
Abstract
Hydrogen Peroxide—Roles in Health and Disease
Although many pre-clinical studies report antioxidant protection against disease in models that promote oxidative stress, it should be remembered that these protective effects have not been observed in large-scale human clinical trials. In fact, such trials have not only failed to protect against disease, but in many cases these antioxidant supplements (especially when vitamin E or beta carotene have been used) have been detrimental, including in the cardiovascular system (7). Such observations, along with cumulative new evidence that H2O2 plays important regulatory roles, have further added to the contemporary view that these oxidants are crucial signaling molecules. Indeed, as antioxidants are anticipated to prevent oxidant sensing and signaling, this could potentially explain the harm they can cause, perhaps by preventing an appropriate homeostatic or adaptive response.
Here we consider the concentrations of H2O2 that are produced in biological systems, assessing whether there is a disconnect between those levels and what is normally added exogenously in experimental model systems to observe a biological response. Many proteins are now known to sense H2O2 and alter their function as a result, providing a mechanism by which an oxidant signal is transduced into a biological response. We also discuss the molecular basis of H2O2 sensing by these proteins, specifically focusing on kinases that have been shown to be regulated by post-translational oxidative modifications. These kinases, CAMKII, PKA, and PKG, are not only well known as important regulators of cardiovascular function, but there is significant evidence showing their redox regulation. Modulation of kinases in this way is significant as this allows direct integration of changes in cellular redox status with phospho-regulatory pathways. In addition to the direct modulation of CAMKII, PKA, and PKG by oxidants, there are several other kinases that can be indirectly modulated via protein oxidation of upstream targets. This includes apoptosis signal-regulating kinase-1 (ASK-1), which is kept inactive through an interaction with reduced thioredoxin (Trx). This interaction is disrupted by reactive oxygen species that oxidize Trx, preventing it from binding ASK-1. This leads to the activation of ASK-1 and subsequent stimulation of ASK-1 dependent apoptosis (49).
H2O2—How Is it Produced?
The reduced oxygen species superoxide is constantly generated in cells as a by-product of metabolism or by specialist oxidase enzymes (Fig. 1). Superoxide is readily converted to the more stable form H2O2 following reduction by superoxide dismutase or spontaneous decomposition (37). Once formed, H2O2 acts as a physiological second messenger signaling molecule by selectively oxidizing target or sensor proteins, as discussed below. In addition to oxidases, the mitochondrial transport chain also generates superoxide through the leak of electrons that combine with molecular oxygen to yield superoxide. Therefore, the metabolic state of the cell alters the rate at which electrons leak from their transport chains, changing the amount of superoxide and H2O2 generated. Out of numerous oxidase enzymes, the NADPH oxidase (NOX) family of proteins are the only with a primary role in generating superoxide and H2O2 (14). Superoxide is also enzymatically generated as a by-product by xanthine oxidase, uncoupled nitric oxide synthases, urate oxidase, aldehyde oxidase, D-amino oxidases (such as monoamine oxidase, indoleamine 2,3-dioxygenase), as well as by any peroxidase that can reduce molecular oxygen to superoxide (18).

H2O2—How Much Is Produced and Accumulates in Cells and Tissues?
The net cellular H2O2 concentration depends on the rate of increase (influenced by influx and the rate of endogenous synthesis) against consumption by conversion to water or other reactive oxidant species (ROS), as well the efflux rate. Measurement of H2O2 in samples has revealed that normal human plasma typically contains 1–8 μM H2O2 (with an average of ∼3 μM) (35, 36), although 35 μM has also been observed. Higher concentrations have been observed in activated macrophages, which can generate up to 6×10−14 mol H2O2/h/cell, generating a local concentration of 10–1002 μM (19). In addition, stimulated neutrophils were shown to release H2O2 that accumulated at 1–12 μM in the extracellular medium (57, 60). Elevations in H2O2 were detected during disease, including ischemia and reperfusion where an extracellular H2O2 concentration of 100 μM was observed in the brain (30). Individual cells lining the gut and urinary tract are routinely exposed to 100 μM H2O2 (18), while respiratory lining cells experience 0.1–1μM H2O2, but can be elevated 20-fold in subjects with inflammatory lung disease (34, 59). Elevations in H2O2 have also been reported in end-stage kidney disease (13 μM) (27), hypertension (7 μM) (35), and ischemia and reperfusion (160 μM) (30). In addition, patients with respiratory lung neutrophilia have μM levels of H2O2 in their breath condensates (3). In contrast to extracellular measurements, direct measurement of intercellular H2O2 is markedly more challenging due to rapid reactivity of this oxidant species (short lived), although some such studies suggest mM H2O2 can occur (57). An important consideration is that the subcellular concentration of H2O2 can vary hugely and be much higher in an individual cell or cellular compartment as compared to a cell-wide average concentration.
H2O2—Exogenous Treatment in Model Systems
Oxidative stress is routinely studied by exposing model systems to exogenous H2O2. In many studies an acute (5–10 min) oxidative interventions is utilized, with ∼50–500 μM H2O2 being required to induce measureable protein oxidation or functional effects reliably. The relevance of this concentration range of H2O2 to health or to disease is hotly debated, albeit we would contend that the studies discussed above justify such levels, at least pathophysiologically. It should be remembered that the exogenous H2O2 concentration will not be the same as in the intracellular environment due to impedance by the cell membrane (2). Although such concentrations are considered highly toxic by some researchers, in our isolated rat heart studies there was no detrimental impact when they were exposed acutely to 100 μM H2O2. Although cardiac contractility was quickly reduced to ∼85% of basal left ventricular developed pressure and the coronary vessels vasodilated, the preparation remained stable thereafter (8, 12). Similarly, isolated ventricular myocytes tolerate similar levels of oxidants, including H2O2, without acute cell death. It is also notable that many high profile studies exposed cells and tissues to 100 μM H2O2 and above. Biteau et al. used 500 μM H2O2 during the discovery of sulphiredoxin (5). Woo et al. also demonstrated the reversibility of peroxiredoxin oxidation, using H2O2 at 100–500 μM (64). Sestrin-mediated repair of hyperoxidized peroxiredoxin was demonstrated in cells exposed to 100–500 μM H2O2 for 6 hours (11), and redox control of forkhead proteins illustrated using 100–1000 μM H2O2 (42). The H2O2 concentration in the cell induced by TNF reaches levels proportionally similar to the concentration induced by exogenous addition of 1–3 mM H2O2, although it can be fatal after a certain threshold (32). In addition to the concentration, we should be aware of the total amount of H2O2 to which cells are exposed. Even though an identical concentration of H2O2 is used, increasing the volume applied to the same amount of cells could result in increased oxidative damage because scavenging H2O2 requires a certain amount of electrons, which have a limited availability within the intracellular environment.
As highlighted above, membranes limit H2O2 entry into cells such that the intracellular concentration is less than that applied, perhaps 1/7th (2). On entry to the cell, the H2O2 encounters a buffered reducing environment, as well as several antioxidant enzymes that further lower the concentration. For example, cells including cardiac myocytes are replete in catalase which decomposes H2O2 with a Km >1 M (43). As this enzyme has evolved with such a high half constant for decomposing H2O2, it may suggest extremely high H2O2 levels occur within some intracellular locations. Such high H2O2 levels may be anticipated near the site of synthesis or at specific subcellular locations with an oxidizing redox potential such as peroxisomes, the endoplasmic reticulum, or outside the cell. The H2O2 buffering system of the cell also comprises of other peroxidases other than catalase, which includes glutathione peroxidase. Glutathionine peroxidases are found in most mammalian cells and use glutathione as an electron donor to reduce H2O2 to water. Mammalian cells also have many highly abundant isoforms of peroxiredoxin that decompose H2O2, and are considered the primary controllers of basal H2O2 levels, crucial to the control of redox signaling.
Peroxiredoxins have a Km for their substrate H2O2 in the 10–20 μM range and have been suggested to act as gate keepers of oxidant signaling, as higher concentrations of H2O2 induce hyperoxidation of their catalytic cysteine to sulfinic acid which prevents H2O2 decomposition. This hyperoxidation event allows cellular peroxide to rise and so oxidize proteins that would not normally with active peroxiredoxin present, known as the ‘floodgate’ hypothesis (65). This ‘gate’ is subsequently closed by the reactivation of the peroxiredoxin, which is achieved by an ATP-dependent enzyme called sulfiredoxin (5). This repair of sulfinated peroxiredoxin is supported by structural studies of this enzyme in a complex with sulfiredoxin (31). For such a repair mechanism to have evolved is logically consistent with peroxiredoxin being a natural, instead of an artefactual, occurrence. With this in mind, it is intriguing that peroxiredoxin hyperoxidation is not observed until cells or tissues are exposed to ∼100 μM H2O2 (39, 52). This suggests such H2O2 concentrations are biologically relevant. Although exogenous H2O2 of ∼100 μM and above is generally required for peroxiredoxin hyperoxidation, the intracellular concentration remains an important question. 2.4 μM Prx I efficiently reduced 100 μM H2O2 in vitro (66), but when H2O2 was cumulatively increased to 200, 500, or 1000 μM, this progressively inhibited peroxidase activity as a result of hyperoxidation. The transition point at which the peroxidatic thiol hyperoxidized was when H2O2 was 10-fold the peroxiredoxins Km for substrate (i.e., at 200 μM). At this point, H2O2 was in a 40-fold molar excess over the peroxidase, suggesting the peroxidase hyperoxidation in cells may require a similar excess of H2O2 over peroxiredoxin. The molar concentration of peroxiredoxin is therefore an important question. Peroxiredoxins are highly abundant, constituting 0.1–0.8% of the total protein in some cells, with >106 copies per cell. Heart cells are replete with peroxiredoxins; perhaps present at ∼40 μM (53). On this basis, we could expect that the concentration of H2O2 substrate of this enzyme (i.e., H2O2) occasionally to be of the same order, or perhaps an order of magnitude or more above. As peroxiredoxins decompose H2O2 with a rate constant of up to 107 M −1 s−1 (46), this suggests the cellular concentration of oxidant is somewhat above that of enzyme that efficiently metabolizes it. Overall, these considerations may suggest that treatment of cardiac cells and tissues with 100–500 μM H2O2 is justified in terms of physiological significance.
H2O2 Sensing by Cysteine Thiols
A primary way that H2O2 is sensed and transduced into a biological response involves the oxidation of protein thiols (PSH) that have a low pKa (Fig. 3). These post-translational oxidative modifications can control the activity of target proteins in much the same way as related control mechanisms such as phosphorylation. Low pKa protein thiols exist substantially in the deprotonated thiolate anion (PS-) state at physiological pH. This ionized state profoundly increases the rate of oxidation by H2O2 and also provides a basis for selective H2O2 signaling. If H2O2 indiscriminately oxidized any protein thiol with which it came in contact, this would result in uncontrolled modifications and would be inconsistent with this oxidant serving as a messenger molecule that conveys information. Further selectivity may also be achieved by co-localization of the source of H2O2 with the redox-regulated target protein, as has been shown to occur in the regulation of S-nitrosylation by nitric oxide formation (25).
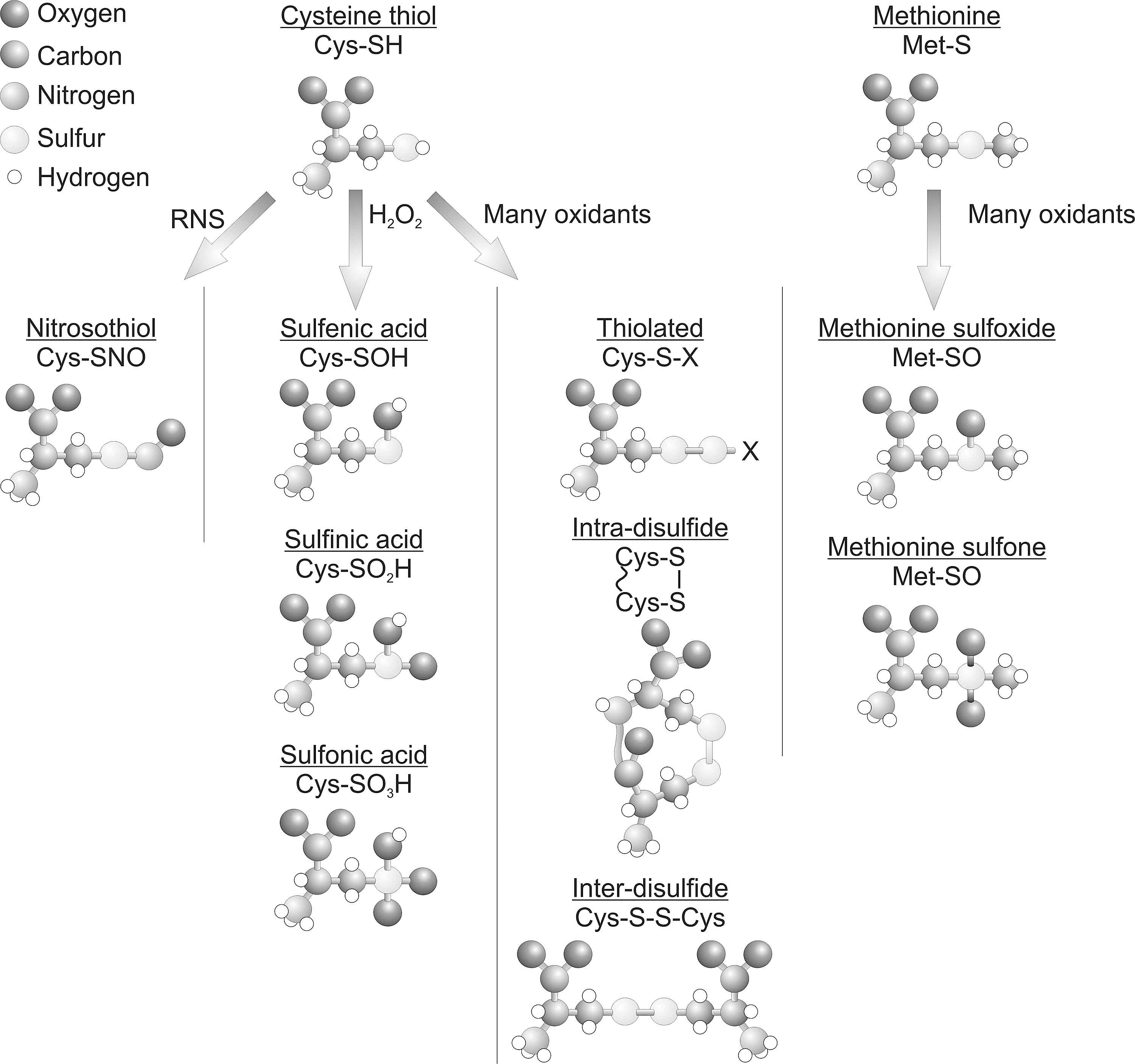
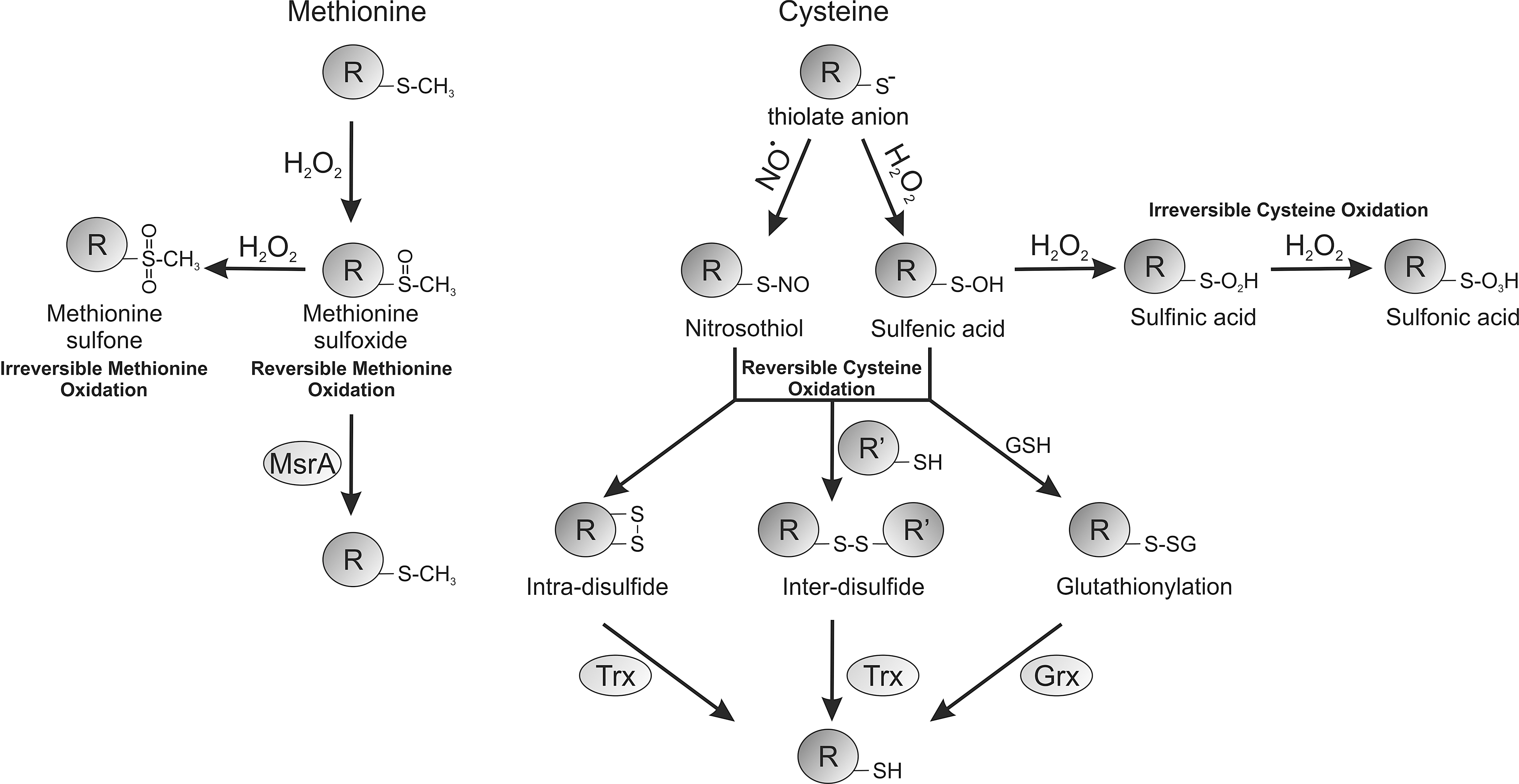
Figure 2 shows how reactive protein thiols can be oxidized by H2O2 to yield the sulfenic acid derivative (PSOH), which is generally unstable and can further oxidize stepwise to the sulfinic acid (PSO2H) and then to the sulfonic acid (PSO3H) form. Sulfenic acids can also be redox recycled, normally by forming a disulfide intermediate before conversion back to a thiol, commonly by glutathione- or thioredoxin-dependent reducing systems. Thiol over-oxidation to sulfinic or sulfonic acids were thought irreversible modifications, but at least in the case of the sulfinated form of the 2-Cys peroxiredoxin family of enzymes, this can be reduced back to the catalytically active thiol, as discussed above (5). Budanov et al. have also shown that a sulfinic acid form of peroxiredoxin 1 can be reduced by p53-regulated sestrin 2, although subsequent studies have questioned this finding (63). Other proteins form the sulfinated state, including DJ1, glyceraldehyde phosphate dehydrogenase, and protein tyrosine phosphatase 1B (PTP1B) (39). PTP1B can also form a relatively stable sulfenyl-amide, where a sulfenic acid reacts further with the amide nitrogen within the backbone of this protein (50), which being reversible may serve to prevent cysteine over-oxidation. Disulfides commonly form when proteins sulfenates encounter a thiol (Fig. 3), and can serve as a general mechanism to protect from over-oxidation. This is because disulfide-oxidized proteins can return to their basal reduced state when a cell recovers from an oxidant stress, whereas in most cases a protein sulfinate or sulfonate would require de novo synthesis. The type of disulfide formed depends on the thiol with which sulfenates react. The thiol can come from a small thiol-containing molecule, generating a disulfide adduct in a process known as S-thiolation. The small thiol is most commonly glutathione due to its abundance, resulting in S-glutathiolation of the target. In proteins with an appropriate structure, a sulfenate may be proximal to a neighboring (known as vicinal) thiol, giving rise to interprotein or intraprotein disulfides (9, 22). Protein thiols are also post-translationally modified and potentially regulated by many other molecules, including electrophilic lipids, foodstuffs, and drugs or their metabolites (15,16). Examples include the lipid 15-PGJ2 (15-deoxy-Δ12,14-prostaglandin J2), which can modify a cysteine thiol on soluble epoxide hydrolase, leading to attenuation of its activity. This is through a Michael addition reaction whereby the α,β-unsaturated carbonyl group extracts electrons from the adjacent alkene, making this bond thiol reactive. An example of drugs that target cysteine thiols includes a group of ribosomal S6 kinase inhibitors (including hypothemycin) that adduct through a Michael addition reaction and could be potentially used in the treatment of cancer (68). Although cysteine thiols contribute to a large extent to the regulation of protein function by H2O2, the amino acid methionine can also plays an important role by undergoing oxidation. Methionine can be oxidized in the presence of H2O2 to form a methionine sulfoxide (PSOCH3), which can be catalytically reduced by methionine sulfoxidases or further oxidized to an irreversible methionine sulfone (PSO2CH3). The oxidation of methionine by H2O2 is particularly pertinent to the catalytic regulation of CAMKII, as described below.
In the molecular mechanism of H2O2 sensing discussed above, the scheme presented involves the direct reaction of the oxidant with a low pKa target thiol. Despite target proteins such as PTP1B having quite a low pKa (∼6.8), it has been questioned whether direct oxidation by H2O2 is likely to occur in cells (62). This is because of the high abundance of peroxidase enzymes with a lower pKa (∼5.5) and other structural features that allow reaction with H2O2 at a much faster rate. However, it is clear that enzymes such as PTP1B are indeed oxidized in systems under oxidative stress. Such oxidations may occur as secondary to inactivation of the 2-Cys peroxiredoxin enzymes, as in the floodgate hypothesis outlined above. However, cells are generally replete in other antioxidant enzymes, including the glutathione-dependent 1-Cys peroxiredoxins and thioredoxin. One potential mechanism involves oxidants first reacting with proteins with highly reactive thiols (e.g., peroxiredoxins or thioredoxin), oxidizing them to a sulfenate or a disulfide, which then reacts with the target such as PTP1B. In this scenario, the oxidant sensing and resulting transduction to another type of signal (in the case of PTP1B this would be phosphate inhibition) are achieved by separate proteins, in which the target is passed the ‘oxidant signal’ by the sensor.
Kinases That Are Redox Regulated by H2O2
Ca2+/calmodulin-dependent kinase II
Ca2+/calmodulin-dependent kinase II (CaMKII) exists as four isoforms (α, β, δ, γ) with δ being the most abundant in heart (54). This serine-threonine kinase phosphorylates many of the same targets as PKA. CaMKII contributes to the control of excitation–contraction coupling by regulating the activity of several Ca2+-handling proteins, but is also a key controller of apoptosis and gene transcription.
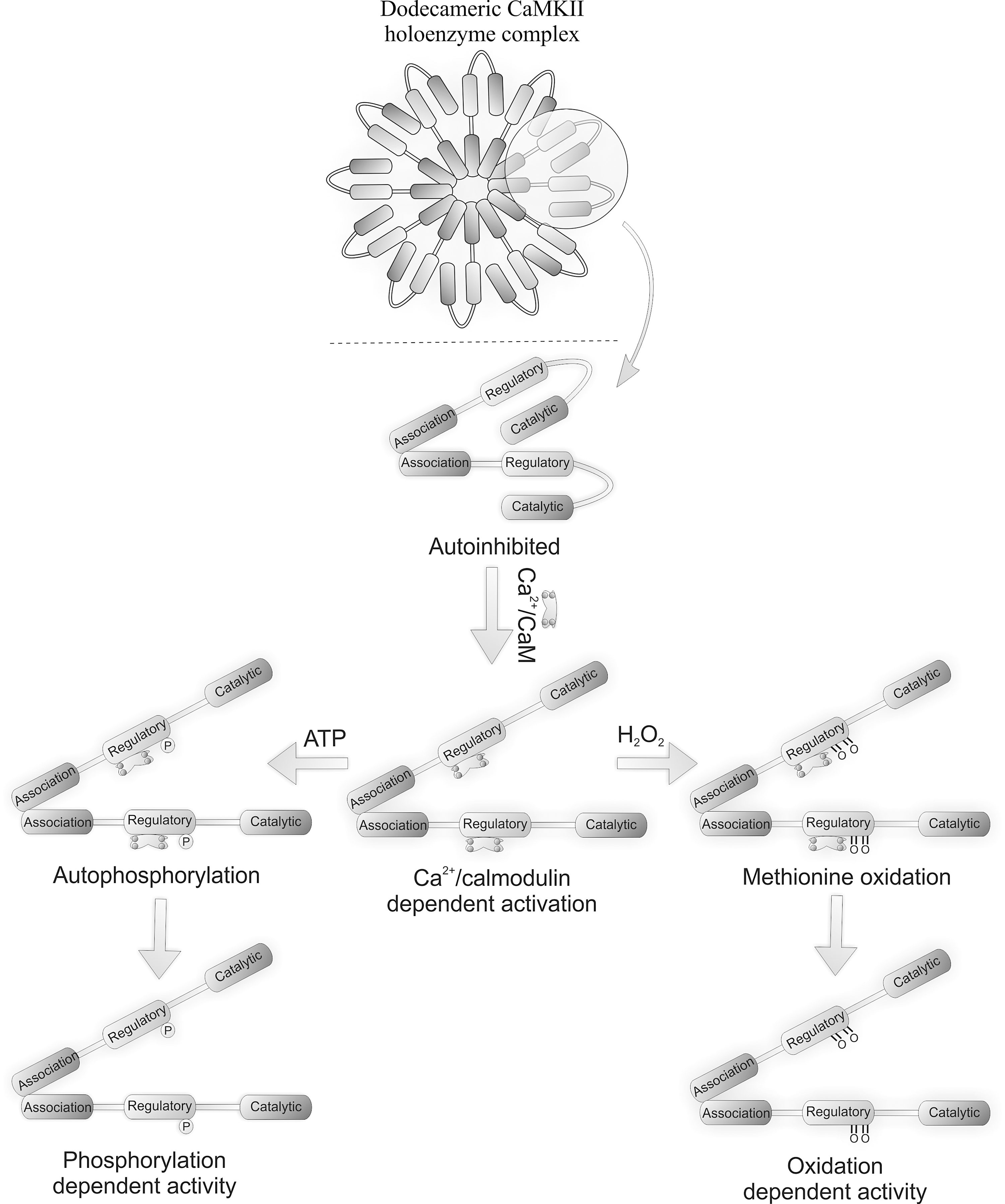
Figure 4 shows the CaMKII holoenzyme consisting of monomeric units that comprise a catalytic N-terminal domain, a C-terminal association domain, and a central regulatory domain. The kinase holoenzyme assembles by monomers interacting through their association domains (28, 54). Figure 4 also shows that each holoenzyme is comprised of either dodecameric or tetradecameric monomer subunits which provide a large number of catalytic domains for interaction with substrates. The CaMKII holoenzyme structure provides a basis for a graded or proportional response to Ca2+. Ca2+, the classical activator of CaMKII, enables catalytic activity via the formation of a Ca2+/calmodulin complex that binds to and stimulates the kinase. Persistent Ca2+-dependent activation leads to inter-subunit autophosphorylation at Thr-287 within the autoinhibitory domain, preventing re-association with the catalytic domain and sustained kinase activity even when Ca2+ returns to basal levels.
CaMKII is activated by oxidative stress, including in the heart, for example during apoptosis (69). Activation during oxidative stress led to the hypothesis that the kinase may be susceptible to direct oxidative activation, which was indeed the case. Ca2+/CaM-activated CaMKII remained active even after Ca2+ was removed if H2O2 was present. Pre-activation by Ca2+/CaM is required for H2O2-induced kinase activation, the targets of oxidation being inaccessible otherwise. The oxidation of two methionine residues underlies the oxidant-mediated activation of CaMKII; a Met282Val mutant being completely resistant to oxidative activation. A Met281Val on the other hand is only partially resistant to oxidative activation. Both ‘redox-dead’ mutants could be activated like wild-type when stimulated by autophosphorylation, whereas a Cys290Val mutant could be fully activated by oxidants like wild-type. A role for cysteine oxidation in CaMKII activation was ruled out, as iodacetic acid (which alkylates thiols) did not prevent H2O2-induced kinase activation. An antibody that selectivity recognizes the dual oxidized M281/282 form of CaMKII was used to show that AngII treatment promoted kinase oxidation in wild-type heart, whereas this did not occur in p47 NOX null mice. AngII-induced oxidative activation of CaMKII mice induced apoptosis in wild-type, but not p47 NOX null, mice. Similarly CaMKII transgenic mice expressing an inhibitory ‘AC3-I’ peptide were resistant to AngII-induced apoptosis (45). Further support for this oxidative activation of CaMKII mediating injury comes from studies replacing wild-type CaMKII with the Met281/282Val form that is resistant to apoptosis and hypertrophy.
Sulfoxidized methionine residues may be reversed back to the reduced state by methionine sulfoxide reductase A (MsrA), as shown in Figure 3. Mice lacking MsrA have increased sensitivity to AngII-induced CaMKII oxidation and apoptosis in heart tissue. Furthermore, MsrA null mice are sensitized to CaMKII oxidation, as well as apoptosis during myocardial infarction. These data strongly support a role for oxidative activation of CaMKII being causatively harmful. Oxidant-activated CaMKII potentially activates the L-type Ca2+ channel (55), with H2O2 increasing Ca2+ influx by this route. Depleting SR Ca2+ stores with caffeine or thapsigargin prevented oxidant-induced L-type Ca2+ channel activation, suggesting intracellular store of Ca2+ are also important (56). The cardiotoxic effects of aldosterone are significantly mediated by oxidative activation of CaMKII (23). Hearts and isolated neonatal myocytes exposed to aldosterone had elevated ROS levels, and CaMKII methionine sulfoxidation was observed on immunoblots. Overexpression of MsrA reduced the detrimental effects of aldosterone. Aldosterone increased the expression of matrix metalloproteinase 9, a known mediator of the cardiac rupture phenotype, in vivo in mice,(26) and this was dependent on CaMKII oxidation, as it was absent in mice overexpressing AC3-I. This highlights the potential benefit and rationale for using mineralocorticoid receptor antagonist drugs to reduce the risk of mortality after myocardial infarction. The proposed mechanisms for classical and oxidative activation of CaMKII are shown in Figure 4.
cAMP-dependent protein kinase
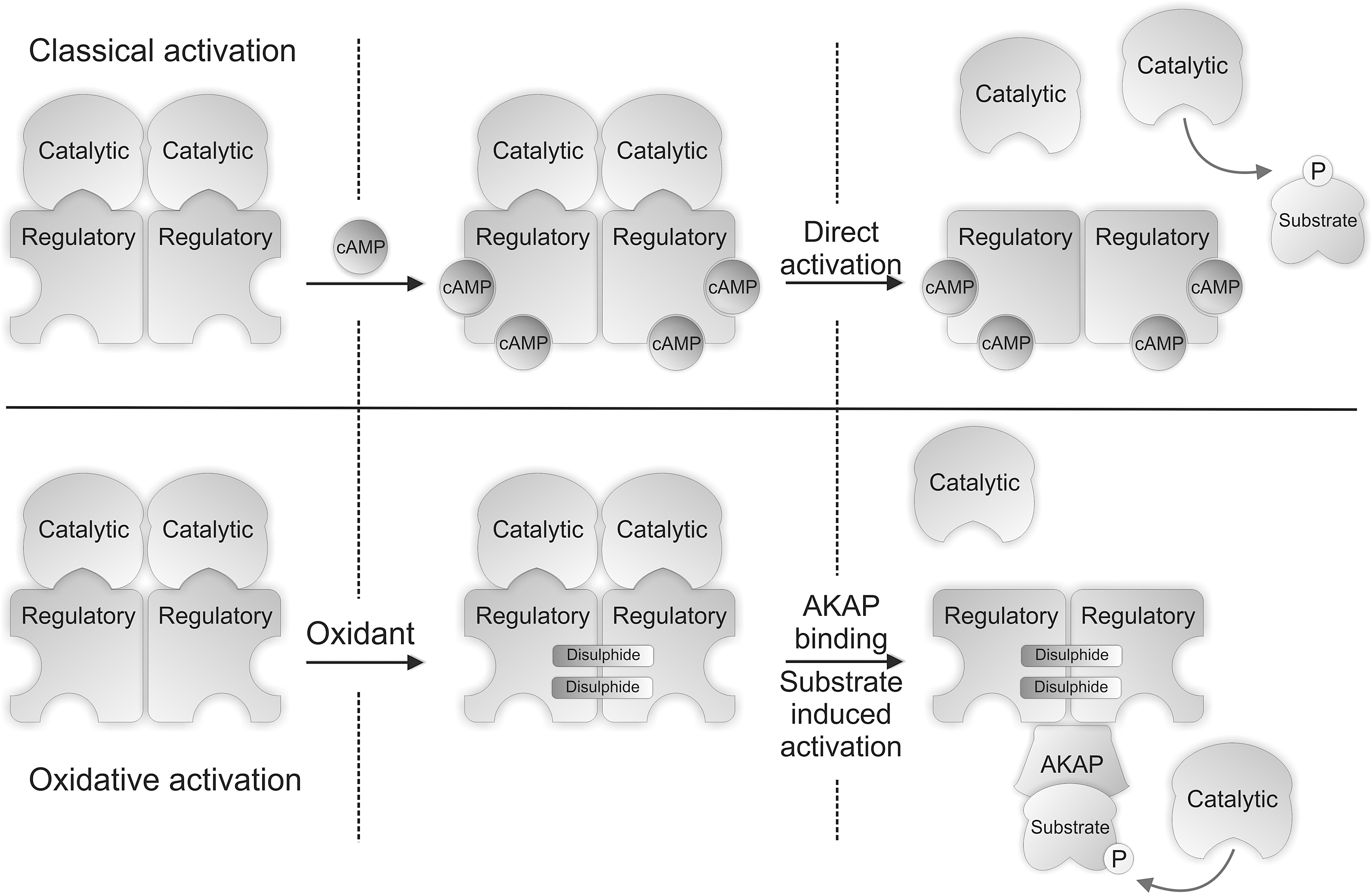
Cyclic adenosine 3',5'-monophosphate (cAMP)-dependent protein kinase is also commonly known as protein kinase A (PKA). PKA is classically activated by elevations in cAMP (Fig. 5) which is synthesized in response to sympathetic nerve stimulation that releases adrenaline or noradrenaline into the circulatory system (4). These hormones bind to β-adrenergic receptors to stimulate adenylate cyclase, via the stimulatory G-protein coupled signaling pathway, which converts adenosine-5'-triphosphate (ATP) to cAMP. This mechanism of β-adrenergic stimulation is a principal component of the physiological ‘flight or fight’ response, enhancing the body's ability to perform exercise. Enhanced physical activity requires increased myocardial contractility, elevating cardiac output to potentiate the supply of blood providing the oxygen and nutrients required for energy generation. cAMP binds to the regulatory subunits of this tetrameric kinase complex which dissociates them from the catalytic, resulting in substrate phosphorylation (33).
PKA is a major regulator of cardiac excitation–contraction coupling, enhancing Ca2+ influx into the cell by phosphorylating and activating the L-type Ca2+ channel. This Ca2+ stimulates the RyR, resulting in further release of Ca2+ from the intracellular sarcoplasmic reticulum store (known as Ca2+-induced Ca2+ release), which binds to troponin C to promote myofilament contraction. PKA also phosphorylates myofilament proteins to achieve a net increase in their Ca2+ sensitivity, which further enhances contractile force during β-adrenergic stimulation. PKA activation in blood vessels also contributes to their vasodilation which enhances the blood supply to the cardiac muscle during the increased energy demand.
PKA is redox-regulated, independently of the classical cAMP mechanism, by a mechanism involving interprotein disulfide bond formation in the regulatory R1α subunits (Fig. 5) (8). The R1α regulatory subunit of PKA was identified as being redox sensitive following a proteomic screen, using diagonal polyacylamide gel electrophoresis, for proteins that form interprotein disulfide complexes. The identification of two disulfides that link the R1α regulatory subunits of PKA (PKARI) was first reported by Zick and Taylor (70), with Cys 16 bonding to Cys 37, forming an anti-parallel dimer containing two double disulfide bonds. These were considered constitutive structural disulfides (38); however these disulfides are not required for dimerization of the 1α regulatory subunits as they are held together by an amphipathic leucine zipper. These disulfides were hypothesized to serve as oxidant sensors, forming the disulfides only during pro-oxidizing conditions. Indeed PKAR1α displayed a H2O2 concentration-dependent increase in disulfide dimer formation in the absence of reducing agent, which was fully returned to the monomeric mass when analyzed with reducing agent present. PKAR1α disulfide bond formation was associated with elevated PKA catalytic activity, which was attenuated by the PKA inhibitor H89, although this antagonist has selectivity issues. Substrate phosphorylation following H2O2 treatment was also independent of cAMP elevation. Subcellular fractionation studies showed that disulfide-oxidized kinase translocated from the cytosol to the myofilament/nuclear fraction and to the membrane. This translocation and increased activity of PKA following oxidation may be explained by increased affinity for its binding partners known as A-kinase anchoring proteins (AKAPs). The inter-protein disulfides between the R1α subunits are located within the AKAP interaction domain and precisely flank the site of AKAP-R1 binding. Recent studies comparing disulfide wild-type R1α binding to D-AKAP with mutants that cannot form a disulfide showed Cys16Ala and Cys37Ala mutations reduced the interaction 3- or 16-fold, respectively (51). PKA interaction with its AKAPs potentially goes beyond the simple concept that this brings the enzyme closer to its substrates, as the presence of substrate itself can increase the dissociation of the catalytic and regulatory subunits (61). This substrate-dependent effect appears to be specific to type 1α form of PKA and may help explain how oxidation of this kinase leads to an increase in the phosphorylation of its substrates without an elevation in cAMP. PKAR1α disulfide oxidation is also induced by CysNO (12), a compound that induced vasodilation in isolated aortic vessels independently of cAMP. PKAR1α disulfide oxidation may therefore have a role in vascular relaxation, especially considering other thiol-oxidizing molecules such as H2O2 efficiently induce vasodilation.
In addition to the finding that PKA1α may be activated by regulatory subunit disulfide formation is a report that oxidation of the catalytic subunit can lead to kinase inactivation. The formation of an intramolecular disulfide bond between Cys- 199 and -343 of the catalytic subunit or its glutathionylation at Cys-199 inactivated the kinase (29). This discrepancy could be due to the experimental models examined, including the type and concentration of oxidant. The R2 and C2 subunits of PKA have also been shown to disulfide dimerize in the presence of oxidants (20). Subsequent studies showed this disulfide inactivates the kinase (17).
cGMP-dependent protein kinase
The regulation of vasotone is crucial for maintaining blood pressure and tissue perfusion with its dysregulation increasing the risk of heart attacks, peripheral artery disease, heart failure, aortic aneurysms, kidney failure, and stroke (24, 40). A major regulator of vasodilation within the vasculature is the kinase cyclic guanosine monophosphate (cGMP)-dependent protein kinase, also commonly called protein kinase G (PKG) (58).
PKG is expressed as two isoforms termed type 1 (PKG1) and type 2 (PKG2). Type 1 is predominately expressed in the cardiovascular system and has two splice variants, PKG1α and PKG1β, which differ only in the first N-terminal ∼100 amino acids. PKG is a parallel-aligned homodimer due to a leucine zipper interaction within the regulatory N-terminal region between each subunit. Unlike PKA, the catalytic and regulatory domains of PKG are fused together as a single subunit. Two molecules of cGMP bind to each subunit of PKG causing a conformational change in the protein structure exposing the catalytic site of the enzyme to induce activation. The proposed structure of inactive PKG1α is a folded conformation, with the catalytic C-terminus being inactive through an interaction with the pseudosubstrate N-terminal autoinhibitory domain (Fig. 6) (1, 10, 44). Hydrogen/deuterium exchange analysis has also identified changes in the structural interfaces of PKG1α that occur during cGMP activation (1), showing the autoinhibitory domain, the hinge region, and the substrate interaction domain in PKG1α becoming more solvent accessible when cGMP binds. This was accompanied by a loss in solvent accessibility of the cGMP binding sites, suggesting a dramatic change in the structure of PKG1α in which the N-terminal and catalytic domain move apart during classical allosteric activation. Gel filtration studies have also shown that when cGMP binds PKG1α that the kinase elongates, consistent with a structural change that would expose the catalytic site (48).

PKG1α is also sensitive to oxidation through the formation of an intermolecular disulfide bond (Fig. 6) (13). Isolated rat hearts perfused with H2O2 showed a dose-dependent increase in the formation of a disulfide dimer complex. A disulfide between the two monomers of PKG1α was first reported in 1980, demonstrating a bond between Cys-42 on each chain (41), but this was considered a structurally constitutive modification. It is clear that under basal conditions PKG1α does not contain a disulfide, but when cellular oxidant levels are increased, the disulfide readily forms. Isolated rat hearts exposed to H2O2 vasodilated to a similar extent to that induced by an NO donor. Follow-up studies measuring tension in isolated rat aortic rings showed H2O2 increased vasodilation in a PKG-dependent manner even when guanylate cyclase activity and cGMP production was blocked, consistent with cGMP-independent activation. In vitro kinase activity assays confirmed disulfide oxidation does indeed increase PKG1α activity independently of cGMP. The kinetics of disulfide activation was measured by Michaelis Menten analysis, showing that disulfide formation increases the affinity of PKG1α for its substrates. This differed from the classical mechanism involving allosteric activation by cGMP, which enhanced the Vmax of the kinase. The enhanced affinity for substrates after oxidation underlies the kinases translocation to subcellular compartments containing targets for phosphorylation.
The transnitrosylating nitrosothiol CysNO, also efficiently disulfide-activated PKG1α (12), contributing to the associated vasodilation of aortic rings, which was significantly guanylate cyclase insensitive. This novel mode of oxidatively activating PKG1α represents a potential mechanism underlying endothelial derived hyperpolarizing factor (EDHF)-induced vasodilation. Indeed, a role for PKG1α oxidation by H2O2 as a mediator of the EDHF response has been substantiated using Cys42Ser PKG1α knock-in (KI) mice. These KI mice cannot undergo disulfide-induced activation due to single atom substitution of the redox sensitive sulfur for an oxygen (47). The KI mice have elevated resting blood pressure, consistent with basal PKG1α disulfide oxidation being an important mediator of physiological vascular tone. In addition, resistance vessels from KI mice had a blunted EDHF response to acetylcholine. Oxidant-induced disulfide activation of PKG1α has also been demonstrated in human coronary arterioles (67). Flow and H2O2 induced dilation in these vessels was shown to be largely dependent on stimulation of BKca channels that could be attenuated by a PKG inhibitor but not a guanylate cyclase inhibitor. Overall, there is now robust evidence that PKG1α oxidation occurs physiologically to regulate vasotone, and these events occur in human tissues.
Conclusion
The formation and ability of H2O2 to modulate intracellular signaling is dynamically regulated by the relative activity of oxidant generating enzymes, peroxidases, and reducing proteins. Changes in H2O2 bioavailability modulates physiological signaling by altering the oxidation status of select target proteins. Its targets in the cardiovascular system include the kinases PKA, PKG, and CAMKII, which upon oxidation undergo a functional change, providing a mechanism by which oxidant generation can couple to phospho-signaling. The oxidation of each of these kinases plays an important role in regulating normal physiological signaling. However, under conditions of oxidative stress, where oxidant formation becomes dysregulated, the oxidation of these kinases may contribute to disease progression due to loss in normal cellular phospho-regulation. Therefore, the use of antioxidants or drugs that can prevent oxidation of select redox-sensitive targets may prove beneficial under certain disease conditions.
Footnotes
Acknowledgments
We would like to acknowledge support from the Medical Research Council, the British Heart Foundation, the Leducq Foundation and the Department of Health via the NIHR cBRC award to Guy's & St Thomas' NHS Foundation Trust.
Author Disclosure Statement
No competing financial interests exist.