
Abstract
Introduction: Constitutive NOS and the Heart

Both NOS1 and NOS3 are constitutively expressed in the cardiovascular system. NOS3 is mostly found in coronary vascular and endocardial endothelial cells and, to a lesser extent, in cardiac myocytes, where it associates with caveolae (13, 68); whereas NOS1 is predominantly localized to the sarcoplasmic reticulum (SR) (254) and, to a lesser extent, mitochondria (28), the Golgi apparatus (174), and sarcolemmal membrane (167). Although the NOS1 gene can be transcribed into five different mRNA splice forms (Fig. 1), there is limited information about their physiological role or subcellular localization in cardiac myocytes (4). The α and μ-NOS1 contain an N-terminal PSD-95/Discs large/ZO-1 homology domain (PDZ domain), which can tether the enzyme to specific myocardial proteins, such as syntrophin (22). By contrast, the β and γ NOS1 isoforms (which lack the PDZ domain) are thought to reside in the cytoplasm (64). The NOS1–2 variant, although similar to the α isoform, is thought to be enzymatically inactive (23). Alternative splicing in intron13 of the human NOS3 gene has also been reported (145); however, its functional significance in the heart remains to be tested. Both constitutive NOS can translocate to different subcellular domains under certain conditions. In particular, NOS1 appears to shuttle from the SR to the sarcolemmal caveolae in cardiomyocytes from failing hearts (18, 53); by contrast, in endothelial cells, NOS3 has been reported to traffic from the plasmalemmal caveolae to intracellular locations, such as the Golgi apparatus and the endoplasmic reticulum (67).
It is no longer thought that NO synthesized within cardiomyocytes acts as a freely diffusible messenger. Instead, constitutive NO release operates within discrete subcellular domains either by stimulating soluble guanylate cyclase (sGC) to produce cyclic guanosine monophosphate (cGMP) or via the S-nitrosylation of specific protein targets. The cGMP-dependent effects of NO are largely mediated by changes in the proteins' phosphorylation state as a result of stimulation of the cGMP-dependent protein kinase (PKG) and/or of changes in the activity of cGMP–stimulated or–inhibited phosphodiesterases (PDE) (72). In the myocardium, constitutive NO production affects the function and phosphorylation state of several proteins that are involved in excitation-contraction coupling (ECC) (e.g., the L-type Ca2+ channel (LTCC) (199, 242), troponin I (135), and phospholamban (PLB) (241, 261), and inhibits both oxygen consumption (143, 253) and β-adrenergic inotropy (82) via a cGMP-dependent mechanism. Furthermore, a direct reaction between NO and thiol groups on cysteine residues causes changes in protein conformation and function that are akin to those induced by phosphorylation (71). Growing evidence supports protein S-nitrosylation as an important mechanism of NO signaling (98), which is implicated in the regulation of the ryanodine receptor Ca2+ release channel (RyR) (65, 85, 244), SR Ca2+ ATPase (SERCA) (17), LTCC (32, 214), and the Kv1.5 channel (166) and the post-translational regulation of β-adrenergic signaling (170, 246). Dysregulated S-nitrosylation of myocardial proteins can result not only from alterations in the expression, compartmentalization, and/or activity of NOS, but also from changes in the activity of denitrosylases such as the S-nitrosoglutathione (GSNO) metabolizing enzyme, GSNO reductase. Indeed, the knockout of GSNO reductase results in enhanced levels of SNO proteins and significantly attenuates experimental asthma and heart failure, while increasing the severity of endotoxic shock in mice (71).
Constitutive NOS Activity and Regulation of Cardiac Function
Paracrine and autocrine actions of NOS3-derived NO
It has long been known that NOS3-derived NO produced in the coronary endothelium modulates the functional characteristics of cardiac myocytes. One of the first demonstrations of this paracrine effect was reported by Paulus et al. in 1995 (172), who stimulated endothelial cell NOS3 by intracoronary infusion of substance P and observed increased left ventricular (LV) diastolic compliance (independent of changes in coronary flow). These effects were later linked to PKG-mediated phosphorylation of troponin I, resulting in a reduction of myofilament Ca2+ sensitivity (135).
The mechanical activation of endothelial cells (shear stress or stretch) and cardiomyocytes (stretch) stimulates the release of NOS3-derived NO (176, 178, 180), and has been proposed to play a role in enhanced stroke volume from a rise in ventricular preload (i.e., the Frank–Starling response). Here, coronary paracrine signaling appears relevant, as denuding coronary endothelium eliminated a preload-stimulated rise in myocardial NO (178). In isolated crystalloid perfused guinea pig hearts, coronary perfusion with L-NG-monomethyl arginine citrate or hemoglobin (NO scavenger) depressed the Frank–Starling reserve (180), though this was not observed in an isolated blood-perfused canine preparation (191). Stretched isolated cardiomyocytes activated NOS3 via Akt-phosphorylation to increase Ca2+ sparks, intracellular Ca2+ transient amplitude, and cell shortening—changes abolished by the genetic deletion of NOS3 or L-N ω-Nitro-L-arginine methyl ester (L-NAME) (176). Lastly, endothelial-derived NO (from bradykinin) can reversibly decrease oxygen consumption by reducing mitochondrial respiration (143, 253), effects that are also attenuated by L-NAME and abolished in NOS3 knockout mice.
Despite these and other studies, in vivo evidence that constitutively expressed NOS3 (in myocyte and endothelial cells) regulates cardiac function under basal conditions remains scant, and evidence from mice genetically lacking NOS3 suggests that any impact is minimal. Basal function is similar between control and NOS3−/− mice, though inotropic and lusitropic responses to isoproterenol (ISO) are enhanced (14, 90). Others have found no differences in rest or ISO stimulated cardiomyocyte function between these models (148, 232). However, stimulation of the β-3 adrenergic receptor (AR) plays an important role in triggering NOS3-derived NO, which, in turn, blunts β1-adrenergic inotropic responses (79) via PKG activation pathways (136); thus, attenuation of β1-adrenergic responses by NO may vary with the intensity of stimulation and the species under investigation.
Actions of NOS1-derived NO
After its detection in SR membrane vesicles of cardiac myocytes in 1999 (254), NOS1 has been shown to act as a major modulator of cardiac function and intracellular Ca2+ fluxes (Fig. 2). In 2002, Ashley et al. reported enhanced contractility and prolonged relaxation in LV myocytes from NOS1−/− mice compared with their wild-type littermates (10). The positive inotropic effect of NOS1 gene deletion on myocardial contraction (both in cardiomyocytes and in vivo) was confirmed by other (25, 55, 199) [but not by all (14, 244)] investigators. Furthermore, Sears et al. reported an increase in Ca2+ current density and larger intracellular Ca2+ transients and Ca2+ stores in the presence of NOS1 inhibition of gene deletion (199). More recently, NO released by NOS1 has been shown to accelerate cardiomyocyte and left ventricular relaxation by increasing PLB phosphorylation of via a cGMP-independent inhibition of serine/threonine protein phosphatases (261).

NOS1-derived NO is also involved in the regulation of RyR function. NOS1 gene deletion has been reported to decrease RyR S-nitrosylation; however, the impact of this finding on RyR open probability remains a matter of debate. Gonzalez et al. reported an increase of diastolic Ca2+ leak from the RyR in NOS1 knockout mice in the absence of differences in channel phosphorylation (85); whereas Wang et al. (244), using the same model, reported a decrease in RyR open probability.
A further mechanism by which NOS1 can modulate ECC is through its interaction with xanthine oxidoreductase (XOR). Khan et al. observed that both enzymes coimmunoprecipitate and colocalize in the SR. In the absence of NOS1, XOR is activated and increases its production of superoxide, which, in turn, reduces myofilament Ca2+ sensitivity (117). Finally, NOS1 has been shown to interact with the sarcolemmal calcium pump (also known as plasma membrane calcium/CaM-dependent ATPase [PMCA]); overexpression of PMCA4b inhibits NOS1 activity and blunts the β-adrenergic contractile response in cardiac myocytes (167).
In addition to its effects in cardiac myocytes, NOS1 acts as a local modulator of the autonomic control of the cardiovascular system. In the heart, NOS1-derived NO increases acetylcholine release from cholinergic neurons via a sGC-cGMP-dependent mechanism. By contrast, NO generated in sympathetic ganglia reduces the synaptic release of noradrenaline [reviewed in Herring and Paterson (97)]. In atrial parasympathetic ganglia, NOS1 is up-regulated by exercise training where it contributes to the enhanced bradycardic response to peripheral vagal stimulation (54). Similarly, an increase in atrial nNOS protein after myocardial infarction has been reported to enhance vagal bradycardia in rats (219). In the central nervous system, NOS1-derived NO exerts an inhibitory role in the regulation of sympathetic outflow, which is blunted in animal models of heart failure (202, 260).
Mechanisms of Constitutive NOS Dysfunction
Since the normal function of constitutive NOS is to generate NO for cell function and adaptation, NOS dysfunction can be considered a state where less NO is generated and/or NO targeting is disrupted. Reduced NO generation by NOS typically coexists with its increased synthesis of O2 −, a condition referred to as NOS uncoupling. This and the interaction of NO with oxidants (to form nitroso-oxidant species such as peroxinitrite [ONOO−]) can further impact the fate of any NO generated. To remain functional, NOS requires critical cofactors, an adequate substrate, and a redox environment that facilitates NO targeting. Since each is altered in heart disease, understanding their mechanisms is important.
BH4 biosynthesis and regeneration and NOS dysfunction
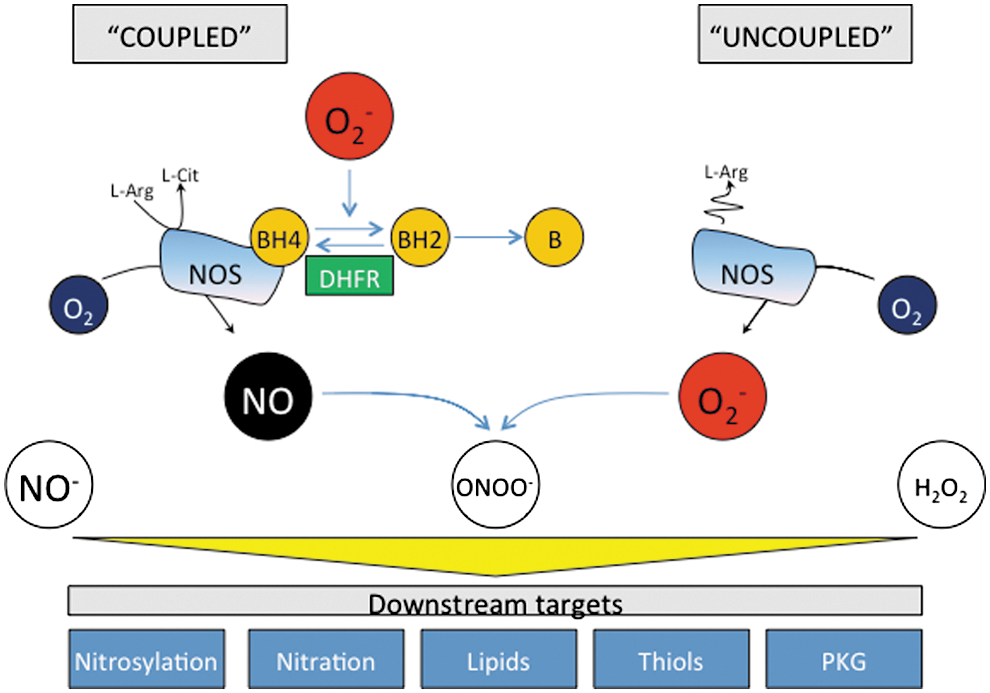
A critical aspect for NOS generation of NO is the presence of the cofactor BH4. BH4 binds close to the heme active site at the interface between the two monomers, interacting with residues from both. Maintenance and stabilization of NOS dimers is dependent on BH4, and BH4 also plays a direct role in the multistep oxidation of L-arginine through the N-hydroxy-L-arginine intermediate and the subsequent generation of NO (211). Depletion of BH4 and/or its oxidation to BH2 shifts the electron transport process, so that rather than generating NO, the enzyme acts as an oxidase (Fig. 3). This condition, known as NOS uncoupling, now appears to be a major mechanism whereby NOS dysfunction translates to pathological disease. It is, therefore, useful to review the mechanisms controlling BH4 synthesis and regeneration.
Intracellular biopterin levels are principally regulated by the activity of the de novo biosynthetic pathway. Guanosine triphosphate cyclohydrolase I (GTPCH) catalyzes the formation of dihydroneopterin 3′ triphosphate (DNTP) from GTP, and BH4 is generated by two further steps through 6-pyruvoyltetrahydropterin synthase and sepiapterin reductase (SPR). GTPCH seems to be the rate-limiting enzyme in BH4 biosynthesis, and the overexpression of GTPCH is sufficient to augment BH4 levels in cultured endothelial cells (31, 49) and in the vascular endothelium and myocardium in vivo (5, 104). Electron paramagnetic resonance spectroscopy studies have shown that BH4 both stabilizes and donates electrons to the ferrous–dioxygen complex in the oxygenase domain, as the initiating step of l-arginine oxidation (86, 187, 235). In this reaction, BH4 forms the protonated trihydrobiopterin cation radical, which is subsequently reduced by electron transfer from NOS flavins (101, 198).
When BH4 availability is limiting, electrons flowing from the reductase domain to the heme are diverted to molecular oxygen instead of to L-arginine, resulting in the formation of superoxide rather than NO. This may lead to the oxidation of BH4 to catalytically incompetent BH2, resulting in a feed-forward cascade of BH4 destruction and further uncoupling of the enzyme (234, 236). Indeed, it has previously been suggested that the relative abundance of NOS3 versus BH4, along with the intracellular BH4:BH2 ratio (instead of the absolute concentration of BH4), may be key determinants of NOS uncoupling (49). This observation is supported by several in vitro and in vivo studies (233, 235) and computational modeling (113) and suggests that mechanisms which regulate the BH4:BH2 ratio, independent of overall biopterin levels, may play a role in controlling NOS coupling that is as important as the well-established role of GTPCH in de novo BH4 biosynthesis. However, others have reported that the absolute level of BH4 and not the biopterin ratio is the principal determinant of NOS activity and uncoupling in endothelial cells and cardiomyocytes (5, 33, 185).
GTPCH protein levels are induced by oxidative stress (204), and bacterial lipopolysaccharide elicits a 2-3-fold increase in GTPCH activity in a variety of rat tissues that constitutively express GTPCH, including cerebellum, liver, spleen, and the adrenal gland. Macrophages, dermal fibroblasts, and tumor cell lines demonstrate a profound increase in GTPCH activity after treatment with IFNγ and tumor necrosis factor alpha (TNFα) (87, 93). BH4 synthesis by GTPCH is subject to feedback inhibition by BH4 and other reduced pterins via a mechanism that requires a regulatory protein known as GTP cyclohydrolase feedback regulatory protein (GFRP) (153). This inhibition of GTPCH by BH4 or GFRP can be reversed by high levels of phenylalanine (153, 259). Given the importance of matching NOS activity with BH4 availability, it is perhaps not surprising to find that stimuli for one also impact the other. For example, aortic GTPCH-1 activity and BH4 levels are enhanced by laminar shear stress by a mechanism shown to involve the phosphorylation of GTPCH-1 at serine 81 (S81) by the α-prime subunit of casein kinase 2 (248). It is proposed that laminar shear, and perhaps S81 phosphorylation of GTPCH-1, disrupts the negative feedback conferred by GFRP in endothelial cells.
In the de novo formation of BH4 from GTP, the conversion of 7,8- DNTP to 6-pyruvoyl tetrahydropterin (PTP) is catalyzed by the zinc-dependent metalloprotein, 6-pyruvoyl tetrahydropterin synthase (PTPS). Although GTPCH is the rate-limiting step to BH4 synthesis in most tissues, PTPS maybe rate limiting in some cells, most notably human hepatocytes, after stimulation with cytokines and other immunological stimuli which induce BH4 synthesis by the up-regulation of GTPCH expression (223). The final steps in the biosynthesis of BH4 are two successive propyl side chain reduction reactions, which are catalyzed by SPR. Studies are only now beginning to elucidate the role of SPR as a site for the regulation of BH4 synthesis; in particular, the genetic knockdown of SPR by RNA interference in endothelial cells is associated with reduction in both intracellular BH4 content and NO production, while in vivo delivery of the SPR gene significantly increases vascular SPR protein expression, activity, BH4 content, NO production, and NO-dependent vasorelaxation (78).
In addition to de novo synthesis, BH4 is modulated by dihydrofolate reductase (DHFR), a key regulator of folate metabolism that also regenerates BH4 from oxidized BH2 (165). Net BH4 bioavailability reflects the balance between de novo synthesis, the oxidation of BH4 to BH2, and the regeneration of BH4 by DHFR; the latter now appears very important for ultimately determining cellular BH4 homeostasis, NO bioavailability, and, ultimately, NOS coupling. Recent studies have investigated the recycling function of DHFR in cultured endothelial cells and mouse models of BH4 overexpression and deficiency (47, 48, 212). Reduced BH4 and NOS uncoupling after angiotensin II exposure are reversed by overexpressing DHFR (34). By contrast, pharmacological inhibition of DHFR activity by methotrexate, or genetic knockdown of DHFR by RNA interference, reduces intracellular BH4 and increases BH2, leading to NOS3 uncoupling in endothelial cells. In cells expressing NOS3 with low biopterin levels, DHFR inhibition or knockdown further decreased the BH4:BH2 ratio and exacerbated NOS3 uncoupling. These data have been corroborated in vivo where NOS uncoupling was induced in the aorta of hph-1 mice after treatment with methotrexate, and prevented by endothelial overexpression of GTPCH (48).
Interestingly, studies have found that DHFR levels and activity decline in experimental models of cardiovascular disease, suggesting that insufficient recycling of BH2–BH4 by DHFR is, at least in part, responsible for the reduced BH4 levels and the accumulation of BH2, leading to NOS uncoupling. For example, DHFR protein levels are significantly decreased in streptozotocin-induced diabetic mice, and diabetes-induced impairment of cardiomyocyte function is exacerbated after DHFR inhibition with methotrexate (186). Furthermore, reduced DHFR activity in adult cardiomyocytes underlies their limited capacity to synthesize BH4 after cytokine stimulation after treatment of rat cardiac allograft recipients with sepiapterin (104). In support of these findings, the up-regulation of BH4 recycling enzymes is sufficient to restore BH4 levels, and effectively “re-couple” NOS3 within the aorta of streptozotocin-induced diabetic mice. Insufficient DHFR activity might also explain impaired vasorelaxation in atherosclerotic vessels from hypercholesterolemic rabbits, despite exposure to sepiapterin as the latter increases intracellular BH2 levels, requiring DHFR to regenerate BH4 (233).
NOS phosphorylation
NOS activity is regulated by a number of protein kinases, and changes in NOS phosphorylation may play an important role in determining and regulating dysfunctional NOS activity in the diseased heart. To date, the majority of evidence stems from vascular studies and the precise role of these post-translational modifications in myocytes remain less clear. In vitro and in vivo experiments have shown that NOS3 activation by phosphorylation at Ser-1177 (human, Ser-1179 in bovine) is mediated by the PI3K/Akt kinase pathway. Transduction of rabbit femoral arteries with constitutively active Akt increased NO-mediated vasodilatation in response to acetylcholine, whereas transduction with dominant-negative Akt attenuates the effect and decreases NO production in endothelial cells (146). Phosphorylation of NOS3 threonine-495 (human) or −497 (bovine) within the CaM binding domain has also been described and appears to be constitutively present in endothelial cells (142). Threonine-495 is a negative regulatory site, and its phosphorylation (probably by protein kinase C [PKC]) is associated with reduced electron flux and NOS3 activity, whereas de-phosphorylation by protein phosphatase-1 enhances the enzyme's activity (152). In cardiac myocytes, application of hydrogen peroxide or angiotensin II transiently increases NOS3 phosphorylation and NO production via an AMPK and Akt-dependent mechanism (196). Pretreatment with PEG-catalase abolished both NOS3 phosphorylation and the positive inotropic effect of angiotensin II, suggesting that myocardial hydrogen peroxide production can modulate both NOS3 activity and inotropy. The angiotensin II-mediated increase in NO was abolished in cardiomyocytes from NOS3−/− mice but unaltered in NOS1−/− myocytes. By contrast, the latter showed a reduction in NO synthesis in response to β-AR stimulation, suggesting that angiotensin II specifically activates myocardial NOS3 activity via a hydrogen peroxide-dependent increase in S1177 and S663 phosphorylation; whereas NOS1 activity is preferentially coupled to β-ARs. Others have reported that the incubation of cardiac myocytes with angiotensin II for 3 h increases NOS1 expression and the speed of relaxation by mechanisms which are dependent on superoxide production by NADPH oxidases (109). Taken together, these findings indicate that myocardial reactive oxygen species (ROS) production may influence both the expression and activity of myocardial constitutive NOSs.
Recent studies have revealed that hearts subjected to various durations of ischemia show a time-dependent reduction in BH4 levels which trigger increased NOS-derived superoxide production. Paradoxically, Akt-mediated phosphorylation, which would usually enhance NOS3-derived NO, further exacerbates the NOS-derived superoxide production, indicating that Ser-1177 phosphorylation is a critical regulator of both coupled and uncoupled NOS3 function (38). However, this may not be relevant with regard to NOS1; although NOS1 contains a potential site for phosphorylation at S1412 at its C-terminal tail that is analogous to the established Akt site found in NOS3 (58, 75, 77), a second phosphorylation site at S847 resides in an α-helix of the autoinhibitory domain of NOS reductase (95, 184). Here, phosphorylation inhibits the binding of CaM and attenuates NOS1 catalytic activity (50, 130).
NOS glutathionylation
Glutathionylation is the specific post-translational modification of protein cysteine residues by the addition of the tripeptide glutathione (GSH), the most abundant and important reducing agent within the cell. Promoted by oxidative stress, glutathionylation can regulate a number of cell processes, including apoptosis, Ca2+ homeostasis, and cellular antioxidant defences (45). Having observed that supplementing dysfunctional NOS with BH4 or L-arginine is often not sufficient to fully restore coupled NOS activity, Chen et al. reported that S-glutathionylation may be a unique mechanism for the redox regulation of NOS3, inducing enzymatic uncoupling and superoxide generation from the reductase domain of the enzyme (40) (Fig. 4). Two highly conserved cysteine residues are critical for NOS3 function, and become glutathionylated under conditions of oxidative stress, promoting NOS3-derived superoxide production. In vitro accumulation of GSSG, induced by exposure of endothelial cells to the GSH reductase inhibitor 1,3-bis(2 chloroethyl)-1-nitrosourea, results in glutathionylation of NOS3; whereas thiol-specific reducing agents reverse both NOS3 glutathionylation and endothelial dysfunction in vessels from spontaneously hypertensive rats (40). There are several proposed mechanisms of S-glutathionylation (1, 21). When the ratio of GSSG:GSH is high, thiol-disulfide exchange with oxidized glutathione (GSSG) may occur. ROS and reactive nitrogen species-derived thiyl radicals can, in turn, react with GSH and protein thiols to allow protein S-glutathionylation. It is believed that the formation of thiol intermediates, such as the thiyl radical, sulfenic acid, or S-nitrosyl, is a more rapid and efficient mechanism for protein S-glutathionylation in vivo and that these mechanisms could play a role in signal transduction. NOS3 thiols can be oxidized by superoxide generated from uncoupled NOS3, exogenous sources, or cellular oxidative stress through the formation of thiyl radicals. Protein thiyl radicals generated from uncoupled NOS3 indeed react with reduced GSH to form NOS3 S-glutathionylation (39).

Classical uncoupling of NOS3, usually induced by BH4-dependent mechanisms, exhibits superoxide generation from the heme iron within the oxygenase domain, which is inhibited by the L-arginine analogue, N-nitro-L-arginine and its methylester, L-NAME. Here, the nitro-substituted arginine analogue occupies the L-arginine binding site, attenuating the reduction of the heme iron by the reductase domain; the ferric heme does not bind oxygen, rendering it incapable of promoting the production of superoxide. Glutathionylation-induced uncoupling of NOS3, by generating superoxide from flavins within the enzyme's reductase domain, is less sensitive to inhibition by L-NAME. This mechanistic difference between BH4- and glutathionylation induced NOS uncoupling has far-reaching implications; in particular, superoxide generation from the NOS reductase domain, initiated by glutathionylation, might act as a “trigger” for BH4 oxidation and haem-derived superoxide production within the oxygenase domain. Since the importance of NOS coupling in cardiovascular health and disease is now well established, therapeutic strategies targeting the restoration of cardiac and endothelial BH4 levels need to consider these issues.
Arginine metabolism: asymmetric dimethyl arginine and arginase
NOS requires L-arginine to function and generate NO, and abnormal arginine metabolism also plays a role in NOS dysfunction. Two pathways have been recently highlighted (237): One is the conversion of L-arginine to asymmetric dimethyl arginine (ADMA) by protein arginine methyltransferase, and the second is its catabolism to urea or ornithine by arginase I and/or II. Both reduce the available supply of L-arginine, and in case of ADMA, further inhibit NOS by competing for L-arginine binding. Enhanced levels of ADMA are accompanied by NOS uncoupling, which is associated with vascular and cardiac dysfunction (7). In vivo levels of ADMA are increased by gene deletion of dimethylarginine dimethylaminohydrolase-1—an enzyme that metabolizes ADMA—leading to increased ADMA, NOS dysfunction, and vascular disease (138).
Increasing the level or activity of arginases also compromises NOS function by limiting available substrate. Arginase is a manganese metalloenzyme which is central to the hepatic urea metabolism, and exists in two distinct isoforms that are widely distributed. Arginase I is the primary form in the liver, while arginase II is more widely expressed. Both are expressed in endothelial cells and vascular smooth muscle that vary with vessel type and species. While first thought to be primarily involved with disposal of amino acid and nucleotide nitrogen, arginase has been recently revealed to play a key role in cardiovascular biology by its regulation of NOS3 and NOS1 (19). Arginase is activated by oxidative stress via a Rho/Rho-kinase pathway in bovine aortic endothelial cells (35, 210), and its up-regulation with aging, ischemic heart disease, hypertension, and heart failure supports its role in NOS dysfunction (194).
The fate of NO: role of redox chemistry
In addition to NOS dysregulation, the bio-availability of NO for targeting sGC or other protein thiols for S-nitrosylation reactions depends on local redox chemistry. As noted, oxidative stress shifts BH4 to BH2, and reactive species such as ONOO− (15) (which are formed from NO and superoxide) can directly impede NOS activity by modification of the central zinc (263). NOS uncoupling provides the ideal microenvironment for this chemistry, as both oxidants and NO are generated by the same enzyme, and this is thought to be an important factor limiting both cGMP-dependent and independent signaling linked to NO (116). ONOO− targets remains far from being well understood, though several proteins have been identified [e.g., SERCA (124)] and [e.g., PKCɛ (12)].
Consequences of NOS Dysfunction in Cardiovascular Disease
NOS, endothelial function, and vascular disease
Given the central role of NO signaling in endothelial function, smooth muscle proliferation, thrombosis, and inflammation, it comes as no surprise that NOS dysfunction is a potent contributor to vascular pathophysiology (96). The importance of NOS3 to vascular homeostasis has been demonstrated by genetic gain and loss of function studies, with deletion worsening neointimal medial thickening and vascular remodeling after injury (160, 258); whereas endothelial-targeted NOS3 overexpression suppresses the same (114) and reduces atherosclerosis in the ApoE-null mouse (231). Significantly, the deletion of NOS3 removed both NO-dependent vasorelaxation and oxidant-coupled hyperpolarization (150). While uncoupled NOS3 generates less NO, its synthesis of O2 −, subsequently converted to H2O2 by Cu,Zn-superoxide dismutase, also results in vasodilation (159, 205). The latter was recently coupled to oxidation of C46 residues in PKG1α to form an internal disulfide bond and activate the kinase in a cGMP-independent manner (27). Mice with a knock-in mutated PKG1α (C46S) cannot undergo this oxidation, and develop hypertension(181). Along with previous NOS3 deletion studies, these results suggest a novel linkage between NOS3-derived oxidants and vasomotor tone. Depressed NOS1 activity also likely plays a role in vascular disease. Interestingly, NOS1 regulates peripheral and coronary flow in humans (200, 201), and its expression rises in early- and advanced-stage atheroma (249) and in the neointima, endothelial cells, and macrophages in vascular lesions. This is likely to play a counter-responsive role, as NOS1 gene deletion models also develop worse vascular pathobiology after vessel injury (131). Although NOS1 nor NOS3 gene deletion alone is sufficient to induce significant vascular disease in the absence of a pathological stress or concomitant disease, combining deletions displays a striking phenotype. Deletion of all three NOS isoforms in mice results in spontaneous coronary artery disease, myocardial infarction, and sudden cardiac death (163, 228), which are marked by intimal inflammation and coronary vasospasm. Thus, defects in one isoform alone may be compensated by others, but combined defects are likely to be markedly pathogenic.
NOS3 also contributes to mechano-signaling in the coronary vasculature, which is important to matching coronary reserve with ventricular demands with exertional stress. The failing and hypertrophied heart has depressed coronary dilator reserve, and NOS dysfunction has long been known to play an important role. Increases in pulsatile flow involve both a rise in phasic shear stress and cyclic wall stretch, both of which are involved with NOS activation via post-translational modifications, notably Akt phosphorylation, that not only involve overlapping (e.g., tyrosine kinase cSrc) but also unique (e.g., VEGFR2 dependent for shear, or independent for pulse-stretch) signaling (140). Reduced vessel compliance—a common feature of aging and many cardiovascular diseases—would suppress a stretch mechano-signal; this may blunt Akt and, thus, NOS3 activation, and dampen cytoprotection against oxidant stress (140). Through this mechanism, dysfunctional NOS activity may be linked to structural changes in the vessel wall, independent of endothelial biology per se.
In addition to modulating vascular tone and proliferation, endothelial-derived NO impacts inflammation. Early studies focused on the role of inducible NOS2 (208) (222); however, NOS3 and NOS1 now appear to be major participants as well (61, 238), providing a more tonic level of NO release. For example, both NOS1−/− and NOS3−/− mice display 2- or 6-fold increases, respectively, in leukocytes rolling at baseline (134, 137), and 9-10-fold increases after inflammatory stimulation.
NOS dysfunction and myocardial ischemia/reperfusion injury and infarction
The cell-specific role of NOS1 and NOS3 in the ischemic and infarcted ventricle is somewhat uncertain, as targeted gene-deletion models are lacking; however, both isoforms are thought to play a role. Mice globally lacking either NOS1 or NOS3 display worse LV function and remodeling after coronary ligation-infarction (55, 195). Both endothelial-and myocyte-restricted overexpression of NOS3 reduces the severity of postinfarction pathophysiology (107, 110). Somewhat analogous findings were reported for NOS1, where myocyte-specific conditional transgenic overexpression of NOS1 led to reduced infarct size, improved function, and reduced oxidative stress after coronary ligation (28). The same authors reported the up-regulation and translocation of NOS1 to mitochondria in response to ischemia/reperfusion; similarly, transgenically overexpressed NOS1 localized to mitochondria coupled to HSP90 translocation (28). Unlike NOS3 for which expression changes in hypertrophy and heart failure are often not observed, NOS1 expression rises in human HF (53) and after myocardial infarction in rats, where translocation to the plasma membrane and association with caveolin-3 (Cav-3) is observed (18, 52). Although this may reduce the capacity of NOS1 to impact oxidative stress, it also leads to retargeting, with increased cGMP/PKG and S-NO mediated inhibition of the LTCC (214) and β-AR signaling (18, 55, 214) that may protect the heart while down-regulating function.
The mechanisms underpinning NO-derived cardiac protection to ischemia-reperfusion injury involves mitochondrial KATP channels via PKG phosphorylation (255), S-nitrosylation of sarcoplasmic and mitochondrial calcium uptake ATPases and of the hypoxia inducible factor 1α (141), and ONOO-induced tyrosine-nitration of PKCɛ (12). The relative role of NOS3 and NOS1 to these mechanisms may depend on redox conditions that impact NOS coupling, sGC-NO responsiveness (226), and potentially PKG activity (27). S-nitrosylation of a variety of target proteins during ischemic preconditioning is thought to act as a cysteine shield, protecting residues from subsequent oxidation and functional damage on reperfusion (126).
NOS dysfunction and cardiac hypertrophy and failure
Changes in NOS3 and NOS2 in experimental and human cardiac failure and pathological hypertrophy bear many similarities with results obtained in the postischemic/infarcted heart. NOS3 expression has been observed to decline in some studies (60, 177), while others find no change, and NOS1 expression typically increases (18, 52). Gain-and-loss-of-function models have targeted pressure overload, and here, the results are somewhat mixed, likely highlighting the importance of redox environment and status of NOS (i.e., uncoupled or not) in determining the response. For example, chronic NOS3 deletion induces concentric hypertrophy (14), and has been found to worsen remodeling after pressure overload (197). Myocyte-targeted up-regulation protected against this loading model (29). However, others employing abdominal aortic banding found increased hypertrophy but less chamber dilation in NOS3−/−(189). With a more severe ascending constriction model, Takimoto et al. (218) found that NOS3−/− mice were paradoxically protected, displaying neither dilation nor progressive LV systolic dysfunction as compared with controls, and linked this to the lack of oxidative stress in mice lacking NOS3. Thus, while NOS3 may usually be protective against maladaptive stress, once uncoupled, it potently contributes to the pathophysiology, itself becoming a major ROS generator. In this condition, its absence results in improved biology.
In vitro studies have demonstrated the uncoupling of NOS1 activity in response to ROS via both reversible oxidation of BH4 (induced predominantly by superoxide) and irreversible heme degradation (induced by peroxynitrite) (213). Less is known about the mechanisms and functional consequences of NOS1 uncoupling in vivo, though it may in part account for left ventricular diastolic dysfunction. In deoxycorticosterone acetate-salt mice, treatment with the partial selective NOS1 inhibitor 7-nitroindazole reduced superoxide in left ventricular tissue, while oral BH4 supplementation maintained or restored PLB phosphorylation. NOS1 inhibition or gene deletion also impairs myocardial relaxation by reducing the PLB phosphorylated fraction (241, 261).
The role of NOS2 (inducible NOS) to pathological LV remodeling in heart failure is still being defined. Targeted overexpression in myocytes is protective against pressure overload (245) and as previously noted, this may relate to antioxidant effects (169). However, whether NOS2 provides a source of ROS due to uncoupling under stress is less clear, as the deletion of NOS3 appears sufficient to suppress ROS activity (218). Furthermore, since much of NOS2 signaling appears less dependent on cGMP and more on S-NO modifications, the impact of up- or down-regulation is likely to depend on the local redox environment, not only affecting the enzyme itself, but also the proteins whose cysteines would be targeted for S-nitrosylation.
The scaffolding protein Cav-3 plays an important role in NOS function in the cardiac myocyte, and evidence supports the role of Cav-3 dysregulation in heart disease. In a study conducted on mouse models of dilated cardiomyopathy (adenosine A1-receptor or TNFα overexpression), Cav-3 expression declined and correlated with functional deficits as well as reduced Akt activation and SR-ATPase gene expression (66). In this study, human-failing myocardium Cav-3 trended to a decline, though some studies of experimental heart failure have reported increased Cav-3 expression (15). Mice genetically lacking Cav-3 develop hypertrophic cardiomyopathy that is accompanied by activation of the p42/44 mitogen activated protein kinase (MAPK) pathway (251), while cardiomyocyte-targeted over-expression of Cav-3 (100) may attenuate cardiac hypertrophy, increasing cGMP levels, a-type and b-type natriuretic peptide (ANP/BNP) expression, and enhancing nuclear targeting of Akt, itself shown to be anti-hypertrophic (227). Cav-3-NOS modulation likely contributes to these effects. For example, in the pressure-overloaded heart, NO-dependent activation of sGC is markedly depressed, in part due to oxidation coupled to a decline in its expression within Cav-3 microdomains (226). In hearts genetically lacking Cav-3, sGC activation by NO or by direct small-molecule activators is also lost (226—indicating a critical role of the protein in normal interactions between NOS-NO generation and sGC-cGMP production. Since caveolae also serve as a major nexus for a broad range of cell signaling, coordinating receptors, kinases, ion channels, and the dystrophin-glycoprotein complex (80), more than NOS3 dysregulation is involved when Cav-3 is abnormal. Loss-of-function mutations in Cav-3 have been linked to several forms of muscular dystrophy, including limb girdle, hyper-creatine kinase, and rippling muscle disease (80). Several case reports that suggest Cav-3 mutations (e.g., T77M) (225) can result in hypertrophic cardiomyopathy, though this remains rare, and whether the disease involves NOS dysregulation is unclear. NOS1 colocalizes with Cav-3 in skeletal muscle but not in normal cardiac myocytes. However, some have observed plasma-membrane translocation of NOS1 in heart failure syndromes, where interaction with Cav-3 may well play a role in altered signaling (16, 53).
NOS and cardiac transplantation
Despite general success with human heart transplantation, graft rejection continues to be a primary cause for postoperative mortality and morbidity (46). Most research has focused on inducible NOS2, which may have positive and negative effects, the latter being coupled to mitochondrial (220), metabolic (139), and apoptotic (215) modulation. NOS3 is also likely involved. While depressed NOS3 is observed and thought to contribute to graft/host vasculopathy (57, 175), data regarding the benefits of gene therapy that enhance NOS3 are mixed (106, 108, 257). This could reflect redox imbalance with a graft rejection; so, NOS3 becomes more likely to be uncoupled if up-regulated.
NOS and angiogenesis
There is evidence that NOS-derived NO controls the process of angiogenesis during heart development (262). This regulation becomes even more important during pathological conditions where the formation of new vessels plays a key role in the remodeling of the heart. Recent findings suggest that NOS3 regulates the migration of endothelial-derived progenitor cells (EPC) which are produced by the bone marrow and mobilize on stimulation to induce neovascularization (173). NOS3−/− mice have a reduced recruitment of EPC and impaired angiogenesis (3). The absence of NOS3 prevents the up-regulation and mobilization of EPC after aortic constriction, resulting in enhanced cardiac hypertrophy and fibrosis, and reduced capillary formation (115). These negative effects are reversed when wild-type bone marrow is transplanted in NOS3−/− mice. EPC are also recruited to the myocardium during ischemia preconditioning (103), although whether this is causing a beneficial effect on cardiac remodeling has not yet been elucidated. In myocardial infarction models, EPC mobilization and angiogenesis can be increased by the administration of estradiol or atorvastatin (105, 132); in both cases, the increase in EPC recruitment is mediated by NOS.
NOS and arrhythmia
Both NOS1 and NOS3 play key roles in electrical activation of the heart, and their dysfunction contributes to arrhythmia. Mice lacking NOS3 display a slower heart rate, and an increase in the transient inward current that has been coupled to aminoglycoside-induced ventricular ectopy and tachycardia (182). While NOS3−/− myocytes have normal resting action potential duration (APD) and LTCC current under basal condition, their response to ISO is altered, with longer APD and augmented I Ca associated with increases in early and delayed afterdepolarizations (243). The latter may be related to a loss-of-negative cGMP/PKG effects on the β-subunit of the LTCC (256). NOS1 plays a key role in Ca2+ cycling, and its absence results in an increase in peak and diastolic calcium and ventricular ectopy (25, 199). Mice lacking NOS1 develop worse arrhythmia and SCD after myocardial infarction (25).
A novel mechanism linking NOS and arrhythmia has been revealed by genetic studies of mutations in the sodium channel Nav1.5. The channel protein forms a complex with NOS1, PMCA4b, and a-syntrophin, and the late Na+ current is enhanced by S-nitrosylation (9, 36, 112, 230). A mutation in SNTA1 enhances S-NO modifications, and is a cause for long QT syndrome (LQT3). Furthermore, genome wide association studies showed a genetic variant in the gene encoding the NOS1 adaptor protein, CAPON, to be associated with prolongation of the QT interval (9) and sudden cardiac death (112). The SNP is in a noncoding region, and mechanisms by which it confers changes in NO-dependent signaling remain underway.
Dysfunctional NOS activity has also been linked to atrial fibrillation (AF). Short-term AF leads to reduced atrial NOS activity and NO bioavailability (30), coupled with an increase in Rac1 activity and superoxide production from NOX2 oxidases. In contrast, long-standing AF and atrial structural remodeling are associated with increased mitochondrial oxidase activity and NOS uncoupling secondary to a reduction in atrial BH4 content and an increase in arginase activity (121, 185). In human atrial myocytes, NO donors prolong APD by decreasing I Kv4.3 and human I to1 (84); whereas endogenous NO increases the inwardly rectifying K+ current, I K1 and shorten the APD by S-nitrosylating Kir2.1 channels (83). These findings suggest that the decreased S-nitrosylation of Kir2.1 channels observed in human samples from patients with AF may represent a compensatory mechanism which attenuates the shortening of APD and atrial refractory period and, thus, atrial electrical remodeling in AF. Although recent data support the use of statins to counter NOX2-dependent atrial superoxide production as a strategy which prevents the new onset of AF after cardiac surgery (6), other findings indicate that this approach is unlikely to be successful in long-standing AF (185).
Therapeutic Approaches for Fixing NOS-opathy
The approaches that are used for ameliorating NOS dysfunction in cardiovascular disease have targeted each of the features we have just reviewed. They fall into six categories: organic nitrates, NOS activators, NOS recouplers, arginase inhibitors, enhancers of cGMP/PKG signaling, and modulators of NOS-ROS interaction (Fig. 5). Although these appear as promising targets, clinical translational remains limited, though several approaches are being actively pursued in patients with heart disease.

Organic nitrates
Organic nitrates are used in the treatments of ischemic heart disease and heart failure by increasing blood NO concentration via their denitration. Nitroglycerin (glyceryl trinitrite [GTN]) is the most common form, and it rapidly improves cardiac hemodynamics by vasodilation, augmenting cGMP in vascular smooth muscle (224). GTN denitration has been previously attributed to reactions with GSH S-transferase, cytochrome P450, and XOR (76); however, recent data indicate (41) that mitochondrial aldehyde dehydrogenase 2 (mtALDH2) is the major enzyme (128, 129). GTN bioactivation may also result in the formation of other NO-related species that activate sGC in a haem-independent manner. While among the initial agents used to reduce cardiac loading in heart failure, organo-nitrates lost favor due to the clinical superiority of other vasodilators (e.g., angiogensin converting enzyme inhibition) and tolerance. The latter is now thought to be due to the chronic suppression of mtALDH2 after long-term exposure (216). Nonetheless, recent evidence of the benefits of this drug class combined with hydralazline in the African-American sub-population of heart failure patients has revived interest (221).
NOS activators
Akt activation of NOS3 has been recently mimicked by administration of the gas, hydrogen sulfide (H2S) (179). H2S stimulates a two-fold increase in NO production coupled to Ser-1177 phosphorylation, and the inhibition of Akt prevents these effects. Current H2S “donors” are short-lived, and will have to be modified to generate a meaningful pharmaceutical approach, but this strategy is evolving (247).
Several methods have been recently described as enhancing NOS3 expression. Two small-molecule NOS3 transcription enhancers, 4-fluoro-N-indan-2-yl-benzamide (AVE9488) and 2,2-difluoro-benzo[1,3]dioxole-5-carboxylic acid indan-2-ylamide (AVE3085) were identified by high throughput screening. Both AVE9488 and AVE3085 stimulate eNOS transcription in vivo and in vitro (250). They reduce atherosclerotic plaque formation in apoE knockout mice but not in the apoE/NOS3 double knockout mice (250), supporting specificity for NOS3. AVE30085 improves endothelial dysfunction in diabetic mice through increased NO production and reduced oxidative stress in the vascular wall (37). These compounds may prove useful by enhancing NOS3 expression, though the status of NOS—that is, redox modulation of coupling or uncoupling—could impact their net benefit, and more studies are required.
Another approach that increases NOS3 expression is the polyphenol antioxidant trans-resveratrol (240), which augments NO production (122). Trans-resveratrol also prevents NOS3 uncoupling and lowers oxidative stress while preserving endothelial function (20), and it can up-regulate GTPCH-1 (252), suggesting multiple mechanisms whereby it may prove useful for treating NOS-opathy. Lastly, triterpenoids, such as betulinic acid isolated from birch tree bark, have been shown to improve endothelium-dependent relaxation in rats with L-NAME-induced hypertension by their ability to reduce oxidative stress (74). Betulinic acid may also up-regulate NOS3 expression and reduce NADPH oxidase expression in human endothelial cells through a PKC-dependent mechanism (209).
Lastly, the racemic mixture d- and l-nibivolol, a third-generation selective
NOS recouplers
BH4 is a critical regulator of NOS coupling and function, suggesting that it may be a beneficial therapeutic agent in vascular disease. Pharmacological supplementation of BH4 improves endothelium-dependent relaxation and NOS coupling (155, 157). BH4 aids electron transfer from the NOS reductase to the oxidase domains, assisting in the production of NO and L-citrulline from L-arginine. It stabilizes NOS in its active homodimeric form (183). Moens et al. (157) found that the administration of BH4 to mice with well-established left ventricular pathological remodeling, fibrosis, and dysfunction induced by pressure overload led to the amelioration of these abnormalities and the reversal of maladaptive remodeling. This occurred in association with “re-coupling” of NOS, which accounted for most of the ROS generation in the control (placebo treated) group. Intriguingly, reversal of the pathology/pathophysiology by BH4 was not coupled with enhanced PKG activation. Rather, the data suggest retargeting of NO due to an improvement in myocardial NO/ROS imbalance. More recently, BH4 was shown to improve left ventricular diastolic dysfunction in a mouse model of hypertension via an increase in coupled NOS1 activity (206). BH4 has also been used to treat ischemia/reperfusion (62), and recent data have coupled this beneficial effect with angiogenesis (203).
However, despite initial enthusiasm based on experimental studies, clinical trials of BH4 to treat disorders linked to uncoupled NOS activity (i.e., hypertension, sickle cell disease, cardio-renal syndrome, pulmonary hypertension, and coronary artery endothelial dysfunction) have all been disappointing [reviewed in (156)]. Part of the problem may be differences in the tissue uptake of exogenous BH4, which appears to be species dependent. Another factor is that orally consumed BH4 is rapidly oxidized, and should be reconverted by DHFR to be effective. Even in mice, where exogenous BH4 is effective for treating heart disease, increasing the dose led to a paradox reversal of benefit, re-appearance of ROS, and this was coupled with a decline in the BH4/BH2 ratio toward baseline (155). Variability in both uptake and redox modification of exogenous BH4 depending on the patient and disease would greatly complicate its clinical pharmacology.
There are several alternatives to BH4 itself. Statins can increase BH4 bioavailability in endothelial cells by up-regulating GCH1 gene expression (94), and could assist in NOS uncoupling. Folic acid has been studied, and it can improve the binding affinity of BH4 to eNOS, enhancing the reduction of BH2 to produce BH4, and chemically stabilizing BH4 (158). However, the doses required are unclear, and initial efforts have not been encouraging (59).
Arginase inhibitors
The importance of arginase in disease conditions has been based in large part on the use of small-molecule inhibitors, such as N(omega)-hydroxy-nor-L-arginine, which reverses hypertension in the spontaneously hypertensive rat (11). Other inhibitors [e.g., S-(2-boronoethyl)-l-cysteine] have been used to demonstrate the impact of arginase up-regulation on suppressing NOS signaling in aged vessels (19), and to ameliorate vascular aging (42, 111, 120). Commercial development of clinical arginase inhibitors has begun (Arginetix, Inc., now Immune Control), and assessment of the efficacy of this approach may follow.
cGMP-enhancing approaches
Another approach for tackling NOS-opathy is to concentrate on the cGMP-dependent component of the pathway. While recognizing that this represents a component of NO signaling that may or may not be central depending on the condition, nonetheless, studies have revealed the benefit of enhancing cGMP and thereby PKG activation in the hypertrophied and failing heart. There are three primary means for achieving this: exogenous administration of NO donors or stimulators of an alternative natriuretic-peptide-coupled cGMP synthetic pathway, the direct activation/stimulation of sGC itself, or the inhibition of cGMP hydrolysis by selective PDE. All three have been clinically utilized and/or investigated. The use of NO donors and NPs has the longest history. The former remains limited due to tolerance that has multiple causes, including oxidative stress and the associated down-regulation of aldehyde dehydrogenase-II (216), PDE up-regulation (119), arginase activation (118), and NOS uncoupling. NP administration has not only been primarily used to reduce ventricular load, but may also have benefits on the heart, though its use has been limited to date due to requirements for intravenous administration. This may be changing, however, in the near future (239).
Activators and stimulators of sGC represent a fairly new class of drugs, and several are now in advanced stages of clinical trials. Some such as cinaciguat (BAY 58-2667) function through an NO-independent mechanism, whereas others (stimulators, riociguat, BAY 63-2521) improve the binding of sGC to NO. Low-dose intravenous Cinaciguat decreased diastolic blood pressure and heart rate without lowering systolic blood pressure, whereas at higher doses, mean arterial pressure declined as plasma cGMP increased (73). In a Phase II study, patients with pulmonary hypertension treated with riociguat displayed increased exercise capacity and reduced pulmonary vascular resistance (81). Other trials are ongoing to examine their role in heart failure.
Lastly, studies have also revealed how the inhibition of cGMP-selective PDE5a with drugs such as sildenafil or tadalafil (both widely used to treat erectile dysfunction and pulmonary hypertension) can improve cardiac hypertrophy/failure pathophysiology and remodeling. Sildenafil suppresses as well as reverses existing hypertrophy/dysfunction in mice subjected to pressure overload (162, 217). This and similar agents also protect against I/R injury (192, 193), doxorubicin toxicity (127), and precondition stem cells to improve postimplant survival and function (99). They are also being studied in muscular dystrophy, where both skeletal and cardiac failure have been linked to the loss of dystrophin and NOS1 activity (2, 125). There have been several clinical trials, showing the improvement in heart failure symptoms, cardiac remodeling and function (89), and peripheral vascular reserve and endothelial function (88). A multicenter clinical trial (RELAX) that studies the effects of sildenafil in patients with heart failure and a preserved ejection fraction is near completion; results are due in late 2012.
ROS/NO interactions
The recognition that nitroso-redox imbalance can adversely impact the heart has led to several therapeutic approaches. Broad antioxidant strategies, such as with Vitamin E and C supplementation, have been disappointing. Some found HF risk may even increase, as in the HOPE-TOO trial conducted in patients with vascular disease or diabetes-mellitus-prescribed vitamin E (144). Other trials using folic acid, B-vitamins, and other agents have so far not improved cardiovascular risk.
A few other strategies have been proposed, though clinical data remain controversial. For example, XOR is proposed to negatively impact the nitroso-redox balance in the cardiovascular system (229), and XOR inhibitors such as allopurinol or oxypurinol can improve cardiac function and/or remodeling in the experimental failing (190) and infarcted (154) heart. This may be related to targeting of the NOS1-XO interactome (117) and amelioration of vascular oxidative stress to improve endothelial function (133). However, to date, human HF trials of XOR inhibition have not shown clinical benefit (92, 164). Another approach was the combined use of hydralazine and isosorbide dinitrate. A retrospective analysis of earlier trials led to the hypothesis that, while not particularly effective in the Caucasian patients with HF, the hydralazine-isosorbide dinitrate combination may benefits to African Americans. The clinical trial, reported in 2004 (221), showed a surprising efficacy on mortality and led to the first racial-targeted FDA approval of a therapy. One hypothesis was that hydralazine acted as an anti-oxidant and suppressor of nitrate tolerance, and combined with isosorbide dinitrate, could favorably impact the nitroso-redox imbalance (51, 91, 161). Why this should work only in African Americans—if indeed this is the relevant mechanism—remains unclear, as one would imagine that NOS/ROS imbalance is not unique to this population. The greater incidence of hypertension in African Americans and, thus, potential loading impact remains an alternative explanation.
Conclusions
In summary, NO is a critical regulator of cardiovascular homeostasis, and abnormalities in the performance of its primary constitutive synthases play an important role in heart disease. Growing evidence points to post-translational modifications underlying many of the NOS-opathies, resulting in an imbalance of nitrosative and oxidative environments. This impacts both ongoing NO generation and its chemistry as well as the subsequent capacity to stimulate cGMP and PKG activation or modify targeted0 protein residues. Several new avenues to ameliorate NOS activity or its downstream pathways have shown promising results, but more trials are obviously needed.
Footnotes
Acknowledgments
This study was supported by Fondation Leducq, National Institutes of Health, and the British Heart Foundation.