
Abstract
Introduction
Live images of cultured mammalian cells allow for easy observation of the dynamic movement of mitochondria, which undergo frequent fusion and fission (11, 35, 44) (Fig. 1). If mitochondrial fusion is blocked, the opposing mitochondrial fission leads to the formation of short and fragmented mitochondria, thus increasing the number of individual mitochondria. In contrast, inhibition of fission induces elongation of mitochondrial network structures due to active mitochondrial fusion, thereby stimulating the exchange of mitochondrial contents. Thus, balanced fusion and fission changes the morphology and individuality of each mitochondrion to maintain cellular homeostasis (72).

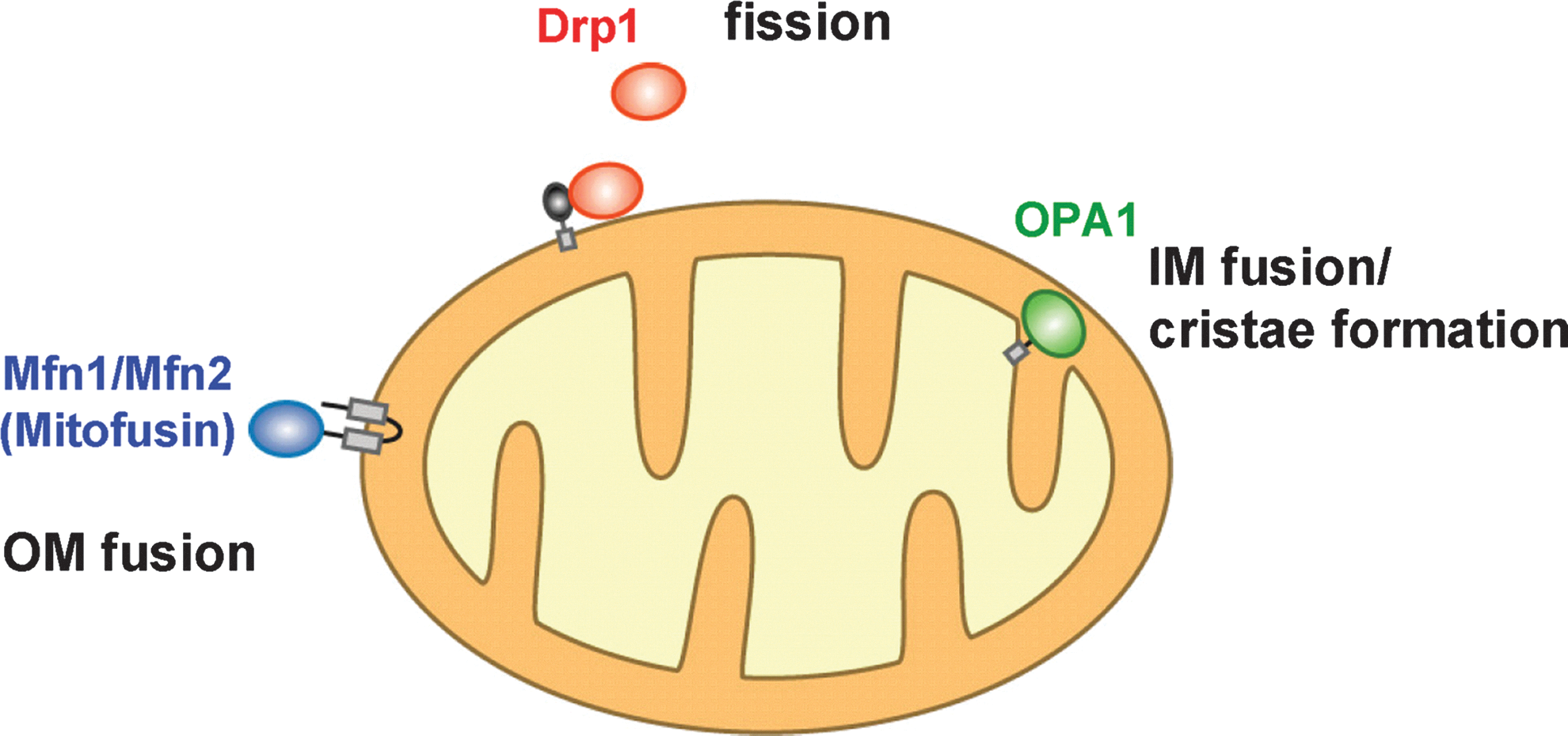
Three types (four proteins) of high-molecular-weight GTPase proteins regulate mitochondrial fusion and fission in mammals (61, 91) (Fig. 2). Two mitofusin proteins (Mfn1 and Mfn2; Fzo1 in yeast) locate on the outer membrane and are involved in outer membrane fusion (11, 22, 69). An intermembrane space-localized GTPase, optic atrophy (OPA) 1 (Mgm1 in yeast), functions in fusion and cristae formation of the inner membrane (62). Cytoplasmic dynamin-related GTPase protein, Drp1 (Dnm1 in yeast), translocates to mitochondria and mediates mitochondrial fission (74). Two mitochondrial fusion factors have been identified as causal gene products of neurodegenerative disorders; mutations in Mfn2 lead to Charcot-Marie Tooth Neuropathy type 2a (97) and mutations in OPA1 lead to Dominant Optic Atrophy type I (1, 17), demonstrating that mitochondrial dynamics have important roles in vivo.
Mitochondrial Fusion in Mammalian Cells
Molecular basis of the mitochondrial fusion reaction
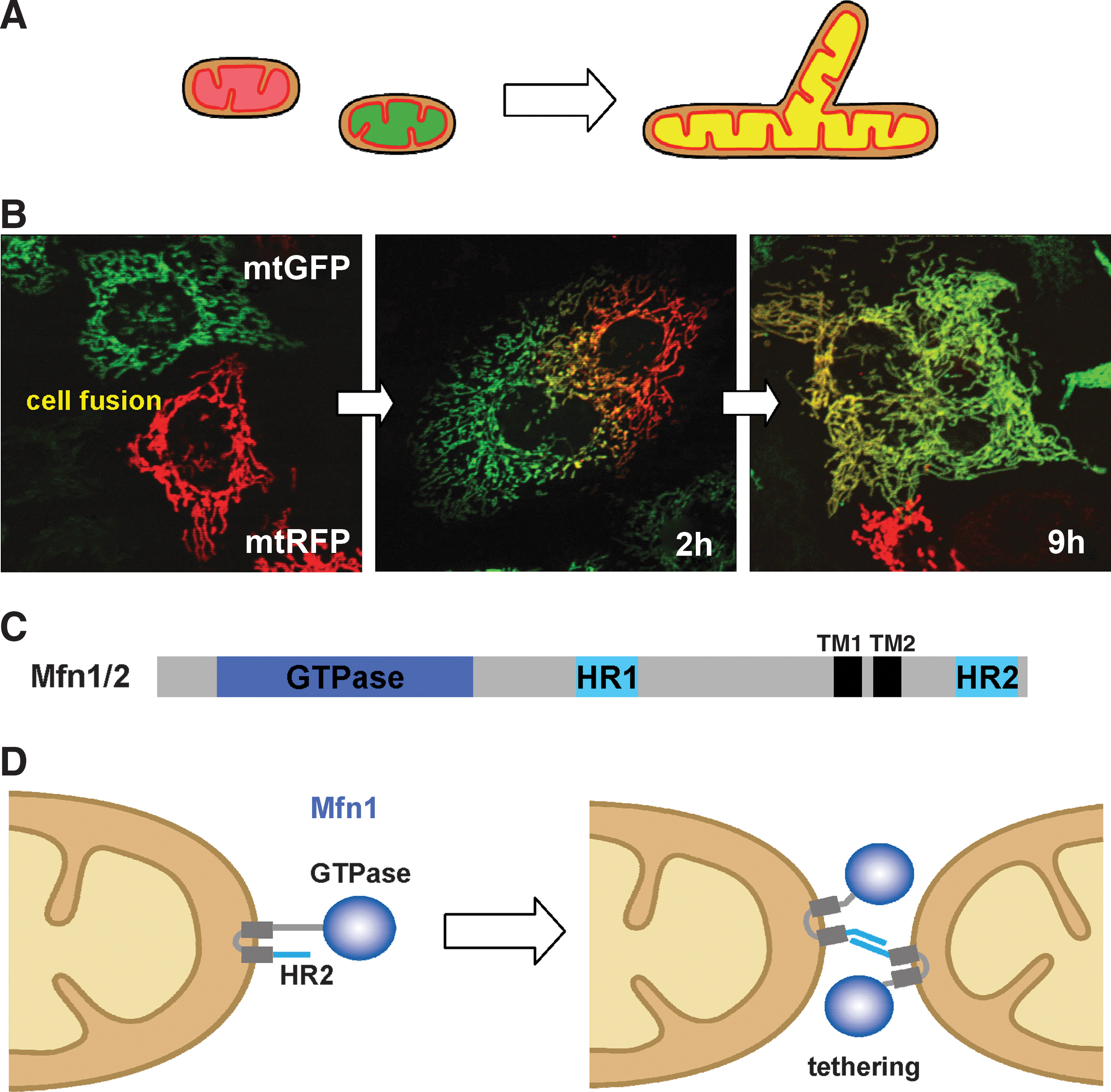
To analyze mitochondrial fusion in living mammalian cells, we and other groups have established assay systems (11, 35, 44) (Fig. 3). Mitochondria are separately labeled with different colored (green or red) fluorescent proteins that are targeted to the matrix (mtGFP and mtRFP, respectively), and the cells are fused using the Sendai virus (35) or polyethylene glycol (PEG) (11, 44). In the fused cells, two fluorescent reporters in the mitochondrial matrix (mt) are mixed in a time-dependent manner. Signals of GFP and RFP almost completely overlap after 9 h of culture in HeLa cells (Fig. 3B), indicating that both outer and inner membranes have fused to facilitate content mixing. The photoactivatable (PA) fluorescence proteins targeted to mitochondria (i.e., PA-mtGFP) are also useful for monitoring mitochondrial dynamics, such as by chasing a single mitochondrion in living cells and measuring the connectivity of mitochondria (38). These experiments revealed that mitochondrial fusion GTPases (Mfn1, Mfn2, and OPA1) and the active membrane potential are essential for mitochondrial fusion (11, 22). Analysis using Mfn1/2-double knockout (KO) and OPA1-KO mouse embryonic fibroblasts (MEFs) revealed that Mfn proteins and OPA1 mediate sequential steps in mitochondrial fusion, and interestingly, outer membrane fusion is only weakly coupled to fusion between the inner membranes, in marked contrast to the case in yeast (77).
Although mitochondria are moved along microtubules by motor proteins (80, 81), microtubules are not essential for mitochondrial membrane fusion (35). Notably, general and widely used methods for measuring mitochondrial fission have not yet been developed.
Two mitofusin isoforms in mitochondrial fusion
Mfn1 and Mfn2 are mammalian homologs of yeast Fzo1 and Drosophila Fzo (11, 22, 69). They contain the N-terminal GTPase domain and are anchored to the membrane by two C-terminal transmembrane segments (TM1 and TM2), extruding both the long N-terminal GTPase domain and a short C-terminus to the cytoplasm (Fig. 3C, D). They also contain two regions of hydrophobic heptad-repeat coiled-coil motifs, HR1 and HR2, localized upstream of TM1 and downstream of TM2, respectively. In mammals, Mfn1 and Mfn2 show differential tissue expression patterns, suggesting that they have isoform-specific functions (22). They form oligomers with homotypic (Mfn1-Mfn1 or Mfn2-Mfn2) or heterotypic (Mfn1-Mfn2) complexes. In RNA interference (RNAi) HeLa cells or in MEFs in which each Mfn isoform has been knocked out, mitochondrial fusion is severely compromised, suggesting that both Mfn isoforms are required for active mitochondrial fusion (11, 22).
In mitochondrial fusion, Mfn1 proteins on opposing mitochondria form transoligomeric complexes for tethering the outer membranes (33, 42). Structural analysis revealed that the C-terminal HR2 of Mfn1 forms homotypic dimers with an antiparallel coiled-coil structure during mitochondrial fusion, suggesting that HR2 functions as a mitochondrial tether before fusion (Fig. 3D) (42). Biochemical analysis revealed that formation of the transcomplex depends on GTP hydrolysis by Mfn1 (physical homo-oligomer in trans) (33). On the other hand, mitochondrial fusion assays in the PEG-fused cells revealed that mitochondria harboring active Mfn1, but not Mfn2, fuse with mitochondria harboring Mfn2 CMT2A disease mutations, indicating that the hetero-oligomeric complexes in trans are important for the fusion reaction (functional hetero-oligomer in trans) (18).
Recently, the mitochondrial fusion reaction was also analyzed using a cell-free assay system with isolated mitochondria (30, 50, 70). In this system, fluorescence overlap of differentially fluorescence-labeled mitochondria (mtGFP and mtRFP) (30, 50), or fluorescence complementation in the fused mitochondria harboring matrix-targeted split Venus luciferase fusion constructs (70) was observed with microscopy. These experiments revealed that mitochondrial fusion requires a membrane potential and GTP hydrolysis, and that the heterotypic Mfn1-Mfn2 transcomplex efficiently mediates mitochondrial fusion compared with homotypic Mfn1 or Mfn2 complexes (30). Interestingly, soluble nonapoptotic monomeric Bax localized in the cytoplasm, but not oligomerized proapoptotic Bax, stimulates mitochondrial fusion only by homotypic Mfn2 transcomplexes, suggesting that monomeric Bax is the active form for mitochondrial fusion and competes with the oligomerized form of Bax to attenuate apoptosis (30). Yeast cells express only Fzo1 for mitochondrial fusion, and there are no reports on mitochondrial Mfn/Fzo homologs in higher plants. Thus, the fundamental and diverse functions of Mfn proteins, such as how a pair of opposed outer membranes are fused physically after tethering by Mfn1, and the functional difference of Mfn proteins during the fusion reaction remain unknown. Atlastin is a homologue of Mfn proteins. It is anchored to the endoplasmic reticulum (ER) tubules in the similar membrane topology as Mfn proteins and involved in homotypic fusion of the ER tubules. Bian et al. performed crystal structure analysis of the cytosolic segment and demonstrated that upon GTP binding two apposed atlastin molecules dimerize and tether the membranes. GTP hydrolysis then causes conformational changes (rotates by 180°) that pull the membranes together, to induce membrane fusion (4). It is interesting to know whether the similar mechanism is involved in Mfn-dependent mitochondrial fusion.
Interestingly, the function of Drp1 as a fusion stimulator was recently proposed by Huang et al. (32). Intra- or intermolecular interaction of HR1 and HR2 (Fig. 3C) attenuates activity of Mfn2 in the closed conformation, and exogenously expressed Drp1 specifically interacts with HR1 to displace HR2 of Mfn2 (the open conformation), which allows HR2 to interact with HR2 of opposing mitochondria (32, 42), thereby facilitating mitochondrial tethering and fusion. Conversely, Mfn1/2-binding protein (MIB), a member of the medium-chain dehydrogenase/reductase protein superfamily, is essential for cellular function by regulating mitochondrial membrane dynamics as a negative regulator of Mfn, although the reaction mechanism remains to be analyzed (21).
Regulation of mitochondrial inner membrane fusion by OPA1
Treatment of cells with the protonophore carbonylcyanide m-chlorophenylhydrazone (CCCP) disrupts membrane potential to severely inhibit mitochondrial fusion, indicating that the membrane potential is required for fusion (35, 44) (Fig. 1B, left panel; Fig. 4A). OPA1 variants are involved in mitochondrial inner membrane fusion and cristae organization. They are synthesized in the cytoplasm as the preproteins (Fig. 4B), transported into the mitochondria, and anchored to the inner membrane by the N-terminal transmembrane domain (TM) as large molecular size (L-OPA1) and small molecular size (S-OPA1) species. When the cells are treated with CCCP, the large molecular size molecules (L-OPA1) are proteolytically cleaved and the small molecular size (S-OPA1) molecules are rapidly accumulated (19, 34) (Fig. 4C). Ectopic expression of various OPA1 proteins in OPA1-RNAi cells revealed that L-OPA1, but not S-OPA1, recovers mitochondrial network structures, suggesting that L-OPA1 stimulates mitochondrial fusion (34). Expression of both L-OPA1 and S-OPA1 is required for mitochondrial fusion in OPA1-KO MEFs (76). Prohibitins 1 and 2 (PHB1 and PHB2) are essential for cell proliferation and apoptosis resistance by regulating OPA1-dependent cristae morphogenesis (51). Deletion of PHB proteins leads to the selective loss of L-OPA1 in association with a strong compromise of the above phenotypes, and these defects are suppressed by the expression of an uncleavable form of L-OPA1 (51). In stress-exposed cells, mitochondria hyperfuse and form highly connected network stryctures: the response named stress-induced mitochondrial hyperfusion (SIMH) (83). SIMH requires L-OPA1, Mfn1, and the mitochondrial inner membrane protein SLP-2. In the absence of SLP-2, L-OPA1 is lost and SIMH is prevented (83). The mechanism by which PHB proteins and SLP-2 stabilize L-OPA1 in these reactions remains to be analyzed.

OPA1 processing is regulated by several mitochondrial inner membrane proteases: subunits of m-AAA protein complex (paraplegin, Afg3L1, and Afg3L2 in mice; paraplegin and Afg3L2 in humans), i-AAA protease (Yme1L), presenilin-associated rhomboid-like (PARL), and Oma1, and the responsibility of the proteases varies under various conditions (20, 27, 29, 34, 49, 76). In humans, OPA1 is synthesized as at least eight spliced variants, and the alternative splicing affects the processing susceptibility. The exon5b-encoded region provides processing site 2 (S2) and splice variants with this sequence are processed by i-AAA protease Yme1L (Fig. 4B) (27, 76). On the other hand, the exon 5-encoded sequence provides processing site 1 (S1) and variants harboring this sequence are processed by m-AAA proteases or Oma1 (Fig. 4B, C) (20, 29, 34). The m-AAA protease subunits have various functions in constitutive OPA1 processing: paraplegin stimulates processing, whereas Afg3L1 and Afg3L2 in mice negatively regulate the S1 processing. Oma1, a novel ATP-independent metalloprotease in the inner membrane with activities overlapping those of m-AAA protease, is responsible for the stress-induced processing of OPA1 (e.g., in the presence of CCCP) (20, 67), although mechanism of the stress-induced activation of OMA1 remains unsolved. In conclusion, proteolytic cleavage of OPA1 regulates mitochondrial fusion, and stressed mitochondria lose the ability to fuse by OPA1 processing (49).
Physiologic roles of mitochondrial fusion
Mutations in mitochondrial fusion factors Mfn2 and OPA1 result in neurodegenerative disorders, Charcot-Marie-Tooth Neuropathy 2a and Dominant Optic Atrophy I, respectively (1, 17, 97). Mitochondrial fusion factor KO mice are lethal before embryonic day 12.5 (for Mfn1 KO) or at embryonic day 11.5 (for Mfn2 KO), suggesting that both Mfn isoforms are essential for embryonic development in mammals (11). Tissue-specific KO mice using the Cre-loxP system revealed that KO of neuron-specific Mfn2 leads to neurodegeneration (12) and conditional depletion of both Mfn isoforms in skeletal muscle results in muscle atrophy (13). Mitochondria in these tissues and cells are defective in respiration due to the loss of mtDNA or accumulation of mtDNA mutations, suggesting that mitochondrial fusion has essential roles in the maintenance of active mitochondria (13). Distinct from Mfns' role in mitochondrial fusion, Misko et al. demonstrated by live imaging of neurons cultured from Mfn2 KO mice or neurons expressing Mfn2 disease mutations that Mfn2 directly interacts with the Miro/Milton complex and regulates axonal mitochondrial transport (53). During Ca2+ signaling, store-operated Ca2+ channels of the plasma membrane (PM) (CRAC channel) are activated by the depletion of the ER stromal Ca2+ that is sensed by the ER membrane protein, STIM1. Thus, the store depletion triggers translocation of STIM1 to the CRAC channel sites in the ER-PM junctions, leading to store-operated Ca2+ entry. Singaravelu et al. demonstrated that mitochondrial depolarization attenuates this process in a manner dependent on Mfn2; probably a homeostatic mechanism to prevent cellular Ca2+ overload (73).
Induced cardiac Mfn1/Mfn2 double KO mice had abnormal or degenerated mitochondrial cristae and cardiomyocyte respiratory dysfunction, and rapidly progressive dilated cardiomyopathy, indicating that mitochondrial fusion is essential for organelle function and cardiac homeosotasis (14). Zorzano and collaborators recently demonstrated with liver-specific KO of Mfn2 in mice that Mfn2 coordinates mitochondria and endoplasmic reticulum with insulin signaling and is essential for normal glucose homeostasis (71). In this context, Ngoh et al. demonstrated that induction of ER stress upregulates Mfn2 mRNA and protein levels specifically, and conversely, loss of Mfn2 promotes ER stress, suggesting that Mfn2 is an ER stress regulator that is necessary for the homeostasis of the ER (58).
Mfn1 and Mfn2 also function as the positive and negative regulators of antiviral innate immune response, respectively, on the mitochondrial outer membrane in cooperation with mitochondrial antiviral signaling adaptor (43, 92). Whether they are involved in this reaction through their membrane fusion activity is not known.
Pancreatic beta cell-specific OPA1 KO mice revealed that blood glucose-stimulated insulin secretion and ATP production are compromised due to a defect in respiratory complex IV, suggesting that the function of OPA1 in maintenance of the respiratory chain is physiologically relevant to beta cells (94). The molecular mechanisms of how mitochondrial fusion contributes to the maintenance of mitochondrial respiratory activity remain to be analyzed. Interestingly, Scorrano and collaborators have recently demonstrated that OPA1-dependent mitochondrial inner membrane remodeling controls efficiency of steroidogenisis in steroidogenic cells, extending OPA1's function beyond hitherto known function to cholesterol translocation (89).
Mitochondrial Fission in Mammalian Cells
Regulation of mitochondrial fission by Drp1 modifications
Drp1 is a mammalian homolog of yeast Dnm1 that plays an essential role in mitochondrial fission (61, 74). It is a cytosolic protein with the N-terminal GTPase domain, a dynamin-like middle domain, and a C-terminal GTPase effector domain (GED) (Fig. 5B). During mitochondrial fission, Drp1 in the cytoplasm as small oligomers assembles into larger oligomeric structures on the mitochondrial fission sites depending on GTP binding, and then they sever the mitochondrial membrane depending on GTP hydrolysis. The middle domain and GED domain are required for self-assembly into higher order oligomers and for intra- or intermolecular interactions, respectively. Mitochondrial translocation of Drp1 is regulated by various post-translational modifications (9). Here, we concentrate mainly on the regulation of Drp1 phosphorylation reactions because the physiologic relevance of these modifications is becoming increasingly clear.

During cell cycle progression, filamentous mitochondria in interphase are converted to a short and fragmented population during mitosis, and the filamentous mitochondrial structures are recovered after being passed to the daughter cells (78). In the early mitotic phase, Ser616 in human Drp1 (Ser585 in rat Drp1) is specifically phosphorylated by maturation promoting factor (Cdk1/cyclinB complex), which promotes mitochondrial fission. Of note, this mitotic Drp1 phosphorylation and subsequent mitochondrial targeting were recently reported to be regulated by Aurora kinase, a small GTPase RALA and its effecter RALBP1 (40). Under oxidative stress conditions, protein kinase Cδ mediates phosphorylation of Ser579 in human Drp1 isoform 3 (corresponding to Ser616 in the human Drp1 isoform 1), leading to mitochondrial fragmentation and impaired mitochondrial function, which contributes to hypertension-induced brain injury (66).
Importantly, phosphorylation at Ser637 in the GED domain of human Drp1 by cAMP-dependent protein kinase A (PKA) inhibits Drp1 GTPase activity, and this modification releases Drp1 from mitochondria by the inhibition of oligomeric assembly on the membrane, promoting mitochondrial network extension and eventually cell viability (8). This reaction is reversed by calcineurin-mediated dephosphorylation (6). Thus, the PKA-calcineurin system regulates mitochondrial morphology and cell viability by controlling the translocation of Drp1 to the mitochondria. In this relation, Strack and collaborators recently reported that the A kinase anchoring protein 1 (AKAP1) localized on the mitochondrial outer membrane is involved in this reaction; the PKA/AKAP1 complex on the mitochondrial outer membrane thus plays an important role in maintaining mitochondrial integrity and securing neuronal survival (52). In this report, however, phosphorylation of Drp1 by PKA/AKAP1 traps Drp1 in large, slowly recycling complex on the mitochondria, thereby allowing mitochondrial network extension. The mechanistic details by which PKA-phosphorylated Drp1 induces mitochondrial network extension remain to be clarified. In contrast, phosphorylation of the PKA site (Ser637) of Drp1 by Ca2+/calmodulin-dependent protein kinase Iα is reported in primary cultured rat hippocampal neurons, which promotes the binding of Drp1 to mitochondrial Fis1 and stimulates mitochondrial fission (28). A remaining enigma is why phosphorylation at the same site of Drp1 has the opposite effect. In a cellular model of Huntington's disease (HD) expressing huntingtins (Htt) with a longer polyglutamine repeat, the increased dephosphorylation of Drp1 by hyperactivation of calcineurin induces mitochondrial fission and cristae disruption, which leads to an increased response to apoptotic stimuli (16).
Under nutrient starvation, mitochondrial fission is repressed in response to PKA-dependent Drp1 phosphorylation due to increased cAMP levels, leading to the formation of elongated mitochondria with the capacity for efficient ATP production. This response protects mitochondria from autophagosomal degradation and sustains cell viability (25, 68). Modification by the small ubiquitin-like modifier (SUMO) by Sumo E3 ligase MAPL stabilizes Drp1 on the mitochondrial membrane in a Bax/Bak-dependent manner to stimulate mitochondrial fission, apoptisis suggesting that sumoylation is involved in the regulation of Drp1 early during progression (88), whereas this process is reversed by SUMO1 protease SenP5 (98). Other Drp1 modifications, such as ubiquitination by E3 ligase MARCH-V/MITOL, and S-nitrosylation may also contribute to the regulation of mitochondrial fission (9), although the physiologic relevance remains elusive. Furthermore, cytoplasmic endophilin B1 (39), outer membrane GDAP1 (59), and inner membrane protein MTP18 (82) are involved in Drp1-dependent mitochondrial fission, although the underlying mechanisms are not known.
Mitochondrial recruitment of Drp1
Drp1 is recruited from the cytoplasm to the mitochondrial fission sites as punctate structures, coils up mitochondria, and eventually severs them. This process is thought to be regulated by GTP binding and hydrolysis. In yeast, a C-tail anchored mitochondrial outer membrane protein Fis1 has essential roles in Drp1 recruitment in conjunction with the adaptor proteins, Mdv1 and Caf4 (2). Fis1 is evolutionally conserved, but dispensable for Drp1 recruitment in mammals (64). Instead, newly identified factors on the mitochondrial outer membrane are involved in Drp1 recruitment (Fig. 5A). A C-tail anchored outer membrane protein, mitochondrial fission factor (Mff ), was identified from genome-wide RNAi screening for mitochondrial morphology maintenance in Drosophila-cultured cell lines (23). Mff overexpression induces recruitment of Drp1 onto mitochondria and mitochondrial fission, and conversely, its knockdown leads to mitochondrial elongation concomitant with the release of Drp1 from the mitochondria, suggesting that Mff is an essential factor for Drp1 recruitment to stimulate mitochondrial fission in mammals (63). Mitochondrial dynamics 51 (MiD51), also named mitochondrial elongation factor 1 (MIEF1), and the variant MiD49 are N-terminal-anchored mitochondrial outer membrane proteins that function in Drp1 recruitment (65, 95). Overexpression of MiD/MIEF proteins leads to mitochondrial elongation and Drp1 recruitment to mitochondria. Opposite knockdown phenotypes, however, are also reported; one group found that MiD knockdown stimulates mitochondrial tubular extension (65), whereas another group claimed that MIEF1 knockdown stimulates mitochondrial fission (95). The latter group concluded that MIEF1 recruits Drp1 and inhibits GTPase-dependent fission activity of Drp1, but instead it has fusion stimulation activity; thus, MIEF1 is a factor that promotes fusion rather than fission (95). Additional work is required to solve these apparent conflicts. Detailed mechanisms of the Mff and MiD/MIEF1 proteins and their relation in Drp1-dependent mitochondrial fission remain important issues to be addressed in the future. The function of Fis1 in mammalian mitochondrial morphology regulation remains enigmatic and must also be addressed, although its function collaborated with Bap31 in the mitochondria–ER interface as a platform of apoptosis induction is known (37). Of note, Drp1, Fis1, and Mff are also reported to be involved in the peroxisome fission reaction in mammalian cells, although their mechanism of action is mostly unexplored (23, 41). Whether MiD/MIEF proteins are localized to peroxisomes is also not known.
Physiologic roles of Drp1-dependent mitochondrial fission
Mitochondria are thought to derive from endosymbiotic bacteria, and to proliferate by growth and fission of pre-existing mitochondria. In Drp1-RNAi cells, the long mitochondrial structures are maintained during mitosis. Nevertheless, they divide and grow normally, and their mitochondria are passed to daughter cells (78). In an immortalized Drp1 KO fibroblast cell line that we established (Drp1 KO MEFs), the growth rate, respiratory activities, cellular ATP levels, and mtDNA levels are comparable with those of control MEFs, indicating that Drp1 is dispensable for cell viability and maintenance of active mitochondria in MEFs (36). In contrast, another group reported that primary Drp1-KO MEFs are viable, but grow slower than control cells, even though cellular ATP levels are normal (85). In Drp1-deficient cells, mitochondria are unequally segregated to daughter cells by forced fission at the midbody (the cell fission site) in concert with cytokinesis, suggesting that mitochondrial fission is not essential for mitosis, but facilitates the stochastic distribution of mitochondria to daughter cells (Fig. 6, upper row mitosis). Lippincott-Schwartz and collaborators demonstrated that mitochondria exhibit stage-specific cell cycle phenotypes; fragmented mitochondria convert into a hyperfused giant network at G1-S transition. Induced mitochondrial hyperfusion by a chemical Drp1 inhibitor, mdivi-1, in the G1-S-arrested cells increases cyclin E levels to release the arrest to enter into S phase, suggesting that the mitochondrial fusion–fission balance plays important roles in cell cycle regulation (55).

Compared with mitochondrial fusion, the in vivo function of Drp1-dependent mitochondrial fission in concert with other protein factors is still poorly understood. A heterozygous, dominant-negative mutation of the Drp1 gene (A395D in the middle domain) was identified in a newborn girl with severe pleiotropic defects, including abnormal brain development and optic atrophy, who died at the age of 37 days (90). The mutation dominantly affected the mitochondrial and peroxisomal morphologies. To elucidate the detailed physiologic roles of mitochondrial fission in vivo, we and another group generated tissue-specific Drp1 KO mice using the Cre and loxP system (36, 85) (Fig. 6). Although Drp1 is dispensable for viability of MEF cells (Fig. 6, upper row mitosis) as described above, Drp1 KO mice die at around embryonic day 12.5 with developmental abnormalities particularly in the forebrain (Fig. 6, lower row embryonic development). On the other hand, Drp1 heterozygote KO mice have no synaptic and mitochondrial deficiencies, except that H2O2 and lipid peroxidation levels were significantly reduced compared with the wild-type mice (47). Neuron-specific Drp1 KO mice are born, but immediately die due to neurodegeneration. In primary cultured neural cells from Drp1 KO embryos, enlarged mitochondrial clumps are sparsely distributed in the neurites, and the synaptic structures are lost (Fig. 6, middle row neuron). These findings suggest that Drp1 deficiency causes the abnormal distribution of enlarged mitochondria in extremely polarized cells, such as neurites; these spatiotemporal defects may inhibit the ATP supply and Ca2+ signaling, eventually preventing the formation of synapses. A similar situation is observed in T-cell differentiation. Drp1 deficiency affects the delivery of mitochondria to the immune synapse (3). In conclusion, Mff-Drp1 is essential for embryonic development and synapse formation in mice. In addition, a missense mutation in mouse Drp1 in the middle domain essential for intramolecular interactions (Python mice; C452F mutation) leads to cardiomyopathy (2). Determining the physiologic relevance of Drp1 in other tissues that might underlie various human diseases remains a future challenge.
It is recently reported that Drp1-dependent mitochondrial fission initiates follicle cell differentiation during Drosophila oogenesis (54), consistent with the notion that Drp1-dependent mitochondrial fission is essential for embryonic development in mice (36, 85).
Mitochondrial Dynamics and Quality Control in Neurodegeneration
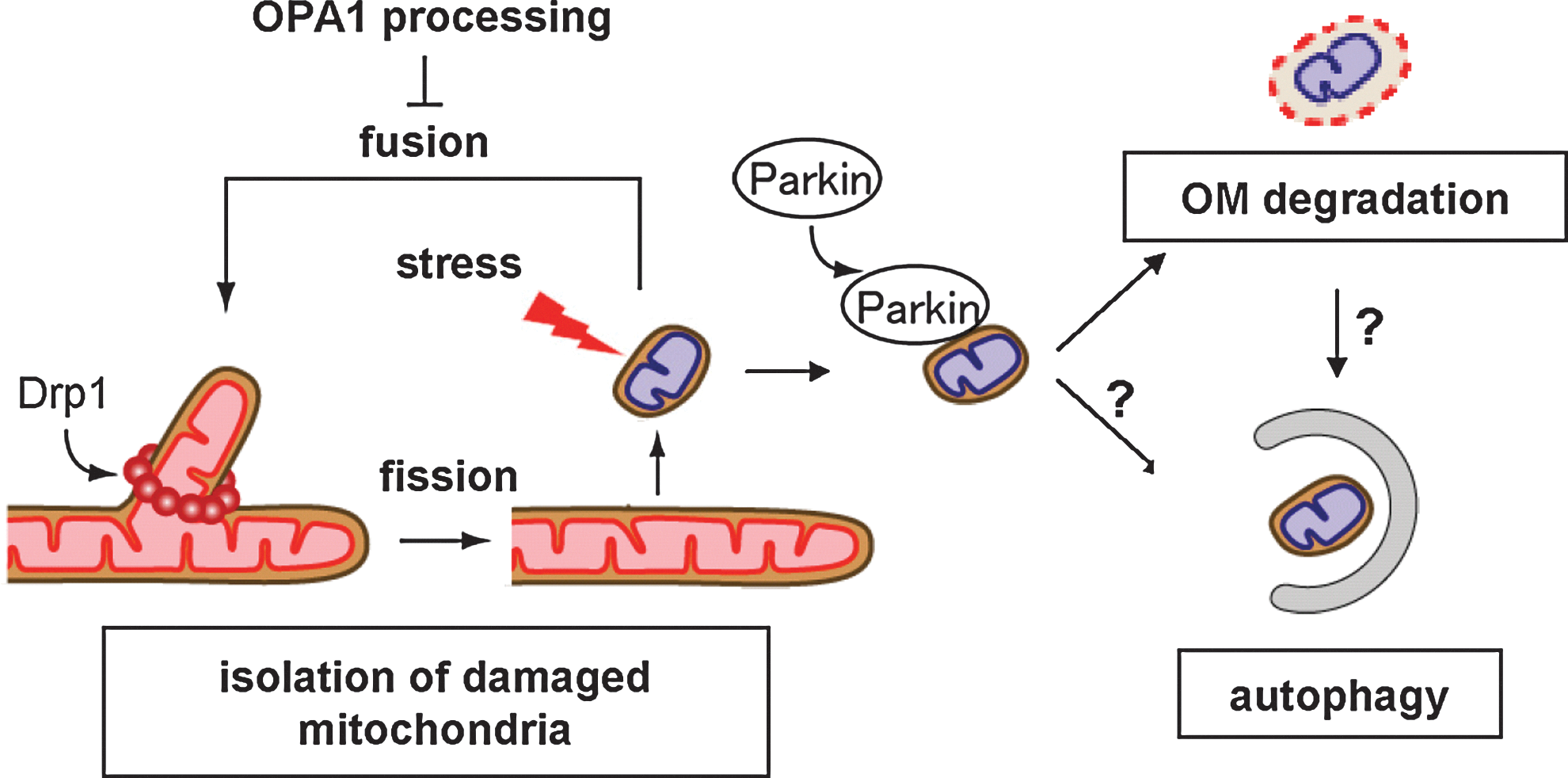
Defective regulation of mitochondrial dynamics is related to neurodegenerative disorders, such as Parkinson's disease (PD), Alzheimer's disease (AD), and HD (10, 46, 57, 75), which appear to be consequences of dysfunction in quality control of the damaged mitochondria via selective degradation (10, 26, 56, 84) (Fig. 7).
The interaction between Drp1 and amyloid beta (Aβ) in the progress of AD was reported as the disease progressed, leading to abnormal mitochondrial dynamics and synaptic damage (10). Nitric oxide is thought to be a key mediator of AD. Recent report demonstrated that Aβ-enhanced nitric oxide production triggers mitochondrial fission and neuronal damage through the C644 nitrosylation-induced activation of Drp1 GTPase activity (15), although the proposed process was challenged (5). Mutant Huntingtin (Htt) (abnormal poly Q expansion in Htt) directly interacts with Drp1 and stimulates its GTPase activity to trigger mitochondrial fragmentation both in vivo and in vitro, thereby impairing the mitochondrial fusion–fission balance and causing neuronal cell death (75). Thus, mutant Htt activates Drp1 directly or indirectly by PKA-dependent phosphorylation [discussed above (16)]. ARSACS is a childhood-onset, autosomal recessive spastic ataxia resulting from mutations in the SACS gene encoding a 4579-aa multidomain protein sacsin, It localizes to mitochondria partially overlapping with Drp1 foci. Exogenously expressed sacsin (1–1368 segment) interacts with endogenous Drp1. Interestingly, mitochondria in sacsin KO mouse cells and ARSACS patients cells display hyperfused balloon-like mitochondria; appears to phenocopy the effect of Drp1 knockdown. Furthermore, sacsin KO mice exhibit age-dependent neurodegeneration of cerebellar Purkinje cells probably due to disturbance of mitochondrial delivery within neurites. These results suggest that sacsin might be required for the regulation of Drp1 activity (24). The leucin-rich repeat kinase 2 (LRRK2) is a large multidomain protein kinase (2527-aa residues) and its mutations are linked with autosomal dominant PD. It is reported that LRRK2 colocalizes with mitochondria and stimulates mitochondrial recruitment of Drp1 by direct interaction and induces mitochondrial fragmentation, mitochondrial dysfunction, and cell vulnerability to stress probably through its kinase activity, and the effects are further exacerbated by PD-associated LRRK2 mutants (60, 87). The mechanism by which LRRK2 kinase induces mitochondrial recruitment of Drp1 to induce mitochondrial fission remains unknown.
In the frequent mitochondrial fusion–fission cycles, mitochondrial fission forms unequal daughter mitochondria into membrane potential-preserved and membrane potential-lowered mitochondria, and the latter fractions are rapidly eliminated by autophagy (termed mitophagy) to maintain overall mitochondrial quality; CCCP-stimulated OPA1 processing from L- to S-isoforms is involved in this segregation (84). Recent studies revealed that the PD-related protein Parkin, a RING domain-containing E3 ubiquitin ligase, is specifically targeted to depolarized mitochondria from the cytoplasm and promotes mitophagy when Parkin-expressing cells are treated with CCCP (57). The PD-related protein kinase PTEN-induced putative kinase (Pink1) stabilized on the outer membrane of depolarized mitochondria is involved in the recruitment of Parkin (57). Mfn proteins are then ubiqutinated by Parkin and degraded by proteasomes; supposedly the response leading to opposed mitochondrial fission to facilitate mitophagy (79, 86, 96). Later, Parkin was revealed to stimulate degradation of many outer membrane proteins by the ubiquitin–proteasome system as a critical step for mitophagy (7, 93) or as a step leading to outer membrane rupture independently of autophagy (93). Further analysis of the pathologic roles of Parkin and mitochondrial quality control is necessary especially in neuronal cells in vivo.
Perspectives
Although a significant amount of relevant data have accumulated on mammalian mitochondrial fusion and fission reactions and several key components modulating these dynamics have been identified, the coordination of these and other as yet unidentified components, and the sequence of events leading to membrane fusion and fission at the molecular level remain to be urgently elucidated. It also remains a challenge for the future to discover the physiologic roles of mitochondrial fusion and fission machines in highly differentiated tissues using tissue-specific KO mice.
Footnotes
Acknowledgments
This work was supported in part by a Grant-in-Aid for Young Scientists (A) (to N.I.), Grant-in-Aid for Scientific Research (B) (to K.M.) [Japan Society for the Promotion of Science], Grant-in-Aid for Scientific Research on Priority Areas (to N.I., H.O., T.O, and K.M.), Grant-in-Aid for Special Promoted Research (to K.M.) [The Ministry of Education, Culture, Sports, Science, and Technology, Japan], Funding Program for Next Generation World-Leading Researchers (to N.I.), and the Kato Memorial Bioscience Foundation (to N.I.).