
Abstract
Introduction
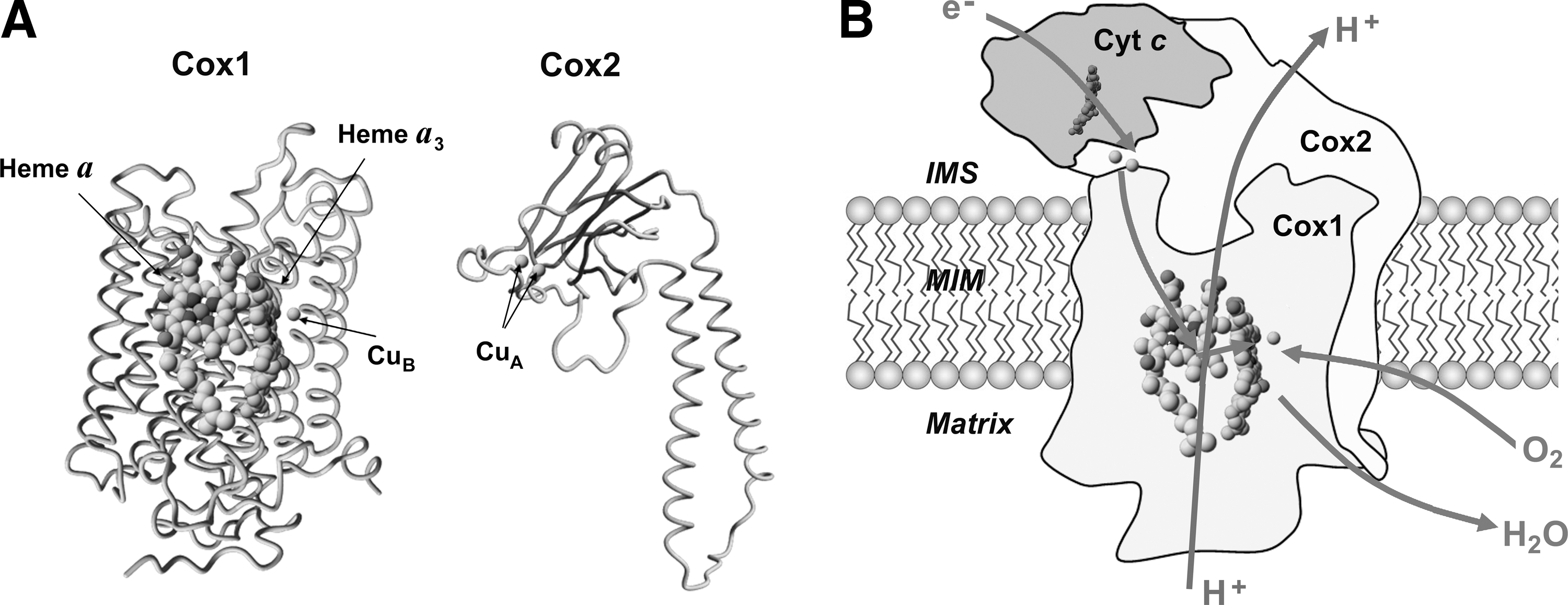
The focus of this review is on the terminal MRC enzyme cytochrome c oxidase (COX, complex IV), a heterooligomeric heme A-copper oxidase that catalyzes the reduction of O2 by ferrocytochrome c. Although COX is one of the major oxygen-consuming enzymes of the cell, ROS generation during O2 reduction is negligible. The enzymatic catalytic core is formed by three mitochondrial DNA-encoded subunits that enclose three metal centers (Fig. 1A). Cox1 contains a low-spin heme a site and a binuclear center formed by a high-spin heme a3 and a copper atom (CuB). Cox2, which interacts with cyt c, contains a mixed valence dinuclear copper center (CuA). Oxygen reduction requires four electrons, which enter COX through the CuA site, are transferred to heme a, and subsequently to the binuclear active site, where O2 is bound and reduced. The reduction of O2 to water proceeds via one electron at a time, and thus involves several successive reduction steps. The COX catalytic cycle has been recently reviewed in detail (104). Importantly, in the MRC, COX retains all partially reduced oxygen intermediates until full reduction is achieved (104), which avoids ROS generation. In COX, electron transfer is coupled to proton pumping across the inner mitochondrial membrane (Fig. 1B), a function that may be modulated by the core subunit Cox3, thus contributing to establish the proton gradient required to synthesize adenosine-5′-triphosphate (ATP). In addition to the core subunits that are conserved in the prokaryotic enzyme, mitochondrial COX contains 8 (yeast) to 10 (mammals) nuclear-encoded subunits. They play roles in regulating COX assembly and function and are believed to act as a shield to protect the catalytic core (31).
Investigations in recent years have shed light into the sophistication surrounding COX biogenesis, disclosing several redox-controlled or redox-dependent processes and regulatory mechanisms to minimize oxidative damage. Besides its structural subunits, COX assembly requires an extensive and growing number of ancillary factors, which act at all the steps of the pathway (31). The assembly process is thought to be linear, consisting on the successive incorporation of subunits to membrane-inserted Cox1. Recent data have suggested that an additional alternative pathway may exist to incorporate COX subunits and subunits of other MRC complexes directly into a macromolecular structure to form supercomplexes or respirasomes (72). COX is known to display intrinsic heterogeneity concerning its subunit composition. Cox5 exists in two oxygen-regulated isoforms, and two newly identified COX-associated proteins, Rcf1 and Rcf2, are required for growth in hypoxia and for oligomerization of a subclass of COX complexes into respirasomes (18, 95, 101).
Here, we will discuss recent data on COX biogenesis steps and regulatory processes involving ROS signaling to regulate COX subunit composition, mitochondrial oxidative folding, and redox regulation of copper delivery to COX as well as translational control to prevent formation of pro-oxidant intermediates (Fig. 2). We focus on the processes described in the yeast, S. cerevisiae, with information from prokaryotes and higher eukaryotes when appropriate.

Redox Control of COX Isoenzyme Assembly
Oxygen-regulated COX isoenzymes involving subunit Cox5/COX4
COX plays an important role in MRC regulation to adapt to changing environmental oxygen levels through a mechanism involving a nuclear-encoded subunit, termed Cox5 in yeast and COX4 in mammals (31). Structurally, Cox5/COX4 have interacting surfaces with two core subunits, Cox1 and Cox2 affecting COX redox centers and the stability of the holoenzyme (31). Both Cox5 and COX4 exist in two interchangeable isoforms termed Cox5a/b and COX4-1/2, respectively, whose expression is regulated by oxygen (52). Cox5a/COX4-1 are expressed in normoxia (above 0.5 μM/1% of O2), whereas Cox5b/COX4-2 are expressed under hypoxia (below 0.5 μM/1% of O2) (12, 34). In yeast, cyt c also exists in two oxygen-regulated isoforms: normoxic iso-1-cyc (CYC1) and hypoxic iso-2-cyc (CYC7), which act synergistically with the Cox5 isoforms (2, 102). Cox5 isoforms have differential effects on the COX enzymatic activity. The hypoxic isoforms modify an internal step in electron transport between heme a and the binuclear reaction center, which occurs three to four times faster than in the normoxic COX isoenzyme, thus altering the enzyme maximal turnover number (TNmax), but not its Km (2). As a result, by regulating the proportion of each isoform assembled into the holoenzyme, the catalytic efficiency of COX is modulated adjusting the MRC electron transfer rate to adapt to changes in the environmental oxygen tension (44).
Transcriptional regulation of COX5a/COX5b isoforms in yeast
In yeast, the oxygen regulation of all nuclear COX genes is mediated by heme that acts as an intracellular effector of the environmental signal through two heme-dependent transcriptional activators. Hap1 arbitrates the oxygen regulation of aerobic genes and its activity is modulated by heme binding (107). The Hap complex (Hap2/3/4/5) mediates regulation in response to two environmental stimuli, oxygen concentration and carbon source availability (33). How heme modulates the Hap complex activity remains unclear. In normoxia, the expression of nuclear COX genes, including COX5a, is induced by the Hap2/3/4/5 complex, while COX5b is repressed by the transcriptional repressor encoded by ROX1, whose expression is induced by Hap1 (23) (Fig. 3A). A second poorly characterized repressor, the oxygen-independent ORD1, specifically contributes to COX5b aerobic repression (60). Instead, in hypoxia, heme is poorly synthesized and neither the Hap2/3/4/5 complex can activate its target COX genes nor Rox1 represses COX5b expression (97) (Fig. 3A).

Role of hypoxic COX in redox signaling in yeast
COX can act as a nitric oxide (NO) reductase using nitrite as a substrate (15). In normoxia, oxygen binds COX with a higher affinity than nitrite. However, in hypoxia, the NO reductase activity is enhanced, thus producing NO (15). The recent discovery that in hypoxic conditions Cox5b-containing hypoxic COX produces a higher amount of NO than the normoxic enzyme, further emphasizes the role of COX in hypoxic signaling, and suggests a positive feedback mechanism since NO serves as an additional signal to activate expression of several hypoxic genes, including COX5b and CYC7 (15).
Regulation of mammalian COXIV-1/COXIV-2
In mammals, COX4-1 and COX4-2 are respectively encoded in the genes COXIV-1 and COXIV-2, which expression are also inversely regulated by oxygen concentrations (Fig. 3B). Additionally, COXIV-2 expression varies depending on the development stage and tissue (52). For example, a high expression level for COXIV-2 mRNA is paradoxically observed in highly oxygenated adult lung and trachea of human and rat. Highly efficient hypoxic COX could minimize electron leakage and contribute to protect these tissues from oxidative damage (52). The hypoxia-inducing factor 1 (HIF-1) complex, a transcriptional activator that functions as a master regulator of oxygen homeostasis in all metazoan species, regulates COX subunit isoform switch (34). HIF-1 is a heterodimeric complex composed of a constitutively expressed HIF-1β subunit and an O2-regulated HIF-1α or HIF-2α subunit. HIF-1 activity is inhibited by prolyl (PHDs) and asparaginyl (factor inhibiting HIF-1 [FIH-1]) hydroxylases, which use O2 as a substrate to hydroxylate HIF-1α and HIF-2α. In hypoxic cells, hydroxylation is inhibited, leading to HIF-1-mediated transcriptional activation of COXIV-2 and the LON gene, which encodes a protease required for COX4-1 degradation (34), thus facilitating the COX4-1 to COX4-2 subunit switch. However, promoter analysis have revealed that an additional unidentified cis-element, different from the HIF-1 binding site, could mediate the activation of COXIV-2 expression (52).
Regulation of COX5/COXIV isoforms by oxidative stress
During the transitions from normoxia to hypoxia, yeast cells undergo an increase in mitochondria-generated ROS levels, mostly at the complex III site, which play a role in hypoxic gene induction (28, 36). Recent studies by our group published in this forum issue (64) indicate that expression of MRC hypoxic genes, including COX5b and CYC7, is regulated by environmental- and stress-induced mitochondrial ROS by a mechanism involving Rox1, but not other transcription factors, such as Ord1. Contrary to what occurs in hypoxia, oxidative stress induces ROX1 expression in a Hap1-independent manner. However, ROS regulates hypoxic gene expression by modulating Rox1 binding to its target promoters (64). In mammalian cells, HIF-1 can be activated by nonhypoxic stimuli involving mitochondrial-generated ROS as essential intermediates (38). ROS sensing HIF-1α stabilization probably involves several pathways waiting to be fully disclosed. In a possible mechanism, ROS could inactivate the PHDs by oxidizing their iron cofactor, thus promoting HIF-1α stabilization and hypoxic gene expression (9). Thus, the very same programs evolved to maximize energy production and O2 utilization in hypoxia seem to be also used to mediate the response to oxidative stress, including the expression of hypoxic COX.
MRC supercomplexes and COX isoenzymes involving Rcf1 and Rcf2
The structural and functional organization of the respiratory chain has been a matter of debate over more than 50 years (17, 37). Around a decade ago, the groups of Schagger (86, 88) and Stuart (22) produced new evidence of stoichiometric assemblies of individual complexes in yeast and in mammalian mitochondria, and suggested an organization of the MRC in supercomplexes or respirasomes. It is currently accepted that both individual enzymes and supercomplexes probably coexist. This plasticity model suggests that OXPHOS complexes (CI to CIV) switch from freely moving to fixed structures and vice-versa to adapt to changes in cellular metabolism (1, 43). Accordingly, the coexistence of the respirasome unit, composed in mammals of at least CI+CIII+CIV, together with the intermediate supercomplexes CI+CIII and CIII+CIV and free CIII and CIV has been widely described (1, 3, 22, 29, 40, 85 –88, 105). The functional role of the respirasomes in cellular bioenergetics remains unclear, but they have been hypothesized to offer structural and functional advantages to the system, such as the prevention of destabilization and degradation of the OXPHOS complexes, the enhancement of electron transport efficiency through substrate channeling, or the decrease of electron or proton leakages (63). Recent data support a role in controlling the formation of reactive oxygen intermediates (18, 101).
In addition to their structural subunits, the biogenesis of OXPHOS complexes requires a large number of ancillary proteins, generally termed assembly factors. While most factors involved in the assembly of complex I have been discovered in human patients carrying disease-causing versions (100), most factors involved in the assembly of complexes III and IV have been discovered in the yeast S. cerevisiae (31, 93, 94). In S. cerevisiae, COX assembly factors (such as Shy1, Cox14, or Cox25/Coa3) have been found associated with structural subunits of both complexes (30, 71) in what probably represent CIII-CIV supercomplex assembly intermediates. However, the first respirasome-specific assembly/stability factors, the conserved Rcf1 and Rcf2, were identified only recently. Rcf1 is a member of the conserved hypoxia-induced gene 1 (Hig1) protein family (18, 95, 101). Three independent groups have reported that yeast Rcf1 is important for growth in hypoxia and the assembly of the peripheral COX subunits, Cox12 and Cox13. Additionally, Rcf1 interacts independently with CIII and CIV although the interaction with COX seems to be stronger and occurs through Cox3, predicted to be at the CIII-CIV interface. Both Rcf1 and Rcf2 interact with CIII-CIV supercomplexes, but they do not copurify. This has suggested that Rcf1 promotes the oligomerization/stability of only a subclass of CIV monomers that do not contain Rcf2 on CIII2-CIV2 supercomplexes (18, 95, 101). These studies have suggested that CIV displays intrinsic heterogeneity with regard to its subunit composition and that distinct forms of respirasomes can be formed by CIV variants (101). Noteworthy, the impairment of CIII2-CIV2 supercomplexes in the yeast rcf1 null strain was found associated with an increase in ROS generation and sensitivity to oxidative stress (18, 101), suggesting the supercomplexes somehow minimize electron leakage.
The reciprocal effects between ROS and supercomplexes remain to be fully understood. Notably, recent studies on mouse complex III cells knocked out for the Rieske iron–sulfur protein (RISP) subunit have suggested that ROS produce a deleterious effect on the stability of CI, CIV, and supercomplexes, at least in the context of defective OXPHOS (27). In support of that conclusion, treatment of the mutant cells with an SOD mimetic compound (MnTBAP) was shown to partially stabilize CI and supercomplexes (27).
Redox Control of Copper Delivery to COX
Redox environment within mitochondria
Mitochondria are the major ROS-generating cellular organelle, and therefore redox regulation within the mitochondrial compartments is essential for several processes, including COX biogenesis (39). While the redox environment of the cytosol (−286 mV) and mitochondrial matrix (−296 mV) is reducing, the intermembrane space (IMS) is more oxidizing (−255 mV) (51). Consistently, the reduced glutathione/oxidized glutathione (GSH/GSSH) ratio, one of the major redox regulatory systems in the cell, was estimated to be 250:1 in the IMS (compared to 900:1 in the matrix or 3000:1 in the cytosol) (51). It remains unclear though, whether regulation of the IMS redox environment is really contributed by GSH. In human cells, glutaredoxin 1 (GRx1), which catalyzes reversible protein glutathionylation, specifically localizes to IMS (76). However, in yeast, the IMS does not contain Glr1, the GSSH reductase that regenerates GSH from GSSH (75) and redox deregulation induced by unbalancing the GSH/GSSH ratio in the cytosol and the matrix was reported to not influence the IMS redox environment (51). This conclusion has been partially challenged in a very recent article, which has convincingly demonstrated that the glutathione pools of the IMS and cytosol are dynamically interconnected via porins, while no communication was observed between the IMS and matrix glutathione pools (58). By modulating redox pathways in the cytosol and IMS, the authors found that the cytosolic glutathione reductase system is the major determinant of the glutathione redox potential in the IMS, thus explaining why they find it similar to the glutathione redox potential in the cytosol (58).
Although the precise mechanisms involved in the IMS redox regulation remain to be fully characterized, this compartment harbors several redox pathways, including a pathway for oxidative protein folding, and redox chemistry important for COX biogenesis.
Mitochondrial import of COX copper chaperones through an oxidative folding pathway
Because the vast majority of mitochondrial proteins are encoded in nuclear genes, the cells have devised several protein transport mechanisms for delivery to the correct submitochondrial compartments. A small fraction of IMS proteins use a conserved disulfide relay system that involves the redox-regulated import receptor Mia40 and the sulfhydryl oxidase Erv1 (Fig. 4) [reviewed in Refs. (26, 41, 92)].
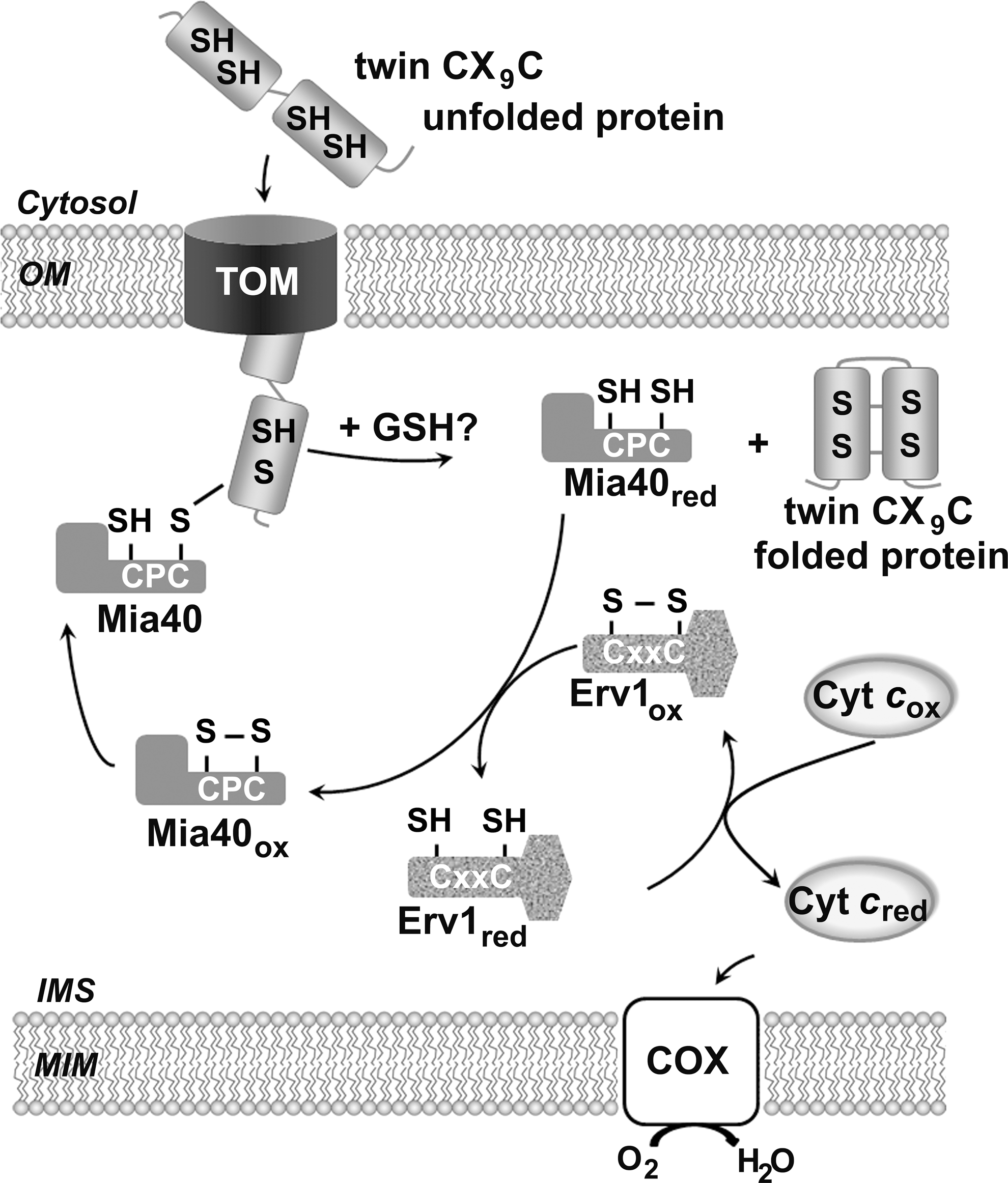
The Mia40/Erv1 system promotes the oxidative folding of newly imported cysteine-rich small precursor proteins, thus ensuring their retention within mitochondria. Most import substrates contain either twin CX3C (73) or CX9C (16) structural motifs, which upon oxidation form two disulfide bonds. Focusing on the twin CX9C proteins, they are mostly required for COX biogenesis (66). In the current model, Mia40 substrates are synthesized in the cytosol and cross the outer mitochondrial membrane in an unfolded state through the translocase of the outer membrane complex. The IMS localized Mia40 traps the substrate proteins by forming an intermolecular disulfide bond. Subsequently, it introduces disulfide bonds in the substrate to promote their native conformation. Upon substrate release, Erv1 reoxidizes Mia40 and delivers electrons to cyt c for further delivery to COX, thus connecting oxidative folding to respiration. The IMS redox environment probably influences the import by the Mia40 pathway [reviewed in Refs. (26, 41, 92)]. Supporting this possibility, glutathione increases the in vitro and in organello Mia40-dependent oxidation of Cox19, a CX9C substrate, by counteracting the formation of long lived Mia40 substrate intermediates (10) and has been shown to contribute to the partially reduced redox state of the Mia40 in vivo (58).
In addition to Mia40, thirteen conserved small CX9C proteins are found in the IMS (66). At least ten of these proteins are known to play roles relevant to COX assembly/stability (66). With a couple of exceptions (Cox17 and Som1), their exact functions are largely uncharacterized. However, several CX9C proteins have been proposed to function on copper and/or redox transfer within the IMS for ultimate delivery to COX (47).
Redox-regulated copper transfer to COX
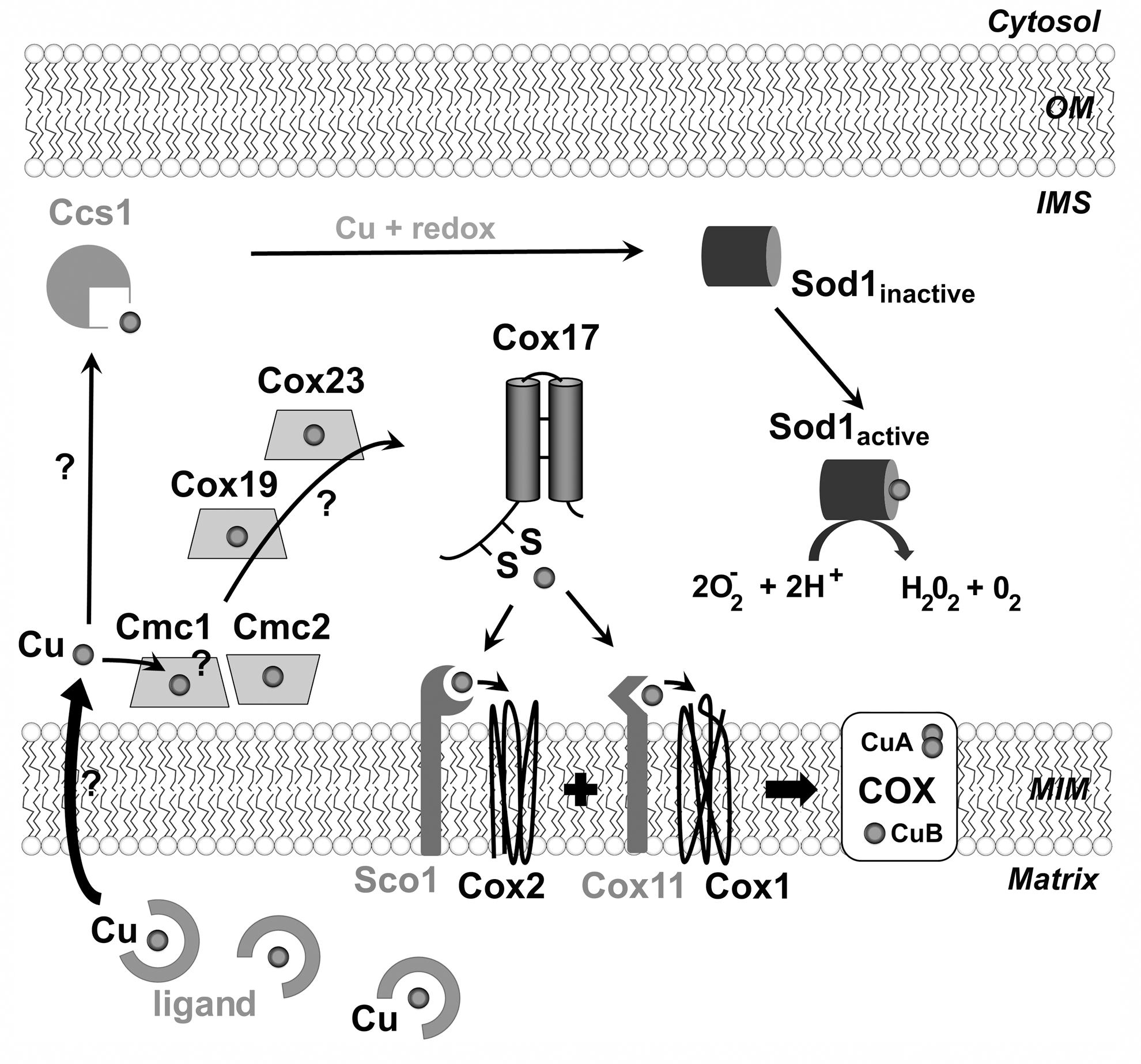
The metallation of the CuA and CuB centers in COX uses copper from a matrix-located pool (21). By unknown mechanisms, copper is transferred to the IMS and reaches Cox17, a hydrophobic CX9C protein containing two additional cysteines proximal to the CX9C motif forming a CC Cu(I) binding site (77). Cox17 transfers copper ions to two metallochaperones, Sco1 and Cox11 that facilitate copper insertion into the Cox2-CuA and Cox1-CuB sites, respectively (Fig. 5) (13, 35, 42, 49, 65). Cox11 and Sco1 are anchored to the mitochondrial inner membrane through a transmembrane α-helix and expose the copper-binding site in the IMS, where copper transfer occurs [(11, 56, 81) and reviewed in (21, 47, 84)].
The CuB site is formed by one copper ion coordinated by three histidine ligands and in close proximity to the heme a3 moiety (67). The soluble C-terminal domain of Cox11 forms a dimer that coordinates one Cu(I) per monomer. The two Cu(I) ions in the dimer exist in a binuclear cluster, coordinated by three conserved cysteine residues (13). The mechanism by which Cox11 transfers copper to the CuB site remains to be disclosed.
The CuA dinuclear center in Cox2 is coordinated by a CxExCGx2Hx2M motif and exists as a [Cu(II)/Cu(I)] complex. A soluble truncated Sco1 variant binds copper donated from Cox17 in vitro (14). NMR studies suggested that human COX17 transfers reducing potential and Cu(I) to enable SCO1 metallation (4). Sco1 directly interacts with Cox2 (65) and has an essential Cx3C metal-binding motif, which is able to bind both Cu(I) and Cu(II) (50). It remains to be elucidated whether Sco1 mediates the transfer of both different valent ions or, alternatively if two Cu(I) are inserted in Cox2 by Sco1 and the active site is successively oxidized. The structural similarity of Sco1 with disulfide reductases has suggested that it could be involved in the reduction of cysteines in the Cox2 copper- binding site (19), facilitating copper incorporation (4).
In mammals, Sco1 has two homologues, SCO1 and SCO2, which play independent, cooperative roles in COX biogenesis (62). In the current model, after COX2 metallation by SCO2- and SCO1-dependent simultaneous or sequential copper insertion, SCO2 reoxidizes SCO1 cysteines, a reaction that allows resetting both proteins for further rounds of COX2 biogenesis (62). While both Sco proteins are required for copper transfer to COX2, SCO2 is additionally necessary for COX2 synthesis (62), which serves to coordinate COX2 availability and formation of the CuA site during COX biogenesis.
Is there a role for CX9C proteins on copper and/or redox transfer?
Studies on the large number of CX9C IMS proteins have suggested that at least a subset could be involved in copper traffic toward Cox17 (47) (Fig. 5). These proteins include Cox19, Cox23, Cmc1, and Cmc2 (47, 66, 84, 94). Supporting this possibility, Cox19 and Cmc1 were shown to bind copper in vitro (46, 82), Cmc2 to interact with Cmc1 (48), and the respiratory defect of Δcox23 and Δcmc1 cells to be partially restored by exogenous copper although Δcox23 cells require concomitant overexpression of Cox17 (7). Current speculation have suggested a daisy chain transfer mechanism in which copper would be successively transferred from the matrix across the inner membrane by an uncharacterized transporter to the membrane-bound Cmc1/Cmc2 and successively to the soluble Cox19, Cox23, and ultimately to Cox17 (47). Such a mechanism would be expected to involve transfer of both reducing potential and Cu(I) to generate reduced Cu(I)-bound proteins. Alternatively, CX9C proteins could play a role in copper transfer toward Cox17 by modulating the local IMS redox environment. Information concerning the in vivo copper-binding capacity and redox state of these proteins seems imperative to attain an understanding of their functions.
Copper distribution to COX and mitochondrial Sod1
The matrix copper pool used for COX metallation is also the copper source for the IMS-localized portion of superoxide dismutase 1 (mt-Sod1) (21). The active enzyme is a homodimer that has one intramolecular disulfide bond, one copper and one zinc atom bound per monomer. Sod1 activation requires the specific copper chaperone Ccs1 (Fig. 5), which forms an intermolecular disulfide bond with Sod1 to introduce a copper atom and a disulfide bond into Sod1 (21).
Interestingly, in S. cerevisiae, Δcmc1 and Δcmc2 cells (48) and most COX mutants have wild-type levels of mt-Sod1, but with a significantly higher activity, while Δccs1 cells have a significantly increased functional COX (our unpublished results). These observations suggest a partition of the copper pool between the delivery pathways to COX and mt-Sod1. A balance between copper transfer to COX and Sod1 could control the intricate equilibrium between efficient respiration, ROS damage, and necessary signaling and protection against oxidative stress. However, similar to ccs1 null mutant cells, it has been reported that a Δsod1 strain shows increased COX biogenesis and mitochondrial mass, which has been proposed to result, at least, in part, from a ROS-dependent Hap2/3/4 complex activation (89).
Strategies to Regulate COX Biogenesis and Minimize Accumulation of Pro-Oxidant COX Assembly Intermediates
Pro-oxidant intermediates are formed during COX assembly
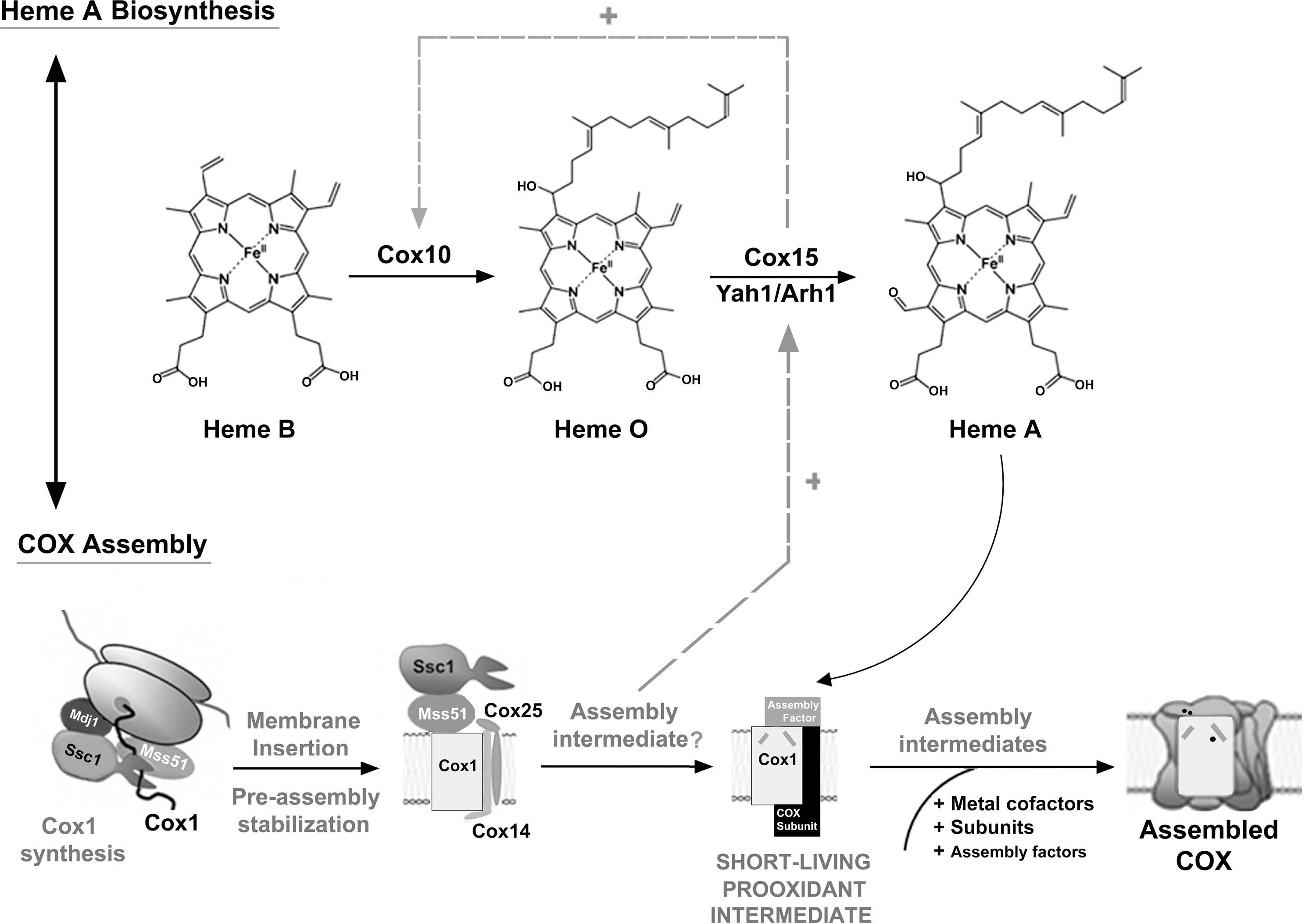
COX biogenesis is extensively regulated to facilitate the formation of the metal centers, while minimizing the formation of reactive assembly intermediates (Fig. 6). COX assembly starts with the synthesis, membrane insertion, and stabilization of the mitochondrially encoded COX subunits (Fig. 2) (94). Briefly, subunit mRNA-specific translational factors activate translation and coordinate the accumulation of the three core subunits (Cox1, Cox2, and Cox3). Following their membrane insertion by the Oxa1 machinery, subunit-specific chaperones (Mss51, Cox14, and Cox25/Coa3 for Cox1, and Cox20, Cox18, Mss2, and Pnt1 for Cox2), stabilize the core subunits and maintain them in an assembly-competent state (Fig. 2). Starting with the Cox1 biogenesis line, Mss51 handles the Cox1-preassembly complex over to conserved assembly chaperones, such as Coa1 and Shy1 (5, 71, 80). This process is concurrent with the maturation of Cox1 by incorporation of its metal prosthetic groups or the formation of the first assembly intermediates, involving Cox1+Cox5+Cox6 (Figs. 2 and 6). Metallated-Cox2 and Cox3 are subsequently incorporated to the growing subassembly, followed by the rest of nuclear-encoded subunits to form the monomeric COX holoenzyme (45, 74) (Fig. 2).

The formation of the metal prosthetic groups within Cox1 are essential during the COX assembly process and will be more extensively described here because they bear the risk of producing highly reactive intermediates.
While Cox1 hemylation is essential for COX assembly, little is known about the players and mechanism that coordinate heme A synthesis and insertion into Cox1. Heme A biosynthesis from protoheme (heme B) involves at least two reactions (Fig. 7). The first reaction is catalyzed by the farnesyl-transferase Cox10 (98), and involves the formation of a heme O intermediate that is converted to heme A in a reaction catalyzed by Cox15, which functionally cooperates in an electron transport chain with ferredoxin Yah1 and the ferredoxin reductase Arh1 (6). Heme A insertion into Cox1 does not occur cotranslationally. Rather, the two heme A sites form downstream of Mss51- and Coa1-containing Cox1 preassembly and stabilization intermediates (53). In contrast, the Mss51-free, Shy1-containing Cox1 assembly intermediate is perturbed in the absence of heme A, thus suggesting that the incorporation of heme A into Cox1 occurs within this subassembly (53).
Cox1 copper metallation was initially envisioned to occur cotranslationally due to the positioning of the CuB site, deeply buried below the inner mitochondrial membrane. Such a hypothesis was supported by studies that show a small portion of Cox11 can interact with mitochondrial ribosomes (56). However, more recent investigations have established that the CuB site formation does not actually occur while newly synthesized Cox1 is interacting with Mss51, but near or at the Shy1-containing Cox1 assembly intermediate, perhaps, simultaneously to the incorporation of the heme a 3 moiety into the heterobimetallic center (53).
It remains unclear in what specific order the CuB-heme a3 center is metallated, although studies in a Rhodobacter sphaeroides Δcox11 strain have shown that the heme a3 moiety can be delivered to this site in the absence of copper (42). Studies in yeast have revealed that null cox11 mutants, as well as sco1 mutants, have increased sensitivity to hydrogen peroxide (54). This sensitivity has been attributed to the presence of a highly reactive Cox1-heme a3 intermediate that accumulates when the assembly pathway is blocked subsequent to Cox1 maturation (54, 57). Insertion of heme a/a 3 in Cox1 occurs before the addition of Cox2 during COX assembly (53), perhaps, through a partially accessible channel on the IMS side of Cox1 that is closed upon Cox2 incorporation (57). Hydrogen peroxide sensitivity is enhanced in strains lacking Cox2, but can be suppressed by overexpression of Cox11 and Sco1 (54). This suggested that these proteins can cap the Cox1 intermediate, thereby precluding peroxide access to the heme a 3 catalytic center (57). Particularly, although a direct Cox11–Cox1 interaction has not been detected, it has been proposed that Cox11 may bind and stabilize Cox1 through transient interactions in a conformer of Cox1 that has less solvent accessibility to the heme a 3 site (57). During normal COX assembly, incorporation of Cox2 would resolve the potentially deleterious Cox1 intermediate.
Noteworthy, the amount of the Shy1-containing Cox1 intermediate found markedly attenuated in Δcox11 cells (53), which could serve to prevent its potential deleterious effects. The model of a pro-oxidant Cox1-containing intermediate is supported by the identification of AFG1, coding for a protease involved in the degradation of mitochondrial-encoded COX subunits, as a multicopy suppressor of the peroxide-deficient phenotype of Δcox11 cells (50).
Additional factors seem to modulate hydrogen peroxide sensitivity in Δcox11 cells. Cox11 has been suggested to have a peroxidase function. This was supported by mutagenesis studies that successfully identified point mutations in Cox11 that specifically affected either COX assembly or hydrogen peroxide sensitivity (99). Additionally, the absence of either Cox11 or Sco1, limits peroxide degradation (99). However, while overexpression of COX11 partially complemented the peroxide sensitivity of a sco1 null mutant, the overexpression of SCO1 in Δcox11 cells did not (99), indicating that Cox11 and Sco1 seem to play not exact, but related roles in peroxide metabolism. Interestingly, studies in Δsco1 and Δcox11 petite strains in which Cox1 is absent showed different effects when exposed to peroxide stress. Unlike Δsco1 cells, whose peroxide tolerance is restored in the absence of Cox1, the Δcox11 petite strain remained sensitive to peroxides (99). Collectively, these observations indicate that although the formation of pro-oxidant Cox1-heme a3 intermediates play a role in peroxide sensitivity, it is not the only factor contributing to the peroxide sensitivity in the absence of Cox11.
Beyond these important details, the cells have developed several strategies to minimize the accumulation of reactive assembly intermediates. Importantly, these very same strategies serve to regulate the concerted accumulation of COX subunits in yeast mitochondria.
Protein quality control mechanisms regulate COX biogenesis and minimize accumulation of pro-oxidant COX assembly intermediates
Most unassembled highly hydrophobic COX core subunits are very efficiently post-translationally degraded by the ATP-dependent AAA proteases of the inner mitochondrial membrane (61). Additional proteases specifically prevent the accumulation of immature Cox1 selectively, such as the conserved metallopeptidase Oma1 that degrades Cox1 in absence of the COX chaperone Coa2 (55). Active degradation will avoid the accumulation of unassembled proteins that could have a tendency to aggregate and disturb membrane homeostasis or to form pro-oxidant species as discussed earlier.
Translational regulation coordinates Cox1 biogenesis and COX assembly and minimizes the accumulation of pro-oxidant COX assembly intermediates
Hierarchically, the assembly line of Cox1 determines the rate of COX assembly. Its relevance is highlighted by the existence of at least two regulatory mechanisms pacing Cox1 synthesis and hemylation to its assembly into COX.
On a first mechanism, Cox1 translation is regulated by the availability of its assembly partners (5, 69, 94) (Fig. 6). This particular type of assembly-controlled translational regulation was initially described for the chloroplast's cytochrome b6f of the green unicellular alga Chlamydomonas reinhardtii (20) and termed control by epistasis of synthesis (CES). A distinctive characteristic of these translational autoregulatory systems is the role of ternary factors, mRNA-specific translational activators, whose availability is regulated by specific gene products (20). In the case of yeast Cox1, the ternary factor is Mss51. Briefly, Cox1 synthesis requires two translational activators, Pet309 (68) and Mss51 (24). While both interact with the COX1 mRNA 5′-UTR, Mss51 plays additional roles in coordinating Cox1 synthesis and assembly (Fig. 6). During and after synthesis, Cox1 is bound to Mss51 in a complex stabilized by two small COX-specific chaperones, Cox14 (5) and Cox25/Coa3 (30, 70), and the mitochondrial Hsp70 chaperone Ssc1 (32). When Cox1 proceeds in the assembly process within Coa1 and Shy1-containing complexes, Mss51 is released, binds Ssc1, and is available for a new round of Cox1 synthesis (32, 69). Pro-oxidant intermediates formed during Cox1 maturation and assembly are short-lived when COX assembly proceeds normally. However, in physiological situations when COX assembly needs to be slowed down, or when COX assembly is impaired, Mss51 remains trapped into the Cox1-containing complex and largely unavailable for its role in translation (5, 30, 32, 70, 71, 78 –80, 91). In this way, the coordination between Cox1 synthesis and the following assembly steps could serve to minimize the lifespan of pro-oxidant assembly intermediates (Fig. 6). Whether the COX1 translational regulatory system is conserved in higher eukaryotes remains unknown. However, human homologues of Mss51, Cox14, and Cox25/Coa3 have been recently identified (96), and so far, the human homologue of Cox14 (C12orf62) has been suggested to coordinate the early steps of COX assembly with the synthesis of COX1 (103).
On a second mechanism, heme A biosynthesis is regulated by downstream events in the COX assembly process (8) to coordinate synthesis to its incorporation into Cox1 (Fig. 7). In most yeast COX mutants, there is a dramatic decrease on heme A steady-state levels. However, by overexpression of COX15 alone or in combination with the ferredoxin YAH1, heme A levels are significantly increased in these mutants, including some in which Cox1 is not synthesized (8). This suggested a feedback regulation of the heme A synthesis when the COX assembly process is stalled (8). In addition to low heme A synthesis, COX mutants also accumulate heme O, indicating that this molecule is stable. heme O levels were very low in Δcox15 cells, a phenotype that was not rescued by the overexpression of COX10, thus suggesting that the first step of the heme A biosynthesis pathway is positively regulated by Cox15 (8). It remains unknown whether increased heme A levels by COX15 overexpression in the absence of COX assembly bypasses Cox1 synthesis downregulation. If that was the case, we could envision a direct connection between COX1 translation and heme A biosynthesis regulations (Fig. 7). However, Δcox14 cells, whose Cox1 synthesis is not downregulated due to the instability of the Mss51-Cox1 early intermediate (5), have heme A and heme O levels similar to other COX mutants (8), thus suggesting that an increase in Cox1 synthesis does not promote heme A biosynthesis. The future identification of the COX assembly intermediate that operate in the regulation of heme A biosynthesis will allow better understanding of the mechanisms that control COX biogenesis to prevent the formation of pro-oxidant intermediates.
Concluding Remarks
Recent investigations have helped defining a substantial network of ROS- and redox-dependent or -regulated processes that operate in the assembly of functional and efficient COX. To further our understanding of these processes, future studies will need to elucidate: (i) the ROS-induced mechanisms of COX subunit isoform switch in yeast and mammals and its impact in cellular physiology; (ii) the effects of ROS in COX assembly into supercomplexes; (iii) how the redox environment in the IMS modulates the function of the Mia40/Erv1 import and oxidative folding pathway and whether the small CX9C proteins play a role in redox control in the IMS; (iv) how copper is transported from the matrix to the IMS and subsequently to Cox17, whether it involves the CX9C proteins, and how the redox environment regulates the process; and (v) how COX assembly-dependent regulation of Cox1 synthesis and heme A biosynthesis are precisely coordinated to prevent the formation/accumulation of pro-oxidant assembly intermediates.
Footnotes
Acknowledgments
Our study was supported by the Muscular Dystrophy Association to F.F. (grant number 158547) and A.B. (grant number 186025), and by National Institutes of Health (NIH) grant GM071775 to A.B.