Cys118–Cys147 disulfide bonds of factor XI are reduced by thioredoxin and PDI (53). The reduced protein is more efficiently activated by thrombin, factor XIIa, or FXIa than the oxidized protein. The Cys362–Cys482 −/+RHHook disulfide bond is only six residues from the scissile Arg369–Ile370 peptide bond. Cleavage of this disulfide bond presumably enhances the efficiency of cleavage of the scissile bond by triggering a favorable conformational change in the vicinity. Patients with antiphospholipid syndrome and thrombosis have higher levels of reduced protein in their plasma than healthy controls. The elevated levels of reduced factor XI may contribute to the thrombosis in these patients.
D. Oligomer formation
1. CD4
CD4 is a T-cell receptor that functions as a coreceptor for binding of T-cells to antigen-presenting cells and is the primary receptor for HIV. It consists of four immunoglobulin-like domains, a transmembrane domain and a short cytoplasmic region. The disulfide bond in the second extracellular domain of CD4, Cys130–Cys159, is a –RHStaple that is reduced on the T-cell surface by thioredoxin, which leads to formation of disulfide-linked homodimers of CD4 (122, 155, 180). The redox potential of the Cys130–Cys159 bond is −241 mV. Domain swapping of D2 has been identified as the most likely mechanism for formation of the dimer, where disulfide bonds form between Cys130 in one monomer and Cys159 in the other (115, 172). One of the cysteine thiolates of the cleaved Cys130–Cys159 bond in one molecule of CD4 bond attacks the Cys130–Cys159 bond in another molecule resulting in a disulfide-linked dimer.
Disulfide-linked dimer formation enhances CD4's coreceptor function (115), but impairs HIV entry and env-mediated cell–cell fusion (20, 121). These findings imply that monomeric reduced CD4 is preferred by HIV, whereas dimeric CD4 is preferred by antigen-presenting cells. Modeling of the T-cell receptor and domain-swapped CD4 dimer bound to MHC class II and antigen also supports the domain-swapped dimer as the immune coreceptor (115).
2. von Willebrand factor
VWF is a multimeric, plasma glycoprotein involved in hemostasis through platelet adhesion and aggregation at the injured vascular wall and as the chaperone for factor VIII (170). The majority of VWF is produced by vascular endothelial cells, and though VWF has been found in megakaryocytes, the physiological significance is unclear. VWF circulates as a series of multimers composed of variable numbers of 500-kDa dimeric units. The VWF monomer contains a number of specific domains' important for function. The order of domains is D1-D2-D′-D3-A1-A2-A3-D4-B1-B2-B3-C1-C2-CK, and each monomer contains binding sites for collagen and for platelet glycoproteins Ib and αIIbβ3 (170). Deficiency of VWF, known as von Willebrand disease, is the most common inherited bleeding disorder (170, 171).
VWF structure is influenced by the blood shear forces, where it reversibly transitions from a loosely coiled ball to an elongated arrangement (176, 182, 189). There is an irreversible structural transition when collagen is present. VWF self-associates into a flexible clump at a critical shear rate and when perfused over collagen forms an irreversible web of fibers that binds platelets (12, 189). VWF secreted by cells also forms fibers or strings when exposed to shear (15, 31, 108). The strings can be several cell diameters in length, and they bind platelets. Fiber formation involves self-association of VWF (12, 15, 173, 196).
VWF self-association and fiber formation involve thiol/disulfide exchange as thiol alkylators or small-molecule thiols inhibit the formation of these structures (28, 31). In accordance with this chemistry, VWF contains unpaired cysteine thiols (28, 31, 50, 108, 125, 222). We recently mapped the location and abundance of the unpaired cysteines in the C-terminal part of VWF and elucidated their role in lateral association.
The VWF C2 domain Cys2431–Cys2453 disulfide bond was reduced in about 75% of a recombinant C-terminal fragment, and fragments containing all three C domains or just the C2 domain formed monomers, dimers, and higher-order oligomers when expressed in mammalian cells (51). Both the Cys2431–Cys2453 and nearby Cys2451–Cys2468 disulfide bonds were found to be involved in oligomer formation (Fig. 17). Reduction of the C2 domain Cys2431–Cys2453 disulfide bond in one molecule, presumably by an oxidoreductase in blood, creates a Cys2431 thiolate anion that attacks the Cys2431 sulfur (of the Cys2431–Cys2453 disulfide bond) in another VWF molecule, resulting in a disulfide-linked dimer. The Cys2451–Cys2468 disulfide/dithiol then mediates formation of trimers and higher-order oligomers (Fig. 17B).
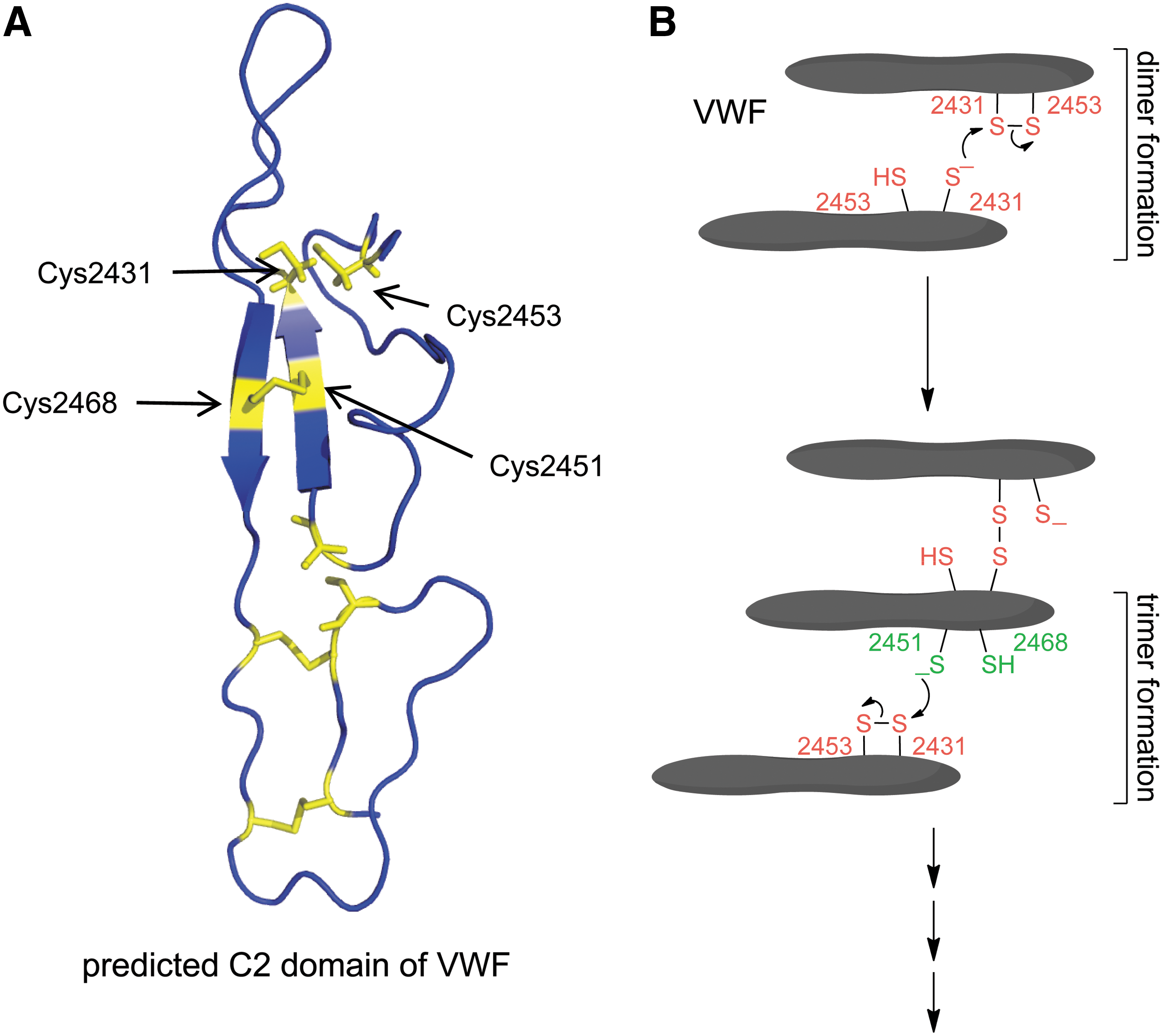
The hemostatic role for lateral association of VWF has not been measured, as defects of lateralization would not be reflected in abnormal results in standard tests of VWF antigen, activity, or multimer patterns. It may be that impaired VWF lateralization is responsible for hemostatic defects in some patients. Mutations in Cys2431 and Cys2451 should result in impaired lateral association that may present as bleeding, while mutations in Cys2453 and Cys2468 should result in enhanced lateral association and thrombosis.
3. Vascular endothelial growth factors C and D
The vascular endothelial growth factors (VEGFs) and their tyrosine kinase receptors play a fundamental role in vasculogenesis, angiogenesis, and lymphangiogenesis (111). There are seven mammalian VEGFs (VEGF, VEGF-B–F, and placental growth factor, PlGF) and three VEGF receptor tyrosine kinases (VEGFR-1–3). The main angiogenic pathway is mediated by VEGF/VEGFR2 signaling, and blocking this pathway was the first antiangiogenic strategy for the treatment of cancer. VEGF-B and PlGF signal through VEGFR-1, which is involved in pathological angiogenesis, recruitment of macrophages to tumors, and lipid metabolism in the heart. VEGF-C and D engagement of VEGFR-3 is involved in lymphangiogenesis in normal and pathological conditions.
The VEGFs form disulfide-linked antiparallel homodimers that are important for their productive interaction with their receptors (Fig. 18A). Ablation of the interdimer disulfide bond destroys activity (195). Covalent dimer formation is constitutive for all VEGFs, except VEGF-C and D, where dimer formation is regulated by a cysteine residue that is unique for these VEGFs and the Caenorhabditis elegans VEGF homolog, PVF-1 (104, 105, 195). The sulfur atom of the unique cysteine residue is only a few Å from the interdimer disulfide bond (Fig. 18A). Mutation of the unpaired cysteine to alanine increases the activity of VEGF-C and D, presumably by stabilizing the dimer (105, 195).
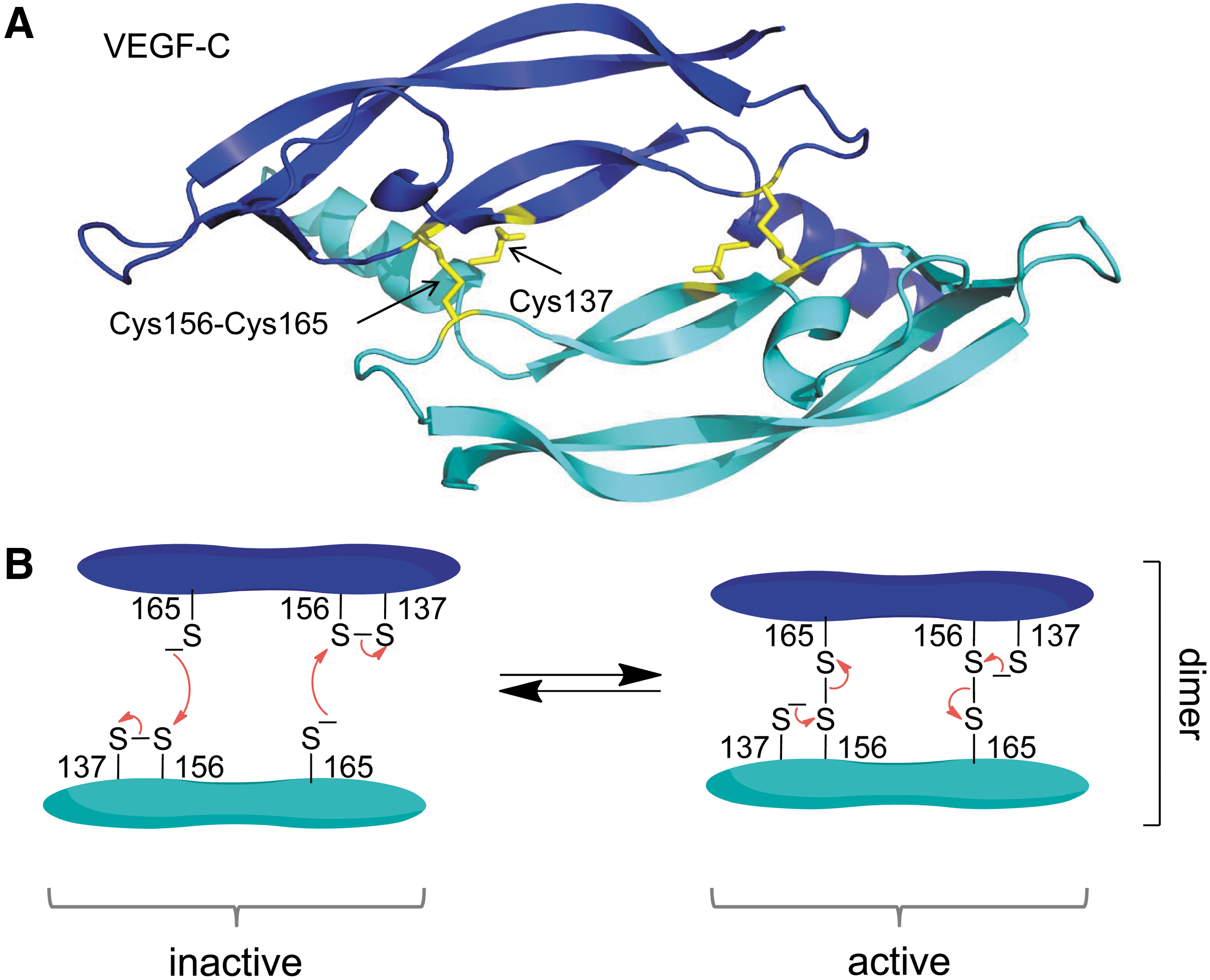
These observations imply that the unique cysteine residue cleaves the interdimer disulfide bond of VEGF-C and D by thiol/disulfide exchange, which inhibits the activity of these VEGFs (Fig. 18B). This process is likely regulated. In VEGF-D, alkylation of the unique cysteine with a small thiol promotes covalent dimer formation (38). Factors that influence the unique cysteine thiol, such as the presence of other thiols or thiol alkylators such as NO, are predicted to control of the biological activity of VEGF-C and D. The inter-dimer disulfide bond of VEGF-C (Cys156-Cys165) is a –LHHook that links a β-strand to a β-loop (105).
IX. How to Identify Allosteric Disulfides
A. Data mining
Computational and bioinformatics tools can be used to identify possible allosteric disulfide bonds in proteins, which can then be tested experimentally. The success of this approach relies on high-resolution three-dimensional structures (X-ray, NMR, or electron microscopy), and a dataset of allosteric disulfides from which common features can be derived.
Structural features of a disulfide bond, such as configuration, solvent accessibility, and the secondary structures that the disulfide links, can be useful measures. We have developed a simple online tool that analyzes disulfide bonds in any three-dimensional structure in .PDB format (
–RHStaple disulfides account for about 9% of all disulfides in X-ray structures and 6% in NMR structures (175). These bonds nearly always link between two antiparallel β-strands or β-loops in a β-sheet and have a very short Cα–Cα distance (68, 174, 175). The–RHStaples also have a higher DSE (mean of 18 kJ·mol−1) than the average disulfide bond (mean of 13 kJ·mol−1) in crystal structures. The higher energy can be due to deformation of the β-sheet itself, as the adjacent strands pucker to accommodate the disulfide bond. A high potential energy is also consistent with a possible functional role, as higher energy disulfide bonds are easier to cleave than disulfide bonds with lower energy (174). Notably, viral coat proteins that mediate attachment and entry of the virus into target cells are particularly rich in –RHStaple bonds (213).
It is important to note that crystal structures may not represent the solution structure of the disulfide bond. We compared protein structures for which there are both X-ray and NMR models and found that the same disulfide bond can exist in different configurations in different models (175). Interestingly, disulfide bonds that form –RHStaples in crystal structures often also form a –LHStaple configuration in NMR structures. The –LHStaple typically has an even shorter Cα–Cα distance and a higher DSE than the –RHStaple (175). It is thought that the high strain energy associated with the –LHStaples makes them relatively rare in crystal structures that have little tolerance for strained structures.
Isomerization of allosteric disulfide bonds may well be an important aspect of their function. Considering the highly directional nature of disulfide bond cleavage (see Fig. 2), it seems probable that a protein reductant might sample a number of alternative isomers of an allosteric disulfide in solution, which reflect the dynamism of the substrate protein, but only react when a favorable configuration is presented. For example, a –RHStaple bond may need to switch to a –LHStaple configuration before it is cleaved. Substrate-binding proteins such as chaperones are predicted to influence the alternative configurations of an allosteric disulfide.
It is likely that allosteric disulfides with a wide variety of configurations that link all types of secondary structures will be identified, so it is important not to dismiss a potential allosteric bond if it does not share any features with the known bonds. Nevertheless, mining of structural databases for traits associated with allosteric disulfides can lead to the discovery of these bonds. For example, since the identification of the –RHStaple bond as a potential allosteric disulfide in 2006 (174), eight –RHStaples in different proteins have since been discovered to be allosteric. The allosteric bonds in TG2 and von Willebrand factor were speculated before the recent functional studies, and the β2GPI allosteric bond was identified by data mining (8).
B. Experimental screens
Proteomic screening methods have been developed to assist in identifying redox-regulated disulfide bonds. A recent study identified many redox-regulated disulfide bonds on the surface of T-cells in vitro and on the surface of mouse splenocytes during inflammation in vivo (127).
For the in vitro study, cultured T-cells were placed in a reducing environment to mimic the conditions of immune activation, which leads to localized mild reducing conditions. One of four different reducing agents was used: the small-molecule tris(2-carboxyethyl)phosphine, and the endogenous protein reductants, thioredoxin, PDI, or GILT. Before adding reducing agents, free thiols present on the cell surface were alkylated with an untagged maleimide derivative. After cells were treated with the reducing conditions, any newly reduced thiols were labeled with a biotinylated maleimide. To isolate all the membrane proteins, lectin-affinity chromatography was used to purify the membrane glycoproteins, and the biotinylated proteins were further purified using avidin-affinity chromatography.
Mass spectrometry was used to identify the biotinylated proteins, which represented proteins that were reduced by one or more of the reducing agents. A wide range of T-cell membrane proteins were found to have disulfide bonds reduced by the protein reductants, thioredoxin, PDI, and GILT, with mass spectrometry identifying 87 proteins as candidates containing redox-labile disulfide bonds. The proteins identified ranged from activating and inhibitory receptors to proteins involved in cell adhesion, antigen presentation, transport, and metalloproteinases. Secreted thiol reductases were also identified, as expected.
The majority of proteins identified were reduced by more than one of the reductants, but ∼30% of the identified proteins were reduced by only a single reducing agent, indicating some selectivity for different disulfides. The actual disulfide bond being reduced in a protein was identified in approximately half of all the identified proteins. The remainder of identified proteins did not contain a biotin-labeled cysteine, which may have been due to lower abundance of the protein and the detection limit of mass spectrometry or peptide ionization problems. It is also possible that these identified proteins do not contain a labile disulfide, but rather were bound to a protein with a labile disulfide bond and were copurified.
For the in vivo study, LPS was administered to mice, which induces a strong immunological response and leads to accumulation of GILT in serum (92). Splenocytes were isolated from LPS-treated and control mice and labeled with biotin–maleimide, and the cell surface proteins were purified as for the in vitro study. Potential allosteric disulfides were found in 37 different proteins, including those involved in B- and T-cell activation (CD8, CD14, CD19, CD22, CD132, and SLAM family receptors), cell adhesion (ICAM1, integrins α2, αX, β5, α3 and β7, and Thy-1), and complement activation (C3, C4-B, and Crry).
Several of the proteins identified on the T-cell surface in vitro were also found on the surface of the activated splenocytes, which indicates that both cell culture and animal models can be useful in identifying proteins regulated by one or more allosteric disulfide bonds. It is likely that many more allosteric disulfide bonds will be identified using this approach as the sensitivity and specificity of proteomics techniques improves.
X. What the Future Holds
The identification and study of allosteric disulfides began about two decades ago (69, 73). The pace of research on this topic has been quite slow, mainly due to the difficulties in identifying and studying this mechanism of post-translational regulation. This has changed in recent years, as experimental methodologies have improved, and the understanding of and appreciation for this means of post-translational protein control have developed. There are now a few dozen good examples of allosteric disulfides (see Table 2), and many dozens more potential bonds that are under investigation. It may be that cleavage of disulfide bonds rivals cleavage of peptide bonds, in terms of the number of proteins regulated by these means. Another few decade years of research are required, though, to answer this question.
Allosteric bonds have been identified in viruses, bacteria, plants, and mammals, so this type of post-translational control appears to apply generally in nature. It is intriguing that disulfide bonds in general have accrued faster in more-complex eukaryotes than in simple species, and that once acquired, they are rarely lost during evolution of the organism (212). There are not enough data as yet to explore the evolution of allosteric disulfides specifically. We predict, though, that the acquisition of these bonds may correlate with sophistication of protein function in complex organisms and contribute to the diversity of life. It is also likely that the proteins involved in cleavage of the allosteric bonds have coevolved with the disulfides.
A research area that we feel will expand rapidly in the next few years is the study of allosteric disulfides associated with human disease. There are already two clear examples of these bonds. The allosteric disulfide in β2-glycoprotein 1 has been linked to unwanted thrombosis in the antiphospholipid syndrome (77), which is associated with accelerated atherosclerosis and recurrent miscarriages, whereas the allosteric disulfide in angiotensinogen has been linked to pre-eclampsia (225), which is a hypertensive crisis of pregnancy. It is likely that some allosteric disulfides will be good targets for new pharmaceuticals. Small compounds or biologicals that selectively target the oxidized or reduced protein and either eliminate the disease form of the protein or stabilize the harmless form will likely emerge in the near future.
Footnotes
Acknowledgments
We thank Jason Wong for the analysis of disulfide bonds in intracellular-versus-secreted human proteins. Protein structure figures have been generated using PyMOL.