
Abstract
Introduction
The clearance of dying cells involves many surface molecules, adaptors, and chemotactic proteins. As such, it is regulated at multiple levels. Despite differences in outcomes (pro- versus anti-inflammatory), there is a certain degree of crosstalk between intracellular compartments once cargo has been internalized. Autophagy, literally “self-eating,” is the process by which cells clear protein aggregates, whole organelles, and other components for the concomitant purposes of nutrient derivation and the mitigation of cell stress that is mediated by potentially cytotoxic aggregates and organelles (e.g., mitochondria) which have persisted past their usefulness, and it is also an important source of antigen derived from degraded pathogens and abnormal self-proteins (19, 64, 128). As a constitutive process involving the formation of double-membrane vesicles that receive input from endosomal compartments and ultimately fuse with lysosomes (70, 119), this process has a dynamic relationship with both immuno-stimulatory and silent phagocytosis. This relationship is made further complex by the active role redox-dependent signal transduction plays in modulating both phagocytosis and autophagy (Fig. 1) (3, 49, 98).

Given the commonalities between the phagocytic and autophagic pathways, including shared molecules and their sensitivity to redox signaling, here we review the most recent findings that link elements of both pathways, specifically through redox-dependent signal transduction. Furthermore, these interconnected cellular processes are placed in the context of cell death and immunity in both health and disease.
Eat-Me, Find-Me, and Damage-Associated Molecular Pattern Molecule Signals
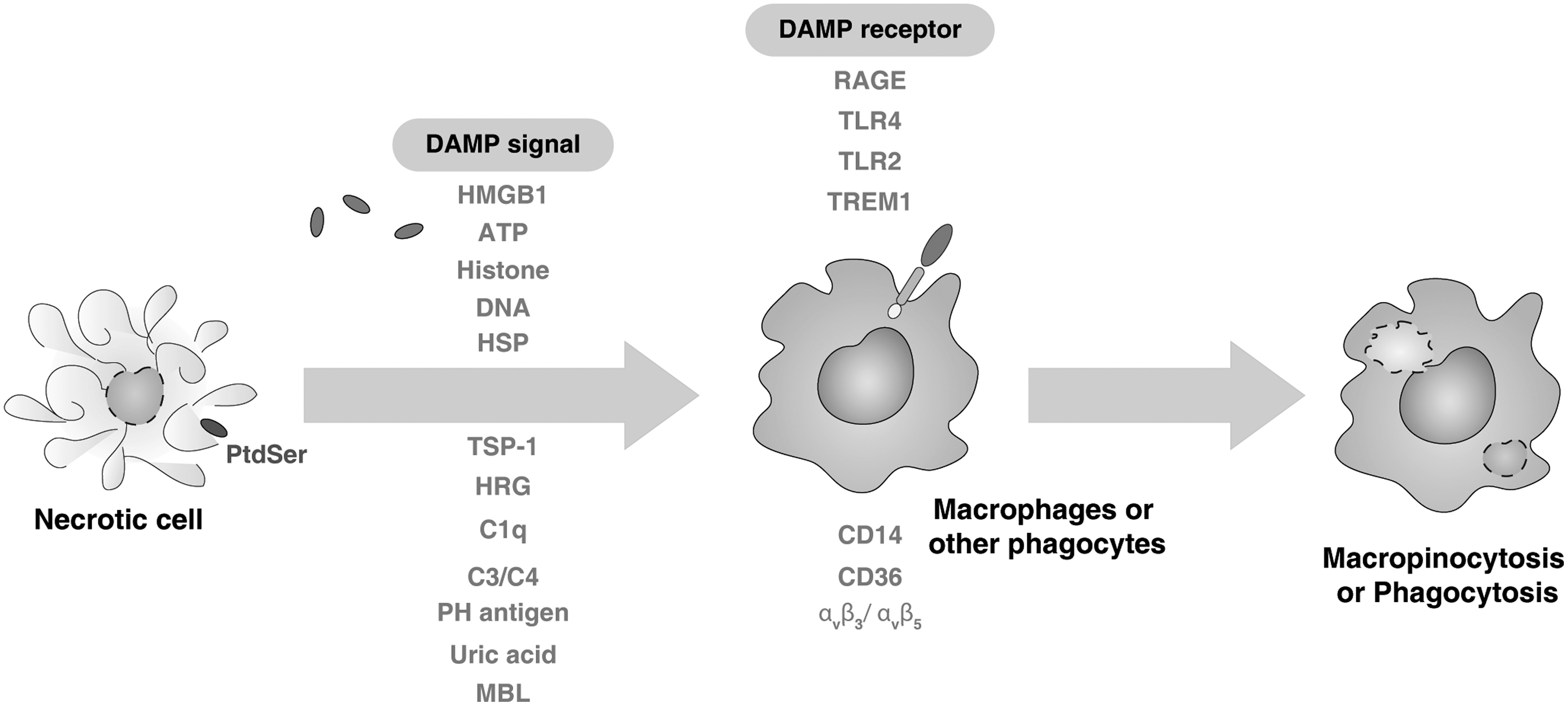
In order for a dying cell to become phagocytosed, it should be rendered recognizable to phagocytes (tissue-resident macrophages, monocytes, and dendritic cells [DCs]). Generally speaking, the initiation of successful phagocytosis relies on four steps (85, 86): (i) the release of “find-me” signals that recruit phagocytes; (ii) the receptor ligation-dependent recognition of “eat-me” signals and the down-regulation of “don't eat me” signals such as CD47, and the complement regulatory proteins, CD200, which facilitate engulfment; (iii) the processing and degradation of corpses involving a series of phagosomal maturation steps; and (iv) the suppression or initiation of inflammatory responses depending on additional innate immune stimuli. Recently, potential find-me signals released by apoptotic cells were reported, namely lysophosphatidylcholine (63), CX3CL1/fractalkine (121), sphingosine 1-phosphate (S1P) (40), nucleotides adenosine triphosphate (ATP), and uridine 5 triphosphate (UTP) (23), which bind to G-protein-coupled receptors, including P2Y2, CX3CR1, S1P1–5, and G2A in macrophages and monocytes (Fig. 2). Unfortunately, current understanding of “find-me” signals is still limited. Interestingly, the iron-binding protein lactoferrin could serve as an anti-attraction (“keep-out”) signal by apoptotic cells to inhibit the migration of granulocytes (12). Although macrophages and DCs are highly motile under basal conditions, certain factors associated with cell death are known to possess chemotactic properties. Many damage-associated molecular pattern molecules (DAMPs) that are released actively during cell stress or passively during necrosis (Fig. 3) or late-stage apoptosis are associated with chemotaxis (72). High mobility group box-1 (HMGB1), the prototypic DAMP, when released into the extracellular milieu attracts both macrophages and DCs by itself, and also binds to chemokines such as CCL19 and CXCL12 (21), thereby promoting more efficient gradient-dependent migration toward them. Other DAMPs, including nucleotides, ATP, and UDP, have also been ascribed chemoattractant properties and are known to be passively released from necrotic cells when the integrity of the plasma membrane is compromised and actively released from apoptotic cells via pannexin 1 channels (14). Given the fluidity of the plasma membrane during both forms of cell death, it is possible that additional, unidentified intracellular proteins may also escape into the extracellular space and recruit phagocytes to sites of injury and apoptosis.
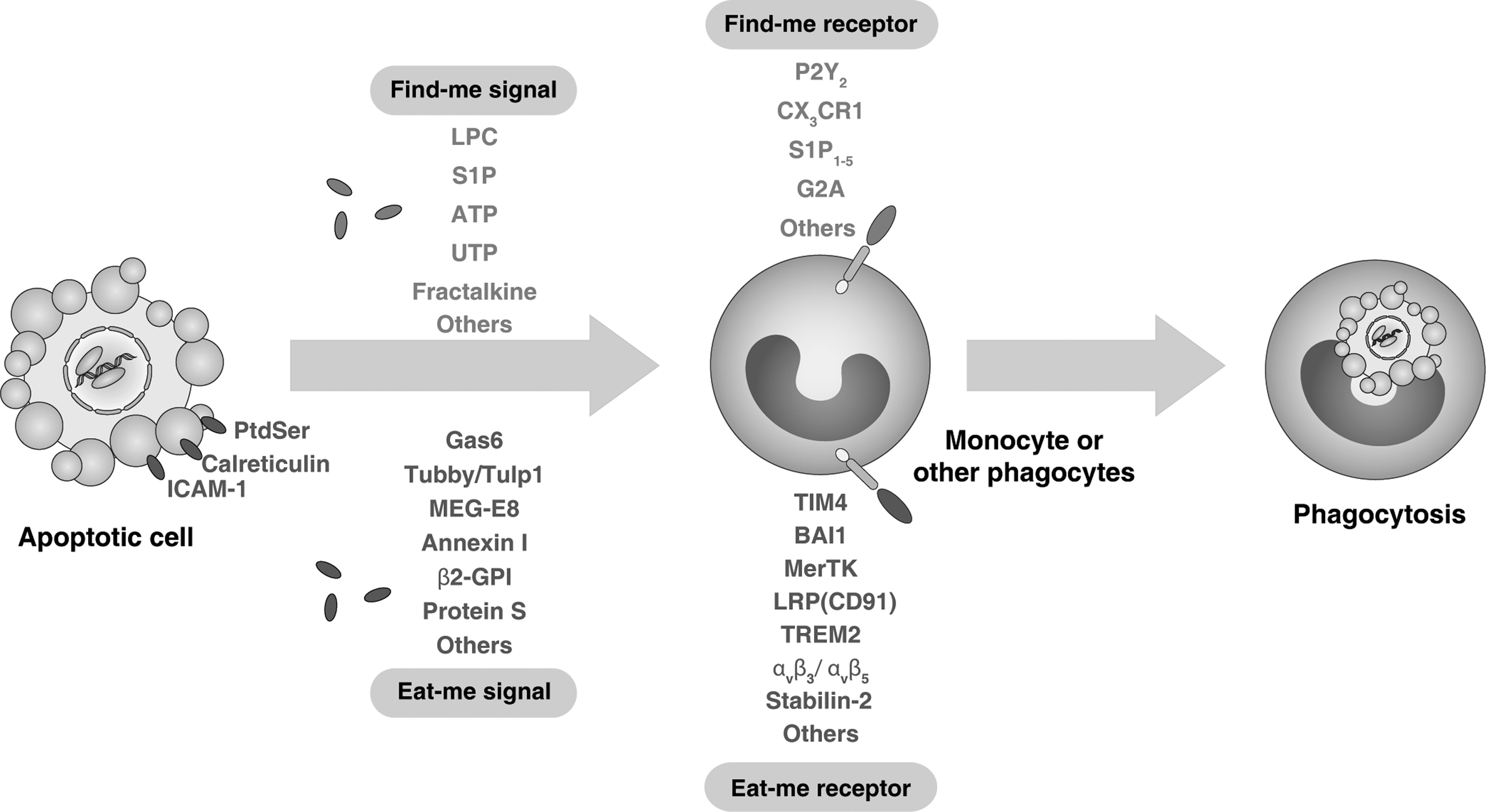
By contrast, “eat-me” signals that facilitate recognition by phagocytes are much better understood (85, 86). Eat-me signals or phagocytosis ligands can be classified into two major categories, membrane-anchored eat-me signals (e.g., phosphatidylserine [PtdSer], intercellular adhesion molecule-1 [ICAM-1], calreticulin) and soluble bridging molecules (e.g., growth-arrest-specific 6 [Gas6], protein S, globule EGF factor 8 protein [MFG-E8], β2-glycoprotein-I [β2-GPI], and annexin I). These signals can be trafficked to the cell surface by canonical and noncanonical secretory pathways, as well as “revealed” by both conformational and enzymatic modification of the plasma membrane (66). PtdSer (which localizes to the inner leaflet of the plasma membrane) becomes displayed on the cell surface when portions of the lipid bilayer flip during apoptosis (25). This allows interactions with receptors and bridging molecules such as T-cell immunoglobulin domain and mucin domain 4 (TIM4), MFG-E8, brain angiogenesis inhibitor 1 (BAI1), and Mer tyrosine kinase (MerTK) expressed on phagocytes, thereby promoting phagocytosis (42, 103). Proteins, such as calreticulin, can also be up-regulated on the surfaces of cells during apoptosis and interact with and activate distinct phagocytic receptors such as low-density lipoprotein receptor-related protein (LRP or CD91) (32).
Unlike apoptosis, cells that release intracellular contents during necrosis or are opsonized with antibodies due to the expression of abnormal self or pathogen-derived antigen are recognized by phagocytes such as macrophages through macropinocytosis mechanisms which are independent of apoptotic cells (Fig. 3) (62). Macropinocytosis is a form of bulk uptake of fluid and solid cargo into cytoplasmic vacuoles, called macropinosomes, and has been studied mostly in relation to antigen presentation. In contrast, necrotic cells can also be phagocytosed by DCs (92). In addition to the activation of complement cascades, an antibody-coated cell or pathogen becomes immediately recognizable to phagocytes which express cell surface receptors that bind to the Fc region of an antibody termed FcR (31). Activation of these receptors initiates the phagocytosis of the target cell, and in concert with pattern recognition receptor (PRR) ligation by pathogen-associated molecular pattern (PAMP) molecules or DAMPs, initiates maturational programs within the phagocyte that stimulate adaptive immune responses via presentation of antigenic peptides in class I and class II major histocompatability complexes (MHC-II) derived from the phagocytosed target cell (43).
Phagocytosis: A Process of Killing Cells or Microbes
Unlike autophagy, phagocytosis has been a recognized cellular process for more than a century (78). Macropinocytosis involves the constitutive “imbibing” of extracellular fluid in a nonspecific fashion, whereas phagocytosis is a highly regulated process that is dependent on precise receptor-ligand interactions. The most well-characterized form of phagocytosis is the internalization of antibody-coated (opsonized) particles by the FcR, and is, therefore, discussed as an example of how phagocytosis proceeds at the molecular level (37, 75). It should be noted, however, that although there are numerous forms of phagocytosis which operate under both parallel and distinct initial molecular regulation, the downstream mechanisms of vesicular formation and fusion are largely conserved.
The ligation of a singular FcR by an opsonized particle is not sufficient to initiate phagocytosis (105). In order to propagate a successful signal that initiates phagocytosis, engagement of multiple receptors is required. This is accomplished by the clustering of receptors that occurs immediately after initial engagement. In turn, this clustering facilitates the ligation of more unoccupied FcR sites and so forth. After this, a tyrosine phosphorylation-dependent signaling cascade (33, 37) is transduced within the phagocyte that causes the recruitment of specialized adaptor proteins such as Grb2-associated binder 2 (Gab2) (39) and CrkII (88), both of which mediate the recruitment of additional scaffolding proteins and initiate the formation of the cellular machinery required for the reorganization of both the actin cytoskeleton (77) and the plasma membrane (125) which are necessary for successful phagocytosis. Significantly, phagocytosis is significantly obstructed when these adaptor proteins are chromosomally deleted in mice (39). Rapid and efficient removal of dying cells and foreign microbes by phagocytes is important during development, tissue homeostasis, and immune responses (Fig. 4). Phagocytosis of apoptotic cells results in tolerance or anti-inflammatory responses. Necrotic cells release DAMPs that stimulate pro-inflammatory responses. Autophagy plays dual roles in promoting or discouraging inflammation. Initially, the plasma membrane serves as the source of lipid membrane for a newly forming phagosome (125), although there is evidence suggesting that additional organelles such as the endoplasmic reticulum contribute to the membrane as the phagosome matures (120). The early phagophore forms after scission from the plasma membrane and begins to mature as it traffics toward the lysosome in a GTPase-dependent fashion (13). The late phagosome acquires a much more acidic pH (59) and fuses with the lysosome via interactions with lysosome-associated membrane proteins (LAMP)-1 and LAMP-2 (51). This newly formed vesicle is termed the phagolysosome and is a degradative organelle in which lytic proteins process the phagocytic cargo and ultimately derive both nutrients and antigenic peptides for MHC-II presentation to immune effector cells (43). Toll-like receptor (TLR) signaling contributes to the regulation of phagosome maturation in macrophages and DCs (10, 11). Moreover, engaging the autophagic pathway via TLR signaling enhances phagosome maturation by microtubule-associated protein 1 light chain 3 (LC3)-associated phagocytosis (LAP) (94). In contrast, other independent studies demonstrate that TLR stimulation does not influence phagosome maturation assayed by defined particles and quantitative methodology; therefore, additional studies are needed to more clearly define the role of TLRs in both autophagy and phagocytosis (89, 129, 130).

Autophagy: A Lysosomal Degradation Pathway
Much like phagocytosis, autophagy is a process that impinges on the formation, maturation, and fusion of vesicles encapsulated by the lipid membrane and facilitates the degradation of selective cargo derived from intracellular components, including whole organelles such as mitochondria and ribosomes, as well as cytotoxic protein aggregates. In addition, foreign pathogens acquired by cytoplasmic sequestration are also degraded by a specialized form of autophagy termed xenophagy (19, 64). There are at least three recognized types of autophagy: macroautophagy, microautophagy, and chaperone-mediated autophagy (Fig. 5) (128). This review focuses on macroautophagy and will hereafter refer to macroautophagy as simply “autophagy.”

The initial steps of autophagy involve the formation of a specialized, double-membranous vesicle termed the isolation membrane, namely phagophore. Recent studies have found that the sources of membrane for these nascent autophagic vesicles can be from the plasma membrane itself, the golgi complex, and even the mitochondrial membrane (119). In addition, under conditions of differing autophagy-initiating events, the primary membrane source may also be different. For example, during cellular starvation-induced autophagy, studies suggest that mitochondria are the primary source from which the membrane is derived (41). As the isolation membrane matures, the protein LC3-I (called Atg8 in yeast) becomes covalently lipidated into LC3-II and incorporated into the membrane as a crucial scaffolding protein (52). Given this role, the conversion of LC3-I to LC3-II serves as a marker for heightened autophagic flux and not surprisingly, cells deficient for LC3 are unable to successfully initiate autophagy (79). Before fusion of the isolation membrane and the formation of the closed vesicle termed the autophagosome, autophagic cargo is recruited via adaptor molecules such as p62 (9, 81), Nix (93, 102), nuclear dot protein 52 kDa (NDP52) (117), neighbor of BRCA1 gene 1 (NBR1) (60), optineurin (126), and galectin 8 (118) (Fig. 5). These molecules contain ubiquitin-binding domains that recognize poly-ubiquitinated protein aggregates, organelles, and bacteria. The autophagosome then traffics to and fuses with the lysosome, forming the autolysosome in which the cargo is proteolytically degraded. Significantly, autophagosomes continuously receive input from endosomes and have been demonstrated to fuse with MHC-II loading compartments, thereby making the autophagosome an essential source of antigen for presentation to CD4+ helper T cells (15). Furthermore, antigens specifically targeted to the autophagosome by fusion with an LC3 construct are much more effectively presented to adaptive immune cells and elicit functionally superior responses (100). Notably, autophagy contributes to dying cell clearance during apoptosis (83).
The molecular mechanisms governing the initiation of autophagy in response to various stimuli are complex and not fully delineated. In a general sense, autophagy can be classified as being either a mammalian target of rapamycin (mTOR) dependent or independent (95). mTOR is a nutrient sensor that is associated with the lysosome which, when inhibited, initiates signaling events that lead to heightened autophagy (87). This is due to mTOR's function of inhibiting Atg1, which is required during the initiation of autophagy (58). As such, many pharmacological agents that induce autophagy operate through this pathway (mTOR inhibitors) and include the drug rapamycin (87) and its analogues. The protein Beclin 1 (called Atg8 in yeast) appears to be central to preautophagic signaling (56). Usually, Beclin 1 is bound to the anti-apoptotic protein Bcl-2. This binding is abrogated during autophagic signaling (82), and promotes Beclin 1 interaction with class III phosphatidylinositol 3-kinase (PI3KC3) to further transduce the message.
Signal Transduction by Reactive Oxygen Species
Reactive oxygen species (ROS) are formed as a metabolic by-product of electron transfer to molecular oxygen, and predate the wholesale oxygenation of the atmosphere 2bya and acquisition of mitochondria 1bya. This generation can occur in a number of ways that are generally associated with mitochondrial function and respiration. Given the drastic physiological and pathological outcomes which are observed through the modulation of ROS, including tumorigenesis and various neurodegenerative and metabolic disorders, it can be concluded that ROS signal transduction plays a critical role in maintaining eukaryotic homeostasis. ROS is generated by both complexes within the electron transport chain (Complex I and III) as well as by nicotinamide adenine dinucleotide phosphate reduced (NADPH) oxidase (Fig. 6) and additional enzymes, which are tightly associated with the mitochondria, including the monoamine oxidases that are critical for the metabolism of monoamine signaling proteins such as dopamine and serotonin (16). In the presence of mitochondrial superoxide dismutase (SOD), O2 •− can be converted to hydrogen peroxide (H2O2), which can then diffuse out of the mitochondria into the cytoplasm. In the presence of high iron concentrations, H2O2 can form the highly reactive hydroxyl radical (•OH) via the Fenton reaction. Catalase is responsible for converting H2O2 to water and oxygen. O2 •− can also react with nitric oxide to form the highly reactive peroxynitrite (ONOO•). Other sources of ROS include the endoplasmic reticulum and peroxisome (44, 101). Interestingly, many signaling events result in dramatic elevations in ROS levels. Ligation of receptors by growth factors such as epidermal growth factor (EGF) induces this response (5), but given the reactivity of ROS molecules, it is difficult to imagine how specific ROS-mediated signaling can occur. However, evidence is beginning to emerge regarding the trafficking of ROS across the plasma membrane. ROS, specifically hydrogen peroxide, appears to cross the membrane in a specific manner via aquaporin channels, representing a method of potential regulation (8). Regardless of this, there are certainly many identifiable targets of ROS signaling. Studies have demonstrated that phosphotases become transiently inactivated after increases in ROS levels, and many cellular processes, including the regulation of responses to hypoxia, inflammatory responses (specifically formation of the NLRP3 or NOD-like receptor pyrin domain-containing 3 inflammasome), phagocytosis, and autophagy are all responsive to ROS signaling (3, 49, 122).

Redox Signaling and Phagocytosis
ROS signal transduction plays a critical role in the cellular physiology of phagocytic cells (3). Oxidant bursts observed after growth receptor ligation also occur in macrophages, neutrophils, and so on, both during phagocytosis and in response to various stimulatory signals. Specific ROS-sensitive targets in these cells have also been identified. Nuclear factor (NF)-κB is a dimeric transcription factor that is involved in the regulation of a large number of genes which control various aspects of the immune and inflammatory response. NF-κB, which is usually localized to the cytosol, is translocated to the nucleus in its active form in response to oxidative stress (34, 65). In addition, mitogen-activated protein kinases (MAPK), including p38 and JNK which require phosphorylation for activation, are responsive to the kinase apoptosis-signal kinase-1 (ASK-1), which is maintained in an inactive state until ROS-mediated oxidation of its binding partner thioredoxin liberates it (47).
However, the most well-known role for ROS within phagocytes is for their bactericidal properties. NADPH oxidase is a multicomponent enzyme that is localized in the plasma membrane of phagocytic leukocytes. The generation of ROS in these cells is mediated by NADPH oxidase, which, when absent, results in an inhibited capacity to clear pathogens (4). It is possible that the ROS generated by NADPH oxidase contribute to host defenses not only through their microbicidal action but also through the modulation of redox-sensitive pathways in phagocytes. A recent study suggests that NADPH oxidase 2 (NOX2) activity decreases proteolytic efficiency of the phagosome through prolonged modification of the lumenal redox environment and oxidation of cysteine cathepsins (90). Anti-oxidant enzymes are required to neutralize phagocytosis and stimulatant-induced oxidant bursts and return ROS levels to basal concentrations in these cells. In addition, as previously mentioned, ROS signaling, particularly through NF-κB, may play an important role in the transcription and translation of NF-κB target genes, which include pro-inflammatory cytokines and chemokines that both recruit and stimulate phagocytes (28, 65).
Redox Signaling and Autophagy
Similar to phagocytosis, autophagy is also responsive to ROS signaling (49, 98). This is beginning to be extensively characterized given the often observed concurrent ROS generation and heightened autophagic flux (Fig. 6). Initially, given the consequences of ROS accumulation on organelles and genomic integrity, autophagy was thought to be enhanced purely as a means to mitigate oxidative stress (76). Although this remains true, and has drastic implications in many pathological states such as cancer and neurodegenerative diseases when the integrity of the autophagic pathway is disrupted, there is increasing evidence of direct regulation of this process by ROS molecules modulating specific autophagy proteins. When LC3 becomes lipidated via direct conjugation to phosphoethanolamine, a deconjugation event should occur in order for the molecule to be properly recycled. This deconjugation is mediated by the protease, Atg4. Studies have demonstrated that in the setting of heightened autophagy, particularly starvation-induced, Atg4 is a target for oxidation by hydrogen peroxide (99). This mechanism for regulation serves as a contributing factor toward the heightened autophagic state observed in a cell treated directly with exogenous hydrogen peroxide. Hypoxia-inducible factor 1 (HIF-1) plays a key role in the regulation of oxygen homeostasis. ROS induces HIF-1α-dependent expression of BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3), which promotes the dissociation of Beclin 1 from its Bcl-2 inhibitors (132). In addition, ROS regulates autophagic signaling transduction pathways that induce autophagy such as mTOR and MAPK.
Negative regulation of ROS-mediated autophagy is facilitated by anti-oxidant enzymes that prevent ROS from being elevated for prolonged measures of time. Interestingly, we know that autophagy is induced in the setting of oxidative stress and that autophagy can serve as its own negative regulator by removing sources of ROS such as mitochondria (mitophagy) and clearing oxidized proteins from the cytosol (a process in which p62 may be required) (97). However, in some conditions, autophagy can contribute to abnormal ROS accumulation by selectively promoting the degradation of the major enzymatic ROS scavenger, catalase (131).
HMGB1 at the Crossroads of Autophagy, Phagocytosis, and Oxidative Stress
HMGB1, which was originally thought to function only as a DNA chaperone that enhances replication, repair, and recombination, was “re-discovered” to be a crucial DAMP that mediates the response to infection, injury, inflammation, tissue generation, and cell migration (2, 71, 113). To perform its role as a DAMP, HMGB1 should transit from the nucleus, through the cytoplasm, to the extracellular environment (7, 71). This process can occur during cell activation as well as necrosis, autophagy, and late-stage apoptosis (57, 96, 109, 116). HMGB1, composed of an A box, B box, and C tail domains, is a redox-sensitive protein. There are three cysteines at positions 23, 45, and 106 (C23, C45, and C106, respectively). C23 and C45 readily form an intra-molecular disulfide bridge, whereas the C106 remains in a reduced form (46). C23 and C45 are required for HMGB1 binding to Beclin 1 during autophagy (109), whereas C106 is required for HMGB1 binding to TLR4 in macrophages (127) and appears to be important for transit to the cytosol.
HMGB1 is a stress sensor, and it is up-regulated or released in response to several forms of stress, including oxidative stress (Fig. 7) (114). Oxidative stress such as H2O2 or knockdown of SOD1 induces HMGB1 release in many cell types (111, 115, 123), whereas anti-oxidants such as N-Acetyl Cysteine and quercetin inhibit HMGB1 release as does knockdown of the autophagy gene, Atg5 (109, 112, 123). The oxidation of C106 (and not C23 or C45) abrogates the ability of HMGB1 to function as an immuno-stimulatory protein when encountered by DCs (57). Exogenous HMGB1 stimulates neutrophil NADPH oxidase activation and ROS generation in a TLR4-dependent manner (26), suggesting the presence of a positive feedback loop for HMGB1 release during oxidative stress. Moreover, means to distinguish the three forms of HMGB1, the “all reduced”, dithiol, and oxidized forms are necessary to understand its disparate role in the nucleus, the cytosol, and outside the cell.
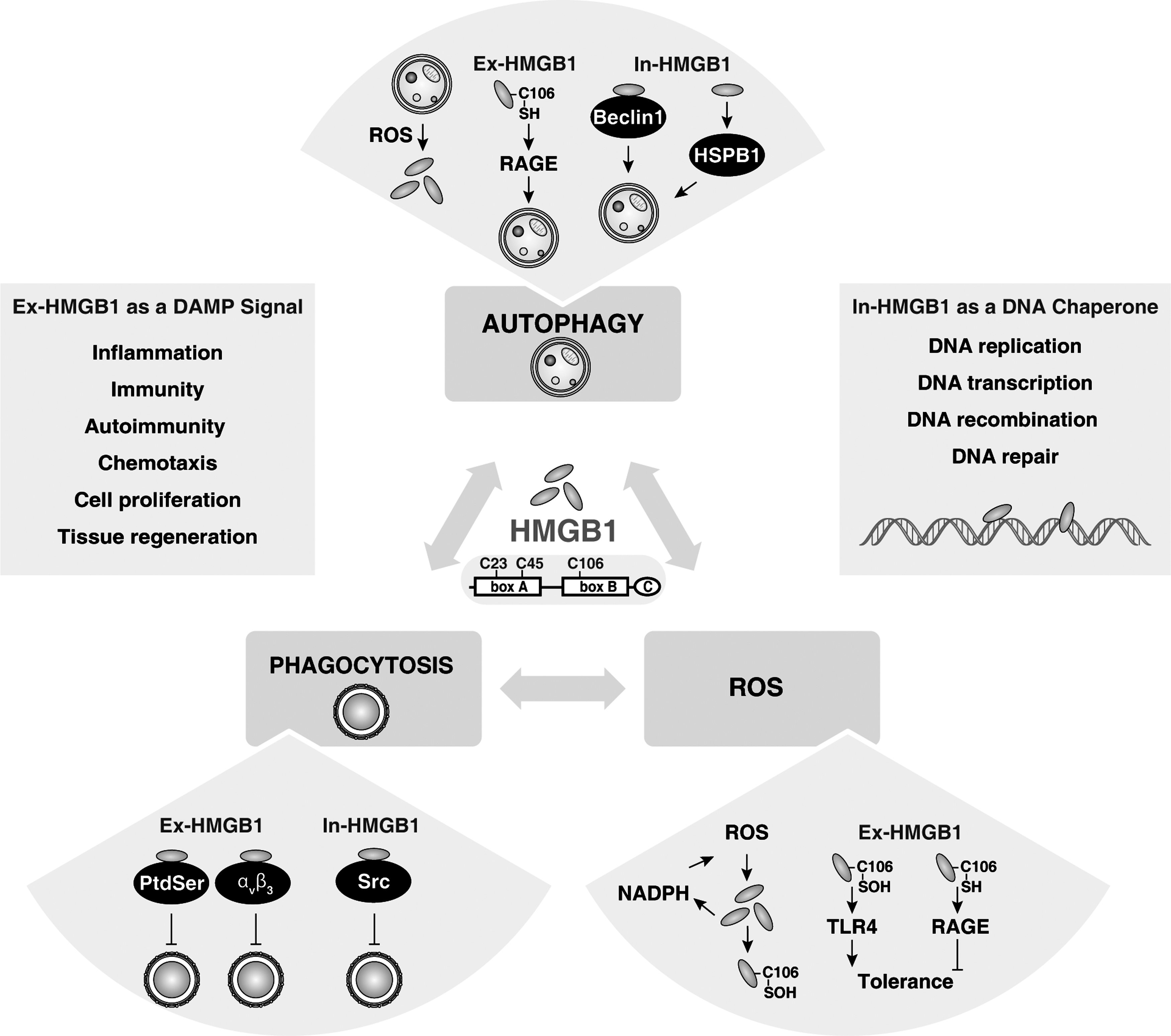
HMGB1 is also an important regulator of autophagy (108) (Fig. 7). Nuclear HMGB1 regulates small heat shock protein β-1 (HSPB1) expression (110). The phosphorylation of HSPB1 is necessary for the regulation of the actin cytoskeleton, which affects the cellular transport required for autophagy in response to mitochondrial injury. Thus, the HMGB1-HSPB1 pathway controls mitochondrial quality by autophagy/mitophagy. Cytosolic HMGB1 is a novel Beclin 1 binding protein that dissociates its inhibitory partner, Bcl-2 (109). The loss of p53 increases cytosolic HMGB1 binding to Beclin 1, thereby promoting autophagy and decreasing apoptosis (69). Extracellular reduced HMGB1 binds the receptor for advanced glycation end products (RAGE), but not TLR4, which inhibits mTOR and promotes the formation of the Beclin 1-PI3KC3 complexes (107). In contrast, oxidized HMGB1 induces the initiation of the intrinsic mitochondrial apoptotic pathway by unknown receptors (107). The induction of autophagy by both intracellular and extracellular HMGB1 is important for tumor development and a novel target for cancer therapy (50, 68, 113, 124). In addition, its receptor RAGE also regulates autophagy in pancreatic cancer cells (53 –55). Targeted ablation of RAGE in mice delays pancreatic tumorigenesis and inhibits IL-6/STAT3-mediated autophagy (53).
Likewise, HMGB1 is also a regulator of phagocytosis (Fig. 7). Extracellular HMGB1 inhibits phagocytosis by binding PtdSer or ανβ3 in apoptotic neutrophils or phagocytic macrophages, respectively (30, 67). These findings provide another mechanism by which exogenous HMGB1 enhances inflammatory responses by targeting phagocytosis-mediated anti-inflammatory responses during apoptosis, although it is unknown whether redox status regulates HMGB1's function in this instance. Moreover, intracellular HMGB1 is a negative regulator of phagocytosis by associating with Src kinase and inhibiting the interaction between Src and focal adhesion kinase (FAK) in macrophages (6). RAGE enhances phagocytosis in macrophages by binding to both PtdSer (29, 45) and free DNA. Future studies are needed to determine why both HMGB1 and RAGE interact with PtdSer, but induce opposite outcomes in the context of phagocytosis.
Autophagy and Phagocytosis in Cell Death, Immunity, and Inflammation
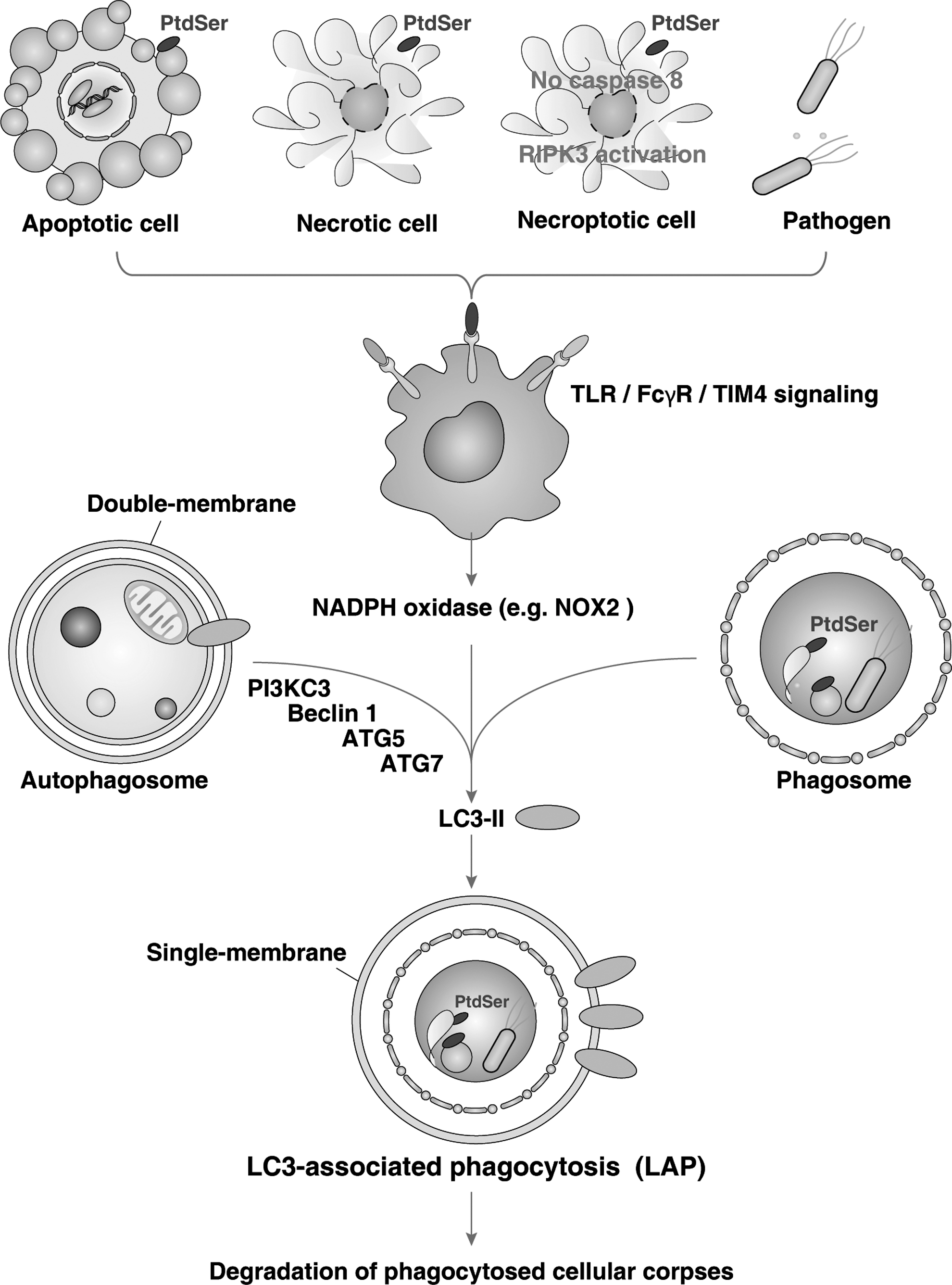
Given the nature of both autophagy and phagocytosis and the respective roles they play in maintaining the homeostatic balance at the level of both the cell and the organism, it becomes easy to accept that a dynamic relationship between both exist, particularly within the setting of cell death. This is so much so the case, that molecules previously thought to be specific to one process, such as LC3 in the case of autophagy, are actually playing similar roles in the other. In macrophages phagocytosing pathogens and cellular debris from apoptotic, necrotic, or the newly described “necroptotic” cells that display PtdSer, LC3 is rapidly recruited to phagosomes which lack the typical double-membrane coat that classical autophagosomes exhibit despite the requirement of Beclin 1, PI3KC3, Atg5, and Atg7, but not unc-51-like kinase 1 (ULK1). (48, 74, 94). This process has been termed LC3-associated phagocytosis or LAP (Fig. 8) (74). As upstream signals, TLRs, TIM4, FcγR, and NADPH oxidase-mediated ROS signal are required for LAP (48, 74). LAP is required for the efficient clearance of cell corpses as well as for phagosomal maturation.
Abnormalities in cell death resolution can contribute to a variety of diseases, including cancer, autoimmunity, and neurodegenerative disorders. There is now a substantial body of literature that explores the relationship between autophagy and apoptosis (73). In fact, autophagic proteins are often directly inhibited by apoptotic proteins. Furthermore, evidence that apoptotic caspases can serve as autophagic substrates suggests another layer of direct crosstalk between apoptosis and autophagy (20). This has compelled many in these fields to orient autophagy opposite to apoptosis and term the process as a form of “programmed cell survival” (55, 56). It has also been suggested that autophagy itself can serve as an alternative form of cell death, although this has not been described in a consistent manner (61). Similar to phagocytosis, autophagy has been demonstrated as helping clear apoptotic cells during embryogenesis (83). They are also linked to nutrient acquisition and energy generation (84, 104), which is important for cell survival during cytotoxic insults.
The primary function of most phagocytes is to destroy pathogens. Phagocytosis plays an essential role in innate immune sensing, and degrading products of internalized pathogens may traffic and be modified as nutrients (104). In addition, phagocytosis is essential in antigen presentation and adaptive immunity (104). Several studies reveal a crucial role for autophagy in adaptive and innate immunity, including pathogen elimination, limiting or promoting virus replication, T- and B-cell homeostasis, and antigen processing and presentation, with the term “immunophagy” (17, 18) referring to all such processes collectively (Fig. 9).

The apoptosis of inflammatory cells and their subsequent clearance by phagocytosis (also called efferocytosis) is key to orchestrating the successful resolution of inflammation and suppressing autoimmune responses. This is in part achieved through the release of anti-inflammatory cytokines IL-10, TGFβ, platelet activating factor (PAF), and prostaglandin E2 (PGE2) (24) and the inhibition of pro-inflammatory cytokines such as TNFα and IL-1β (106). In the absence of LAP, engulfment of dead cells results in increased production of pro-inflammatory cytokines (e.g., IL-1β) and decreased production of anti-inflammatory cytokines (74). A key mechanism of inflammation is the activation of the “inflammasome,” which leads to caspase-1 activation and the maturation and release of IL-1 family cytokines and other inflammatory mediators (Fig. 10). Interestingly, autophagy was previously considered as an anti-inflammatory mechanism when enhanced IL-1β release was observed during sepsis in Atg16L1-null mice (91). However, recent studies note that the induction of autophagy by starvation promotes inflammasome-dependent IL-1β secretion (22). These findings suggest that autophagy plays dual roles in the regulation of inflammation depending on the timing and type of autophagic activation.

Conclusion
The crucial role for both phagocytosis and autophagy in maintaining homeostatic balance and both inciting and regulating host responses to cellular injury makes the identification of cross-talk between the two pathways paramount. Given the wide range of pathologies in which either heightened or inhibited autophagic flux has been implicated as a contributor, the potential uncovering of therapeutic targets continues to be an important task. Cancer biologists and clinicians, in particular, have recognized this and have begun to thoroughly characterize combinatorial approaches to therapy in which autophagy modulators (specifically inhibitors) have been incorporated with promising results. Considering the active role phagocytes play in modulating inflammatory responses in cancers and other diseases, it becomes important to understand how manipulation of the autophagic pathway may affect phagocytic cells.
Making the relationship between phagocytosis and autophagy more complex is the role that ROS signal transduction plays in both processes (Fig. 9). Although direct targets for ROS modulation of both pathways such as NF-κB and Atg4 are beginning to become identified and characterized at the molecular level, much remains unknown regarding the specificity and directionality of ROS signaling. However, it is becoming increasingly clear that signal transduction by ROS molecules occurs in a much more regulated and orchestrated manner than previously thought, and ROS messengers directly modulate both phagocytosis and autophagy.
Footnotes
Acknowledgments
The authors apologize in advance to the researchers who were not referenced due to space limitations. D. T. is funded by the Department of Surgery and the University of Pittsburgh Cancer Institute. They appreciate careful review, fruitful discussions, and criticism by Dr. Michael T. Lotze from the Damage-Associated Molecular Pattern Laboratory.