
Abstract
Introduction

PAH is a multifactorial process, but early disease is related to a dysregulation of critical vasodilator pathways (downregulation of nitric oxide [NO] and prostaglandin signaling) and vasoconstrictor pathways (upregulation of endothelin-1 and reactive oxygen species [ROS] signaling) (75). PAH is associated with decreased bioavailability and responsiveness of NO (14, 32). NO is produced in mammalian cells primarily in the metabolism of

Inhaled NO (iNO) is considered as a potential therapy targeting the NO pathway (33, 39), and a potent and selective pulmonary vasodilator (65). iNO therapy has been proposed for treatment of PAH, but also for persistent PH of the newborn (15), and bronchopulmonary dysplasia in prematurely born infants (21) and post-cardiac surgery (3). Other possible applications of iNO therapy include the treatment of pulmonary ischemia-reperfusion injury (9), the acute respiratory distress syndrome (18), and hypoxemia in the setting of severe chronic obstructive pulmonary disease (26). However, iNO gas has its limitations regarding dose and duration of the exposure, cumbersome and expensive delivery systems, off-target reactions of NO with oxygen to form reactive nitrogen species, and possible rebound PH when the intervention is interrupted (38). These limitations open the door to other molecules that can be a source of NO and nitrosative signaling, including the anion nitrite (NO2 −) (55). Nitrite is a unique reactive nitrogen species, as it is relatively stable compared to NO [51.4 min (67) vs. 0.05–1.8 ms (68) half-life in whole blood] and can readily be reduced to NO, oxidized to nitrogen dioxide, and protonated to form S-nitrosothiol, providing NO-dependent and NO-independent signaling effects that have been reported to modulate vascular remodeling (2, 86).
Rediscovering Nitrite in Biology
Although in the ancient Chinese medicine, the sodium salts of nitrate and nitrite were used as a remedy for heart pains and ischemia (circa 800 AD) (54), only until recently, nitrate (NO3 −) and nitrite (NO2 −) were mostly considered undesired residues in the food chain with potential carcinogenic activity. Nitrate salts have also been used by early civilizations to cure meats, which not only improve their storage time and produce their reddish color but also kill pathogenic bacteria (69). The mechanism of the meat preservation by nitrate was characterized in the 19th century, when it was discovered to be converted to NO (bound to myoglobin). During the 1970s, public concern over toxicity increased, when nitrate and nitrite were associated to the endogenous formation of N-nitrosamines, which are carcinogenic. While remaining extremely controversial, the epidemiological links between nitrate/nitrite exposure, for example, in high-nitrate foods such as leafy green vegetables, remain unclear (25, 79).
The first relationship between nitrite formation and NO production was found by studying immune responses in activated macrophages. Analyses of bacterial-infected (74) or tumor-bearing (36) mice showed that macrophages use
More recently, increasing evidence suggests that nitrite not only is a biomarker of NO formation from NO synthesis but also represents a storage reservoir for NO that can be converted back to NO during physiological and pathological hypoxia (8, 16, 56). It is now known that nitrate and nitrite can be recycled to NO (or other bioactive forms of nitrogen) in blood and peripheral tissue (27, 55), representing an alternative to the classical
The Nitrate-Nitrite-NO Signaling Pathway
The reductive mammalian nitrate-nitrite-NO pathway mediates important signaling events and complements the traditional oxidative
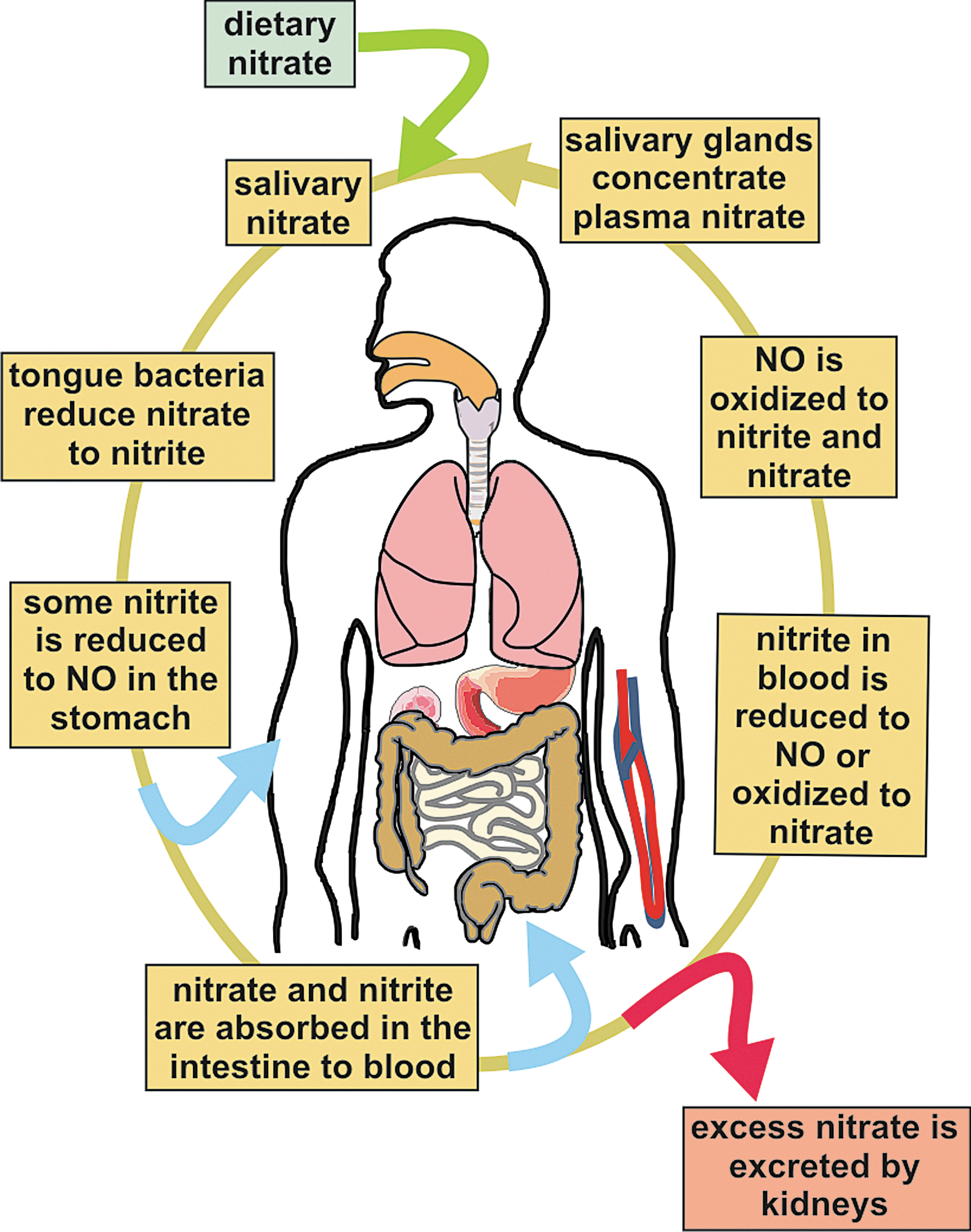
Mechanisms of Nitrite Bioactivation
Nitrite can be reduced to NO in vivo by both enzymatic and nonenzymatic processes (Fig. 4). Biological non-enzymatic NO formation was first reported by Zweier et al. (87) in an ischemic rat tissue where, at low pH, there was NOS-independent NO production. Benjamin et al. (8) showed that in the highly acidic environment (pH 3) of the stomach, nitrite is converted to NO gas. Nitrite production of NO at acidic pH is also enhanced by the presence of reducing compounds such as copper, ascorbate, and polyphenols (82) [Table 1, Eqs. (1)–(3)].

Enzymatic NO formation from nitrite, via facilitated proton and electron transfer reactions, has been reported for a wide variety of metal-containing proteins, including hemoglobin (22), myoglobin (70), neuroglobin (77), cytochrome c (4), cytochrome c oxidase (12), eNOS (78), xanthine oxidoreductase (XOR) (84), aldehyde oxidase (AO) (50), and carbonic anhydrase (1).
Heme-containing proteins
Deoxyhemoglobin
Deoxyhemoglobin (deoxy-Hb) can catalyze nitrite reduction accompanied with NO generation and production of ferric heme (Fe+3). The formed NO can then bind to another deoxyheme to form iron-nitrosyl (Fe+2-NO) (Table 1) (22). As for other heme proteins, the reaction is faster at low pH, indicating the involvement of the protonated nitrite (nitrous acid—HNO2) [Table 1, Eq. (6)]. The reaction is allosterically regulated, being slow in the T-state, but much faster for the R-state deoxy-Hb (37). There is a balance between the faster reaction of nitrite with the R-state oxyhemoglobin and the availability of deoxyhemes to bind to nitrite, leading to a maximal rate of nitrite reduction at about 50% hemoglobin–oxygen saturation. These factors have suggested that the reaction can respond to change in pH and oxygen tension in blood and tissues, and potentially mediate hypoxic vasodilation (28, 29, 37).
Deoxymyoglobin
It reduces nitrite to NO at a faster rate than deoxy-Hb, and has been shown to modulate mitochondrial respiration via inhibition of cytochrome c oxidase (70, 72) (Fig. 5). Confirmatory studies in myoglobin knockout mice suggest a role for myoglobin in regulating respiration and cardiac energetics, and new studies suggest a contribution to hypoxic vasodilation (35). As indicated for hemoglobin, the reaction rates increase with low pH, which is usually linked to hypoxia. Therefore, myoglobin can use nitrite efficiently to produce NO during hypoxic or ischemic conditions (72).

Cytochrome c
It is located at the intermembrane space of mitochondria, where it shuttles electrons from complex III of the mitochondrial respiratory chain to cytochrome c oxidase (complex IV). Under hypoxic conditions and those in which the cytochrome c heme becomes pentacoordinate, cytochrome c can bind nitrite and reduce nitrite to NO (4). The reduction of nitrite to NO by cytochrome c is vastly increased in the five-coordinate state, which is promoted by cardiolipin liposomes (6). This reductive reaction occurs between ferrous cytochrome c (Fe2+) and nitrite and represents a typical electron–proton transfer reaction [Table 1, Eq. (6)] (4).
Cytochrome c oxidase (complex IV)
Nitrite can react with cytochrome c oxidase to form NO (12). This reductive reaction occurs between ferrous cytochrome c oxidase (Fe2+) and nitrite and represents a typical electron–proton transfer reaction [Table 1, Eq. (6)]. Note that nitrite can also be reduced by other proteins, such as myoglobin, to form NO, which in turn can bind to ferrous cytochrome c oxidase and inhibit respiration. The formation of NO and inhibition of cytochrome c oxidase have been proposed as a pathway to reduce oxygen consumption at low oxygen levels, thus promoting oxygen diffusion deeper into ischemic tissues (35, 70, 71). In summary, cytochrome c oxidase has been proposed to function as both a nitrite reductase that generates NO, as well as a target for nitrite-NO-dependent regulation of respiration and oxygen consumption.
Neuroglobin
Evolved from a common ancestor shared by hemoglobin and myoglobin, human neuroglobin is found in brain neurons and also possess nitrite reductase activity. Neuroglobin, like cytochrome c, is a six-coordinate heme protein, with two histidines bound to the heme iron. The binding and reduction of nitrite to NO are favored when the heme is five coordinated. Tiso et al. (77) reported that deoxyneuroglobins where the sixth ligand (Histidine 64) is mutated to a nonheme-binding side chain (H64L and H64Q) can reduce nitrite at a very high rate. The wild-type neuroglobin six-to-five coordination is also regulated in a redox-dependent fashion, with oxidation of two surface redox-sensitive thiols to disulfides increasing the open probability of the heme and increasing the rate of nitrite binding and reduction to NO. Tiso et al. showed in these studies that the nitrite reductase reaction of neuroglobin could modulate mitochondrial respiration, similar to that shown for myoglobin (Table 2) (77). This pathway suggests a possible redox sensor mechanism for neuroglobin-mediated nitrite reduction. (Fig. 6)

Human, at 37°C, pH 7.4 (37).
Sperm whale, at 25°C, pH 7.4 (77).
Human, at 25°C, pH 7.4 (77).
Nitric oxide synthase
While though NOS is responsible for normoxic NO generation from arginine oxidation, it has been recently reported that NOS also catalyzes nitrite reduction to NO under anoxic conditions. Vanin et al. reported that eNOS catalyzes anoxic NO formation in murine microvascular brain endothelial cells, which is abolished by the eNOS inhibitors Nω-nitro-
Molybdopterin-containing proteins
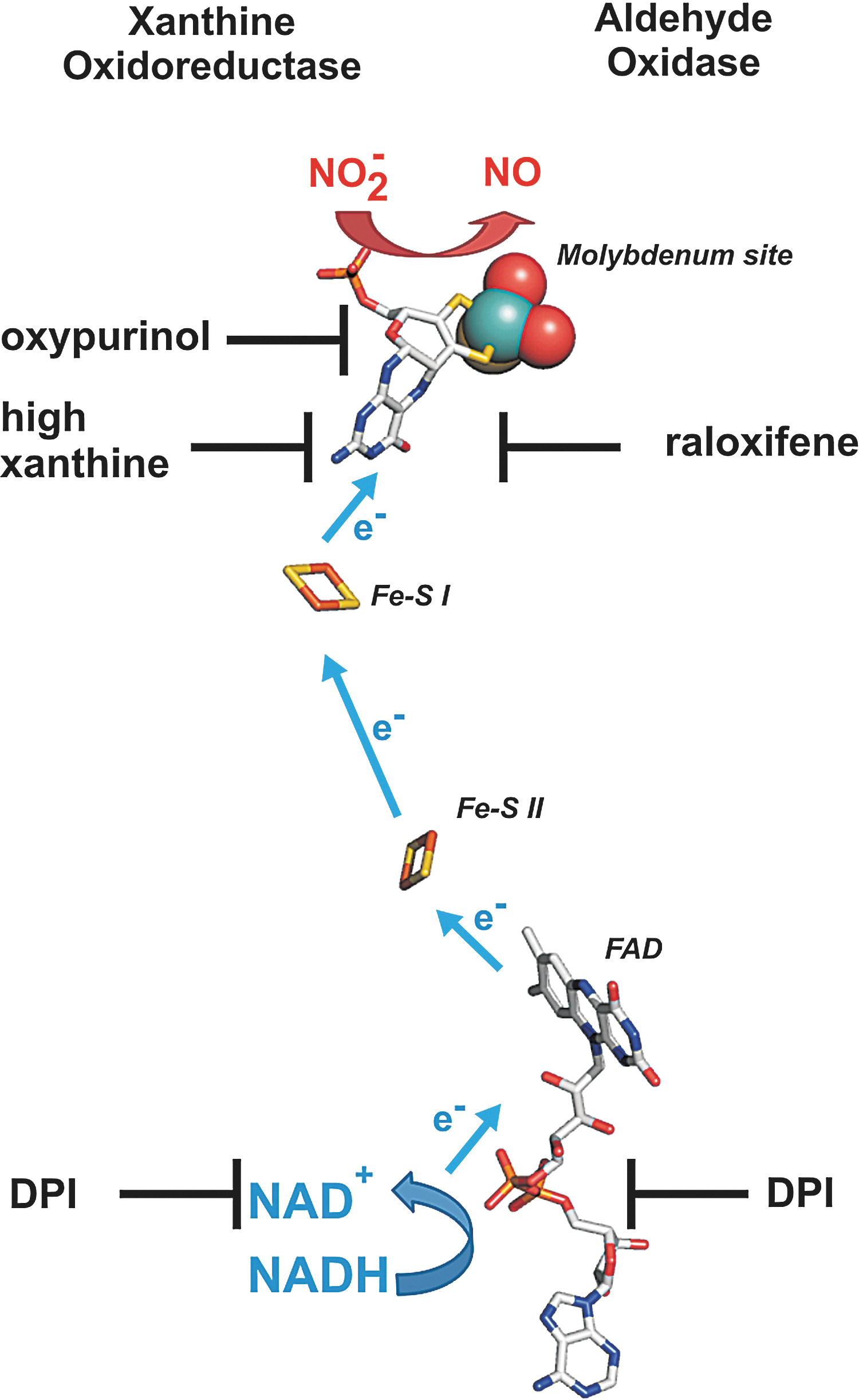
XOR and AO are two dimeric molybdopterin enzymes ubiquitously expressed in human tissues, containing one molybdenum (Mo) center, two nonidentical Fe2S2 clusters, and a flavin adenine dinucleotide cofactor. XOR catalyzes the terminal two steps in purine degradation and exists in cells primarily as a dehydrogenase (xanthine dehydrogenase [XDH]) where substrate-derived electrons reduce NAD+ to NADH. During inflammatory conditions, oxidation of critical cysteine residues or limited proteolysis converts XDH to xanthine oxidase (XO) (80). XO transfers substrate-derived electrons to O2, generating O2 •− and H2O2. However, conversion to XO is not a requisite for ROS production, as XDH displays partial oxidase activity under conditions in which the NADH/NAD+ ratio is increased such as the hypoxemia (34). It is also under hypoxic (0–1% O2) conditions that both XO- and XDH-mediated nitrite reductase activities have been reported (47, 51). This is evidenced by inhibition of XO- and XDH-mediated reduction of nitrite to NO by the XOR-specific inhibitor, oxypurinol, and/or high concentrations of xanthine. This oxypurinol- and xanthine-induced inhibition of NO generation rates results from their binding to the Mo site, which is also the catalytic center for nitrite binding and reduction. A similar effect on nitrite reduction was observed for AO, which is inhibited by raloxifene. As the inhibitor for the flavin site, diphenylene iodonium chloride does not affect nitrite reduction when xanthine or aldehyde served as the electron donor, but considerably reduces rates of NO formation rates when NADH is the reducing substrate. These experiments indicate that nitrite is reduced at the Mo center of XO or AO, but electrons can be provided either at the Mo or the flavin site by various substrates (Fig. 7). It also suggests that electron withdrawal from XOR or AO by nitrite may serve not only to produce NO but also to reduce ROS formation and as such elicit salutary actions regarding NO derived from alternative sources.
Nitrite disproportionation
Another mechanism for nitrite bioactivation is disproportionation, with nitrous acid or NO+ reacting with nitrite to form N2O3 [Table 1, Eqs. (2) and (3)]. This reaction has been proposed to be catalyzed by ferric hemoglobin (5) as well as carbonic anhydrase. Aamand et al. (1) recently demonstrated that NO was generated by nitrite reactions with carbonic anhydrase under both normoxia and hypoxia, which was stimulated by dorzolamide and acetazolamide, two specific inhibitors of carbonic anhydrase (Table 3). They propose that nitrite is structurally similar to bicarbonate and may react with histidines in the enzyme catalytic site to disproportionate to NO. However, more work is needed to fully explore this newly described pathway.
Reducing agent: TMPD=N,N,N′,N′-tetramethyl-p-phenylene diamine; pH 7.0 (12).
Reducing agent: NADH=Nicotinamide adenine dinucleotide; pH 7.4 (52).
Reducing agent: NADH=Nicotinamide adenine dinucleotide; pH 7.4 (50).
As reported in Ref. (1).
Mo, molybdenum; NO, nitric oxide.
Therapeutic Application of Nitrite for PH
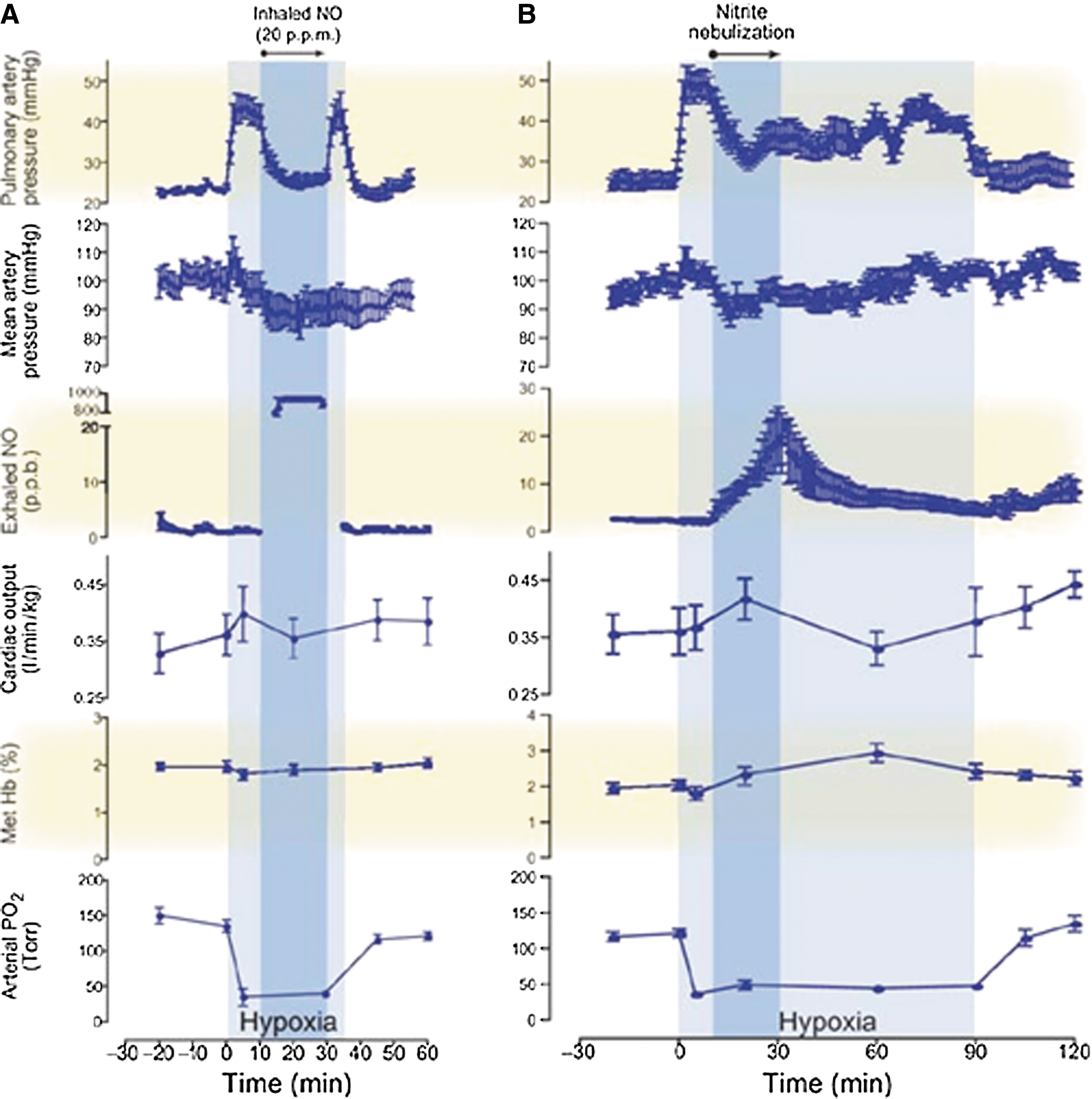
Recent studies have investigated the therapeutic effects of nitrite in PH models (85). Hunter et al. (38) first reported the effects of nebulized nitrite as a selective pulmonary vasodilator in an ovine hypoxia model. In this study, vasoconstriction induced by hypoxia was reduced by inhalation of NO gas (20 ppm) or nebulized nitrite (300 mg in 5 ml of phosphate-buffered saline for 20 min). Nebulized nitrite significantly reduced the pulmonary arterial pressures with lesser effects on systemic mean arterial blood pressure. The kinetic therapeutic responses to nitrite and NO were very different: The nitrite response was slower and lasted longer (over 60 min after inhalation), whereas the effect of NO response was faster, but returns rapidly to the hypoxic baseline when the treatment was discontinued (Fig. 8). The nitrite effect appears to be mediated by its slow conversion to NO, since measurements of exhaled NO gas increased, with values above baseline maintained after the end of the treatment. In addition, consistent with a possible role of deoxyhemoglobin as the nitrite reductase protein, formation of iron-nitrosyl-hemoglobin was also reported (38). More recently, using an ovine model of hemolysis-induced pulmonary vasoconstriction, the effect of nebulized nitrite (0.87 M), iNO (20 ppm), and intravenous sodium nitrite was compared. Both iNO and inhaled nitrite were able to reduce PH, whereas intravenous nitrite was ineffective at the concentrations studied. Nebulized sodium nitrite formulations were also studied in a rabbit model by Egemnazarov et al. (24). In this study, nitrite solutions acidified with ascorbic and citric acid (pH 5.3–5.4) showed a longer-lasting vasodilatory effect, although all formulations were effective in reducing hypoxic pulmonary vasoconstriction.
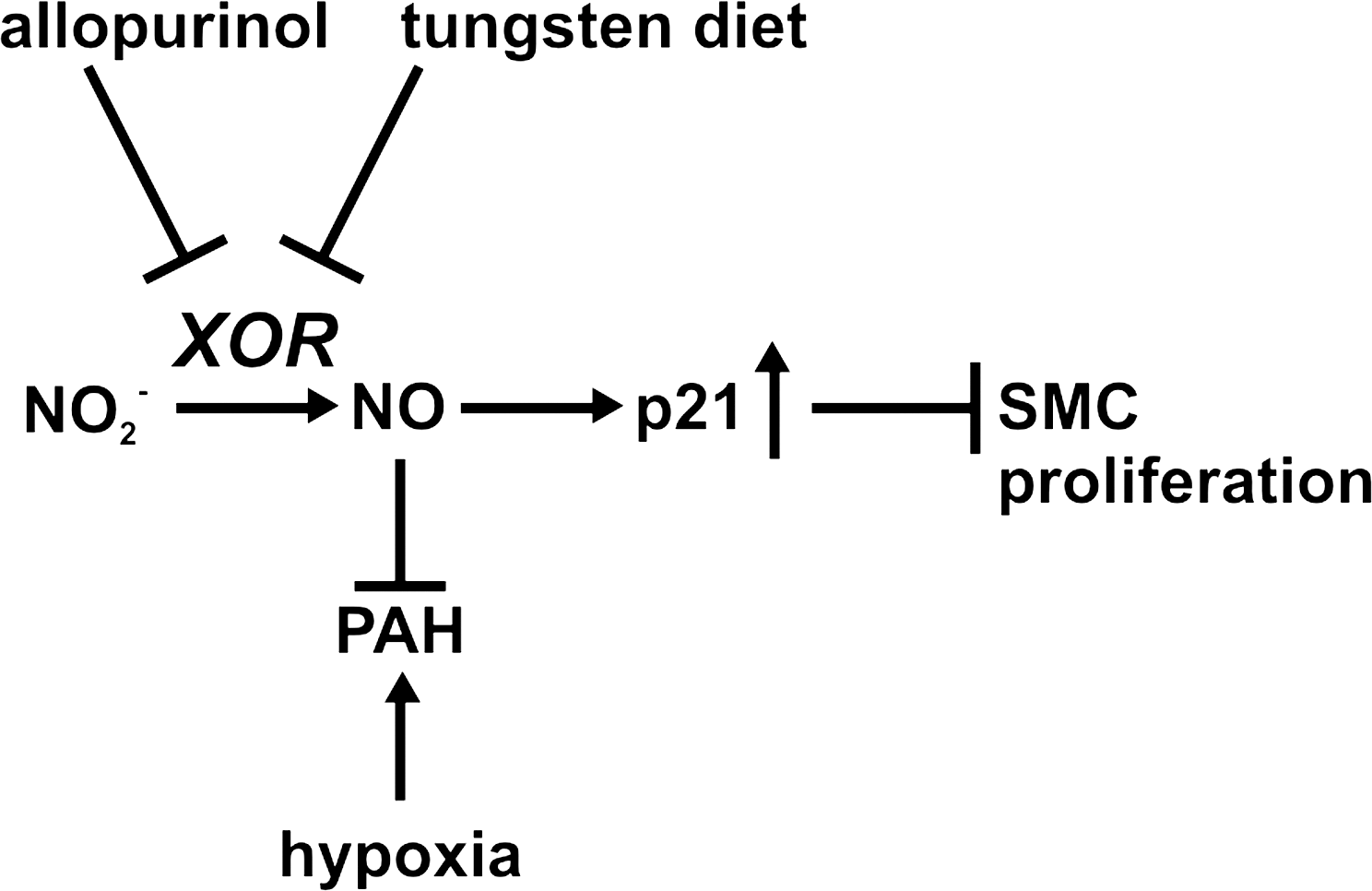
Repeated treatment with nebulized inhaled nitrite has been studied by Zuckerbraun et al. (86) in two animal models of PH: a rat, monocrotaline-induced PH model and a mouse hypoxia-induced PH model. They demonstrated that inhaled nebulized nitrite can prevent and reverse establish PH. In the hypoxic mouse model, intervention after 2 weeks of hypoxia, after the establishment of the PAH, halted its progression and reversed high right ventricular pressures. In the monocrotaline-induced PAH rat model, nebulized nitrite was able to diminish monocrotaline-induced muscularization and hyperplasia of the small pulmonary arteries. Nitrite effects in both models were blocked by the XO inhibitor allopurinol (in vitro) and by tungsten-enriched diet (in vivo), which replaces Mo in the active sites of enzymes such as XO and AO, inhibiting their activity. These data suggest that the protective effect of nitrite against the development and progression of hypoxia-induced PAH is mediated by XOR (86). Additional work suggested that the nitrite effect was mediated by NO–cyclic guanosine monophosphate signaling and downstream induction of the cell cycle checkpoint inhibitor p21, which inhibits smooth muscle cell proliferation (Fig. 9).
Intravenous administration of sodium nitrite has been also explored in the context of PH. Casey et al. (11) investigated the effect of intravenous sodium nitrite (10–100 μmol/kg) as a pulmonary vasodilator in rats under hypoxic conditions. A vasodilatory effect in both pulmonary and systemic arterial pressure was observed, and these effects were inhibited by allopurinol, consistent with other rodent PAH models (86). While these studies suggest a role for XO as a nitrite reductase in rodent models, human studies have been performed testing a role for XO in nitrite-dependent vasodilation. Infusions of oxypurinol for 30 min, followed by coinfusions of nitrite, did not block nitrite-dependent vasodilation in humans (17). Paradoxically, oxypurinol increased nitrite-dependent vasodilation by about 10%. Future studies are required to reconcile findings in rodent disease models compared with human physiological studies.
Other Therapeutic Opportunities for Nitrite in Vascular Disease
In any pathological condition where NO bioavailability is compromised, nitrite administration may provide a therapeutic approach to restore NO levels through the nitrate-nitrite-NO signaling pathway. This approach has a direct relevance for cardiovascular disease, given the well-known vasodilatory effect of NO, and now appreciated vasodilatory effects of nitrite. We will summarize here some of the current evaluations of nitrite in vascular disease.
High blood pressure
As discussed earlier, plasma nitrite levels can be modified by dietary nitrate intake. Larsen et al. (49) studied the effect of dietary nitrate supplementation on healthy volunteers and found a significant reduction in blood pressure and increases in plasma nitrate and nitrite. Other studies have confirmed that dietary nitrate can decrease arterial blood pressure and enhance other parameters linked to NO metabolism, such as platelet aggregation and endothelial function during ischemia. Interestingly, these effects may be greater in men than that in women (46). In general, the dietary effects of nitrate are comparable to effects of vegetable-rich diet, suggesting that the high nitrate content of vegetables can be a part of their observed effects on blood pressure (59). Low doses of intraperitoneal, inhaled, or oral sodium nitrite have been shown to generate NO in blood vessels and to inhibit proliferative responses of smooth muscle cells in murine models of carotid injury.
Angiogenesis
Vascular endothelial growth factor-induced proliferation and organization of human endothelial cells have been shown to be dependent on eNOS and NO production (63), suggesting that nitrite-based therapies could be used to promote angiogenesis. Consistently, chronic intravenous administration of sodium nitrite has been shown to induce angiogenesis in a mouse model of hindlimb ischemia (48), and sodium nitrate therapy stimulated ischemic vasodilation and angiogenic activity after permanent femoral artery ligation (64).
Sickle cell disease
The pathology of sickle cell disease relates to vaso-occlusion by sickled erythrocytes and vasoconstriction due to increase NO scavenging associated with hemolytic anemia (83). Patients with sickle cell disease develop PH as they age, suggesting a potential role for nitrite therapy. A phase Ib clinical study by Mack et al. has shown that infused sodium nitrite was safe and increased forearm blood flow in patients with sickle cell disease (57), although sickle cell patients show a somewhat reduced response to nitrite as compared to healthy volunteers (16, 58).
Myocardial ischemia
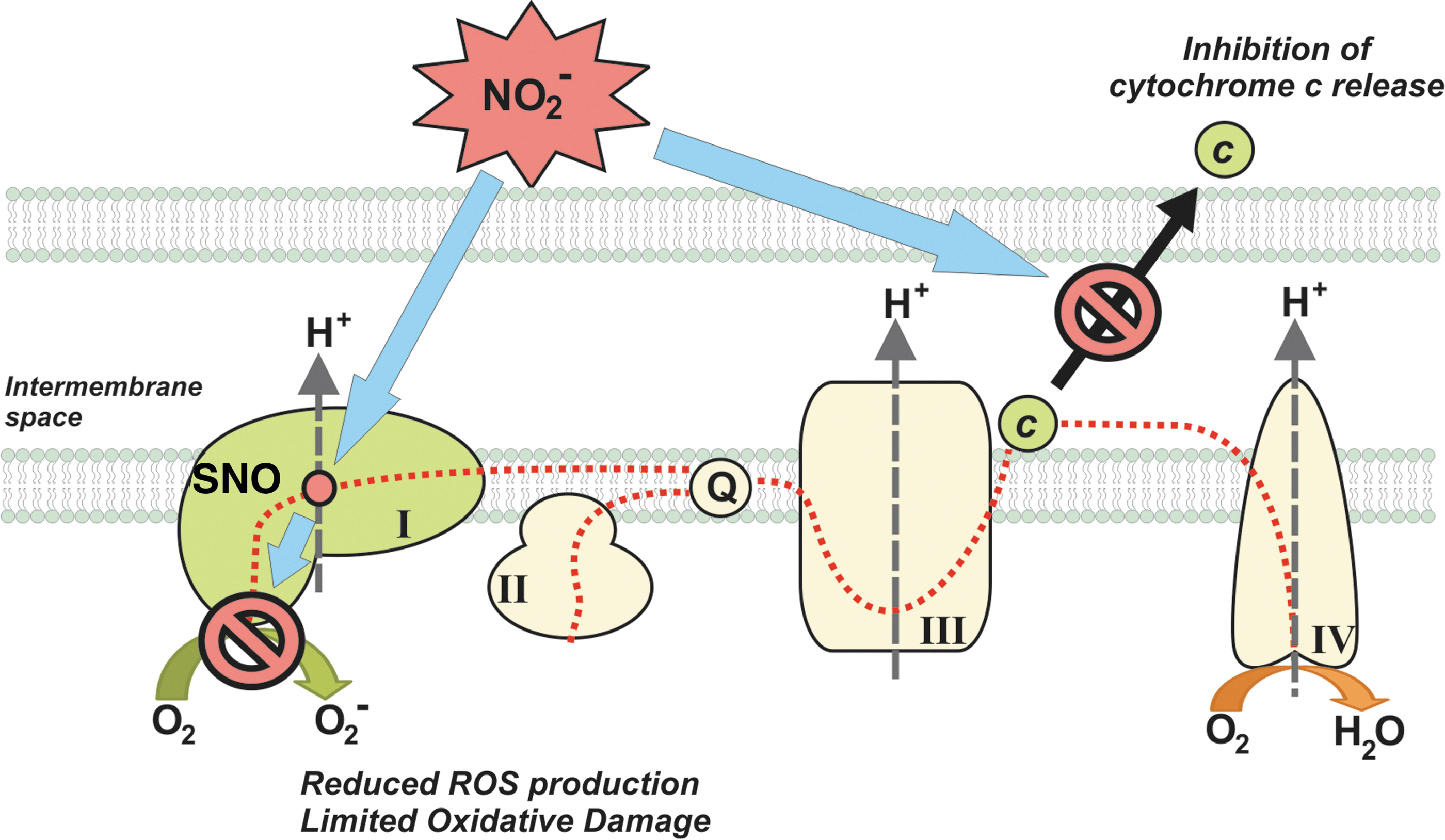
Nitrite has protective effects in different ischemia reperfusion injury models (19). The effects are at least partly due to the ability of nitrite to reversibly inhibit mitochondrial metabolism, via S-nitrosation of complex I, and reduce reperfusion ROS formation (20, 60, 72). This reduction in reperfusion ROS is associated with a prevention of opening of the mitochondrial permeability transition pore and release of cytochrome c (72) (Fig. 10). Several studies have focused on the effects of nitrite treatment on myocardial ischemia. Duranski et al. showed a large effect in decrease of the infarct size when nitrite was applied during ischemia (23). The beneficial effect of nitrite in cardiac infarct has been found be independent of the time of administration during the ischemic phase and can be obtained by dietary intervention (31). The use of intravenous sodium nitrite to ameliorate the consequences of acute myocardial infarction is currently being evaluated in phase I–II clinical trials.
Stroke
Several studies have investigated the use of nitrite on stroke therapy in preclinical models. Early administration of nitrite during reperfusion shows favorable effects in rats (42), although responses depended on the nitrite dosage. High amounts of nitrite provided no protection, and low doses reduced the infarction size and enhanced local cerebral blood flow and functional recovery (44). Long-term, high-dose (100 μg/kg) nitrite treatment showed improvement in the recovery after stroke in a rat ischemia model (45). Some positive effects have been also observed in a rat intracranial hemorrhage model (43).
Nitrite therapy has also been applied in the treatment of delayed cerebral vasospasm after subarachnoid hemorrhage, a subtype of hemorrhagic stroke. This condition is characterized by decreased middle cerebral artery flow due to the narrowing of large-capacitance arteries and often develops days after subarachnoid hemorrhage. Several studies indicate that the vasospasm is related to decreased bioavailability of NO, at least in part due to inhibition of eNOS and NO scavenging by hemoglobin from the subarachnoid clot. Pluta et al. (66) studied the effect of nitrite in a primate model of subarachnoid hemorrhage, and showed that nitrite prevented the development of cerebral vasospasm.
Therapeutic Developments
Nitrite has been proposed and tested successfully in several animal models and patients, with a variety of delivery systems and formulations. A mixture of sodium nitrite and acidifying agents such as ascorbic acid can rapidly release NO. This topical administration has been evaluated for its antimicrobial activity in various skin infections (61) and, with some modifications, in preventing catheter-associated urinary tract infections (10). It is also well established that nitrite can be given orally, but its bioavailability can be difficult to assess, because it is extremely dependent on the oral microbiome and its variable metabolism within the gastrointestinal tract. However, the use of organic allylic nitrocompounds as nitrite donors may overcome this issue (13). A third possibility is the much anticipated use of intravenous infusions of nitrite, but the dose and the duration of the treatment still need to be adjusted (67). One of the adverse effects of inorganic nitrite is the formation of methemoglobin. Nitrite can oxidize the iron at the heme group, modifying it from a ferrous (Fe2+) oxygen-binding form to a ferric (Fe3+) nonoxygen-binding state. If the increase of methemoglobin in circulating blood is higher than 5%, it can lead to cyanosis. However, studies with nitrite intravenous studies only report undetectable or modest increase (67).
As described above, inhaled sodium nitrite is a pulmonary vasodilator that can effectively prevent or reverse PAH in animal models. Studies suggest that it can be delivered safely, and it is ready for clinical translation for PAH patients. Animal toxicology studies with inhaled nitrite in rodents and dogs have been completed and phase Ia and Ib studies in normal volunteers completed. In a phase Ia study, inhaled nebulized sodium nitrite, at doses >17 mg, increases exhaled NO, whereas methemoglobin levels remained <3.5% in all subjects (Bradley et al., unpublished data). A proof-of-concept phase II trial of inhaled nitrite in patients with PAH is currently enrolling sites in the United States and Europe.
Summary and Conclusions
PAH is associated with decreased bioavailability and responsiveness of NO. Despite the potential therapeutic effect of iNO as a selective pulmonary vasodilator, its administration is inconvenient and difficult. The discovery that nitrite is a naturally occurring molecule in the body and may act endogenously as a reservoir of NO has suggested the idea that this anion may represent an alternative strategy for an effective NO-based therapy. NO can only be administered as a gas and cannot be mixed with oxygen, or it reacts to NO2. Nitrite (as a soluble salt) can be effectively delivered as a nebulized liquid, dry powder, intravenous solution, or oral formulation. Also, when compared with iNO, nitrite has a longer half-life. However, two caveats arise for either iNO or nitrite therapies: (i) both may have effects in conditions with soluble guanylate cyclase dysfunction (oxidation or downregulation); and (ii) potential harmful side products may be formed by reaction with ROS (NO2, peroxynitrite, or methemoglobin formation). Nitrite has demonstrated efficacy in multiple animal models not only of cardiovascular disorders but also of inflammatory diseases and bacterial infections. Clinical trials are ongoing and more planned in the near future to demonstrate the therapeutic efficacy of nitrite in patients with PAH.
Footnotes
Acknowledgments
We thank Dr. Jesus Tejero, Dr. Courtney Sparacino-Watkins, and Dr. Eric Kelley (University of Pittsburgh). Dr. Gladwin receives research support from the NIH grants R01HL098032, RO1HL096973, and PO1HL103455, the Institute for Transfusion Medicine and the Hemophilia Center of Western Pennsylvania. Dr. Bueno, Dr. Mora, and Dr. Wang are supported by the Vascular Medicine Institute of the University of Pittsburgh.
Author Disclosure Statement
Dr. Gladwin is listed as a coinventor on an NIH government patent for the use of nitrite salts in cardiovascular diseases. Dr. Gladwin consults with Aires Pharmaceuticals on the development of a phase II proof-of-concept trial using inhaled nitrite for pulmonary arterial hypertension.