
Abstract
Introduction
One of the phenotypic hallmarks of cancer is defective metabolism, resulting in fundamental changes in the manner in which tumor cells produce energy. This defect centers around two major changes. First, it involves increased reliance on glycolysis as a means of producing reducing equivalents, termed the Warburg effect (46). Second, cancer cell metabolism is associated with a dysfunctional mitochondrial electron transport chain, which often results in the aberrant production of reactive oxygen species (ROS) (28, 37).
These metabolic defects often coincide with an atypical redox state that is characterized by an increase in the steady-state levels of free radicals, and a decrease in cellular redox buffering capacity. At the same time, the expression of antioxidant enzymes such as the mitochondrial manganese superoxide dismutase (SOD2) is also decreased in many forms of human cancer. This observation led Oberley and Buettner to put forth the free radical theory of cancer in the late 1970s (35). They speculated that the decreased expression of SOD2 increases the steady-state levels of free radicals which are observed in tumor cells, and serves as a “flywheel” to drive the manifestation of the malignant phenotype by creating genetic alterations. Notably, this model did not address the mechanisms underlying SOD2 down-regulation in cancer cells. A number of laboratories, including our own, have determined that epigenetic silencing plays a significant role in the loss of SOD2 in human cancer. These findings provide a rational foundation for the fusion of the epigenetic progenitor theory with the free radical theory of cancer: Epigenetic down-regulation of SOD2 as an early event can perpetuate a vicious cycle that favors carcinogenesis through downstream alterations in cellular metabolism, genetic stability, and epigenetic plasticity. Earlier, we extensively reviewed the linkages between metabolism and epigenetics in both carcinogenesis and development (5, 15, 16). In this review, we will specifically address epigenetic mechanisms underlying SOD2 transcriptional regulation, the metabolic consequences of SOD2 loss on cellular biology, and how these metabolic events promote further epigenetic instability during cancer development.
Superoxide: The Gateway ROS
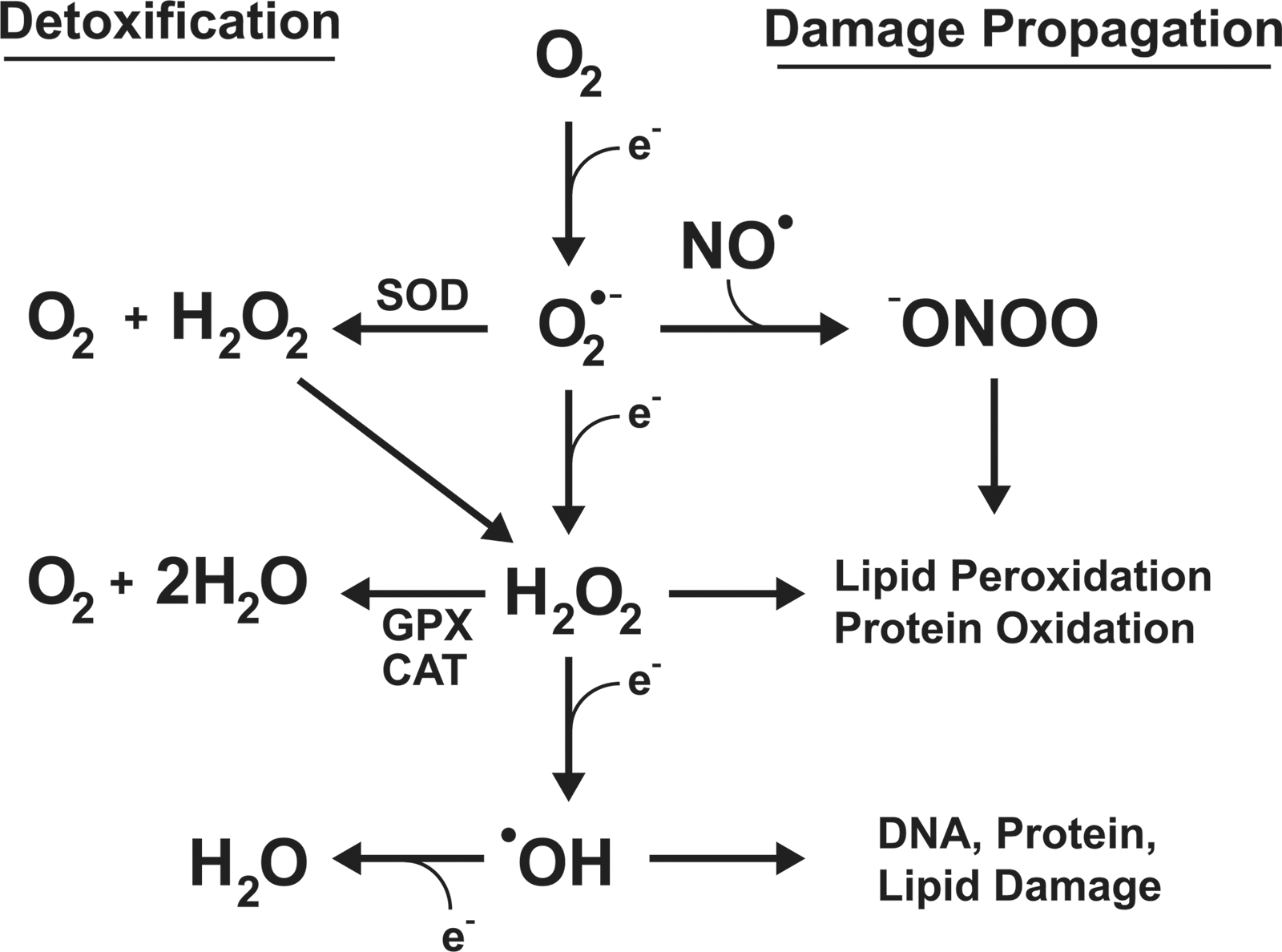
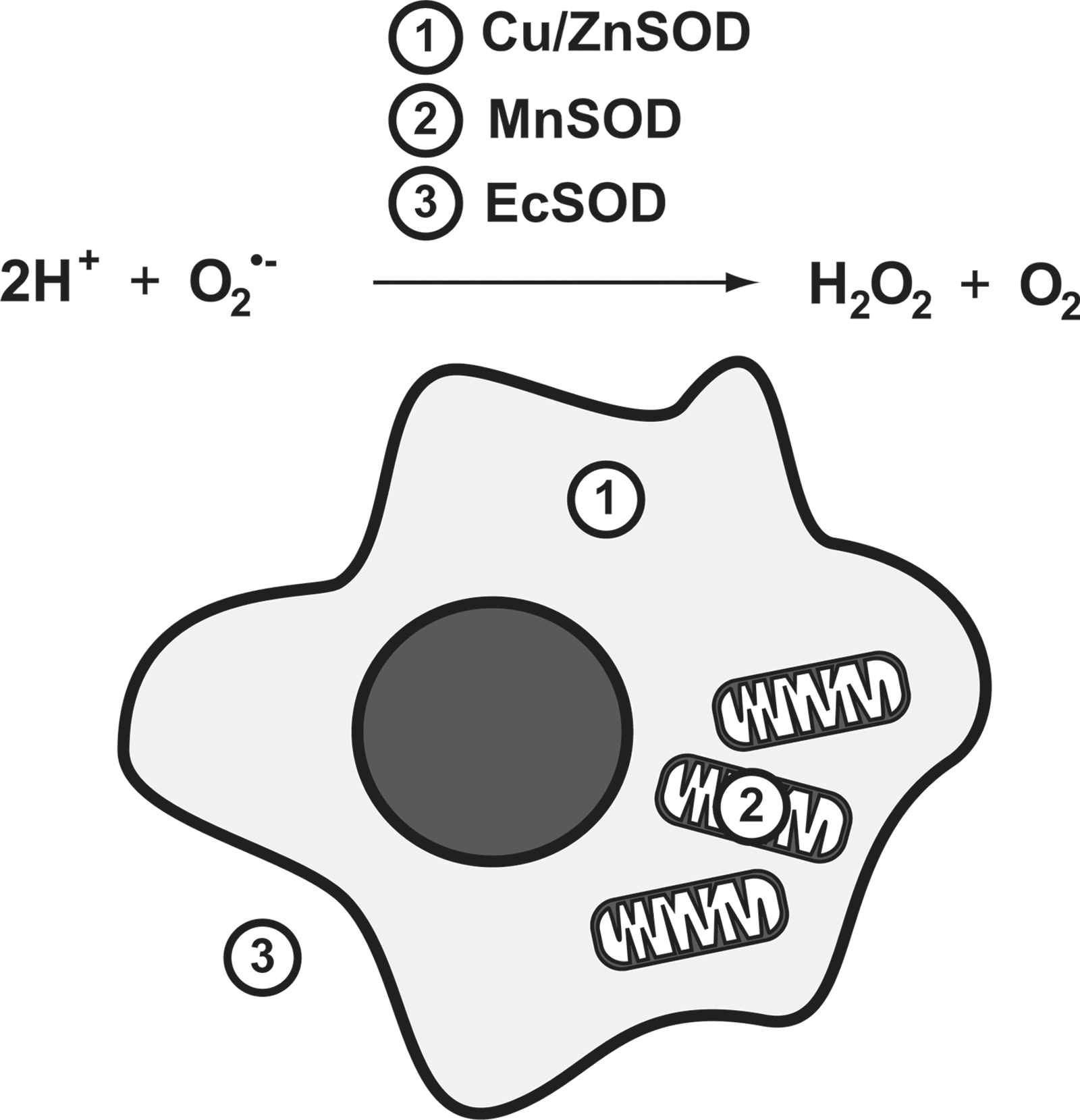
Molecular oxygen (O2), required for aerobic respiration, comes with significant risks for those organisms that have evolved to harness it. At ground state, O2 has two unpaired electrons, and can readily accept others. The electron transport chain of eukaryotic mitochondria takes advantage of this, using O2 as a terminal acceptor for the electrons from NADH and succinates once its energy has been used to generate a proton motive force. The stepwise reduction of O2 to H2O is highly regulated at complex IV, limiting the ability of harmful ROS to escape and damage cellular components. However, electron transport is not a perfect system, and leakage of electrons occurs. When this happens, O2 is rapidly reduced to the superoxide anion (O2 •−). While O2 •− is not an especially reactive agent itself, it serves as the “gateway” for the generation of considerably more ROS, including hydrogen peroxide, hydroxyl radical, and peroxynitrite (Fig. 1). It is, thus, imperative that cells have effective mechanisms for removing O2 •−, as the elimination of this relatively benign species can halt more severe downstream consequences. Humans have evolved three separate superoxide dismutases to address this: the cytoplasmic Cu/Zn SOD, encoded by SOD1, the mitochondrial manganese enzyme SOD2, encoded by SOD2, and the extracellular SOD, encoded by SOD3 (Fig. 2). For the remaining of this review, we will focus on the transcriptional regulation of SOD2 and its potential role in driving carcinogenic phenotypes. For an extensive review of the other human SOD genes, please refer the 2011 comprehensive review by Fukai and Ushio-Fukai (9).
SOD2 Gene Structure and Regulation
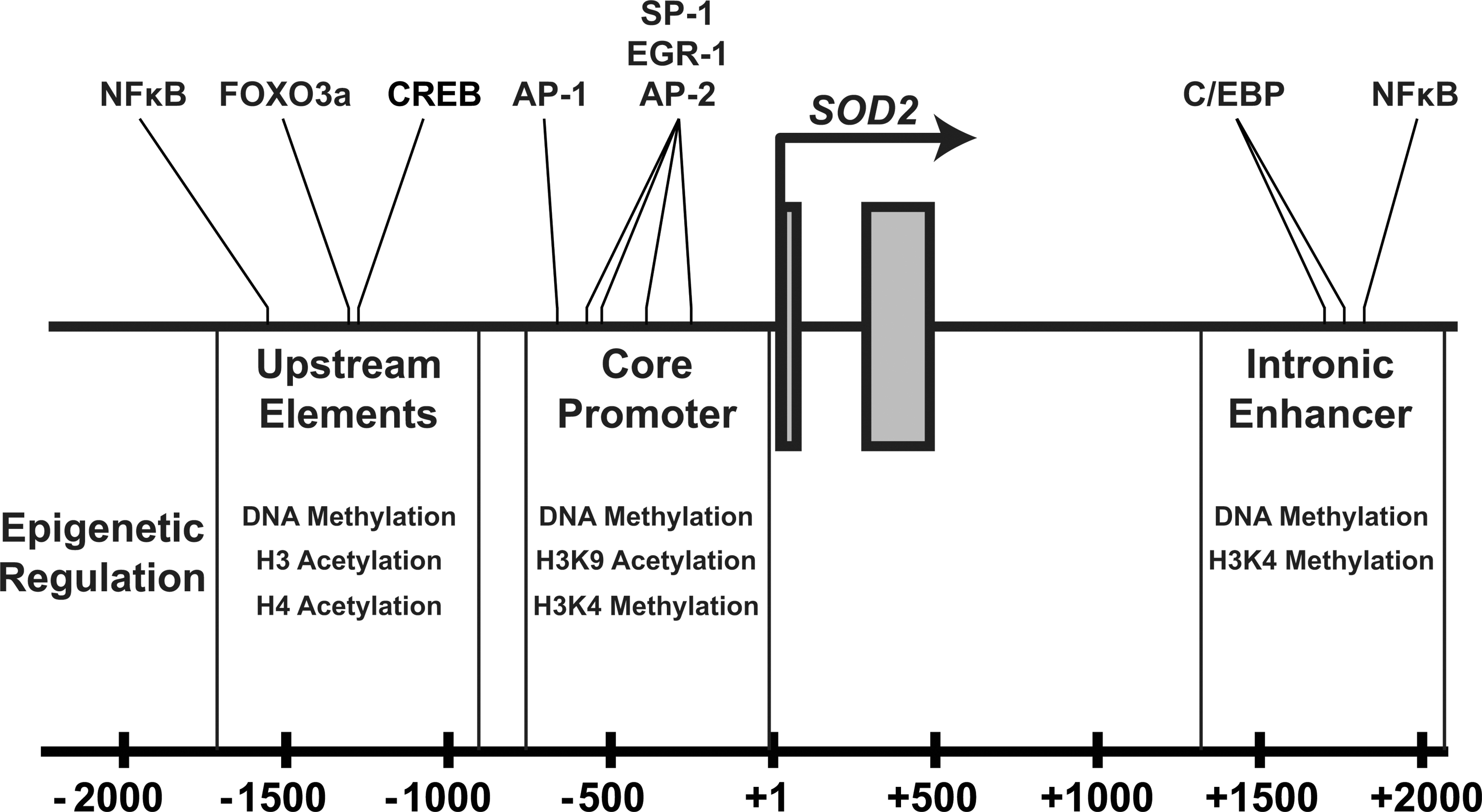
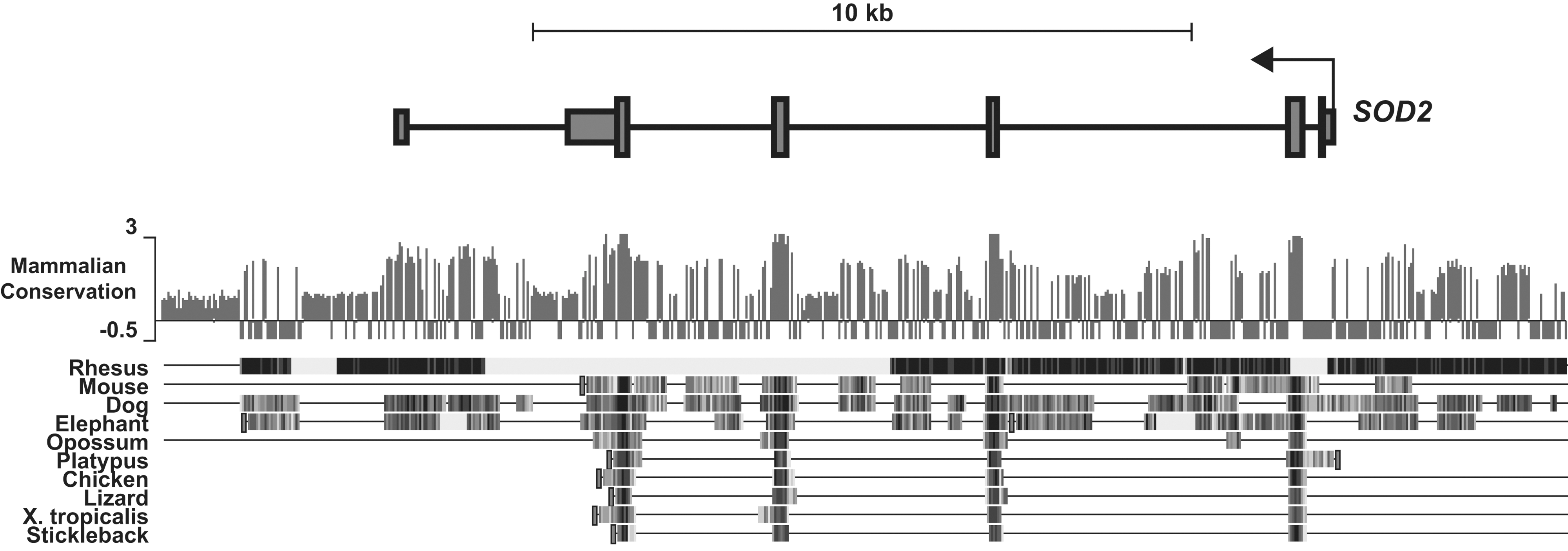
SOD2 has numerous important regulatory regions that contribute to its transcriptional activity. These can broadly be divided into three distinct elements: a GC-rich core promoter adjacent to the transcriptional start site, an enhancer in the second intron (∼1900 bp), and numerous upstream regulatory elements (∼−800 to −1500 bp) that are highly conserved in complex eukaryotes (Figs. 3 and 4). These diverse elements allow the dynamic regulation of SOD2 during changes in cellular metabolism, exogenous stimuli, or abiotic stresses such as ionizing radiation. Our group and others have established the potential for epigenetic control of SOD2's cis-regulatory elements compared with the last 15 years. To date, epigenetic alterations have been identified in all three of these regions in different cell types that exhibit decreased levels of SOD2 (Fig. 3). These epigenetic processes repress SOD2 by rendering one or more of the cis-regulatory elements transcriptionally inert. Other epigenetically silenced genes such as MASPIN and p16 completely lose transcriptional activity in early tumor development (4, 7, 10, 12), whereas SOD2 expression is merely repressed: Tumor cells do not completely lose SOD2 expression, but rather demonstrate less than one half of steady-state SOD2 compared with their normal counterparts (17). Later in carcinogenic progression, when tumor cells become invasive and metastatic, SOD2 expression is re-awakened, and it becomes over-expressed compared with its normal cell counterparts (6). This phenomenon has allowed us to critically evaluate the nature of epigenetic regulatory mechanisms at the SOD2 gene locus. Recently, the advent of next-generation sequencing technologies has enabled the determination of a comprehensive “epigenetic landscape” of SOD2 in various cell types, confirming many of these observations (Fig. 5) (39). Next, we will discuss each of these regions in the SOD2 gene and the fundamental ways in which epigenetic processes regulate their function in SOD2 expression.

The core promoter
The core promoter is the minimal element that is required for the basal expression of SOD2 in vivo. It contains the CG-rich TATA-less sequence that is common to many genes in the human genome (48). While the transcriptional regulation of TATA-less promoters remains enigmatic, the CG rich domain of SOD2 has been well characterized. The core promoter contains several cognate sites for various transcription factors, such as SP-1, EGR-1, and AP-2. Each of these transcription factors plays a role in regulating SOD2 expression in various tissues and model cell lines, and, thus, may contribute to decreased levels of SOD2 protein and activity in cancer. The core promoter is also rife with CpG dinucleotides that can serve as potential sites for DNA methylation. For example, our group has demonstrated that CpG methylation in the core promoter sequence can inhibit in vitro binding of AP-2 to its cognate sequence, potentially limiting its interaction with transcriptional machinery in vivo (21). The Farrar laboratory confirmed methylation of the core promoter using pancreatic cancer cell lines, and demonstrated that the administration of the DNA methyltransferase inhibitor zebularine can rescue SOD2 expression in methylated lines (19, 22). Furthermore, histone modifications in the core promoter region correlate with both chromatin accessibility and the binding of several of these transcription factors. Our group demonstrated in breast cancer cell lines that histone hypoacetylation and the loss of H3K4me2 marks is strongly associated with a loss of chromatin accessibility and decreased transcription factor binding at the core promoter, a phenomenon which was reversible with the administration of histone deacetylase (HDAC) inhibitors (17).
The intronic enhancer
SOD2 levels rapidly increase after exposure to cytokines in a process that requires collaboration between the core promoter and the intronic enhancer (25, 47). In humans, control of the intronic enhancer is mediated by the transcription factors NFκB and C/EBP. Mutations in NFκB binding completely abrogate SOD2 activation by TNFα, suggesting that a majority of the transcriptional activation of SOD2 by cytokines is mediated through NFκB signaling (43). Further studies from our laboratory and data from the ENCODE consortium have shown that this region of SOD2 contains high levels of H3K4me2, a histone modification which is often associated with enhancers (Fig. 5) (17, 39). In addition, work from Jeremy Boss's laboratory demonstrated that epigenetic crosstalk may exist between the intronic enhancer and core promoter. Stimulating cells with cytokines activates the intronic enhancer, simultaneously increasing the level of H3K9ac and facilitating the creation of an open chromatin conformation at the core promoter of SOD2 (13, 30, 38). Despite a thorough characterization, the exact mechanism by which the intronic enhancer influences the core promoter remains elusive. However, data from the ENCODE consortium demonstrate the binding of CCCTC binding factor (CTCF) at the intronic enhancer of SOD2 (Fig. 5). CTCF is an 11-zinc finger insulator protein that regulates communication between enhancer elements and promoter regions, and extensively participates in long-distance chromatin interactions with multiple functions (34). The residence of CTCF at the intronic enhancer of SOD2 suggests the tantalizing possibility that it could be playing a role in regulating concomitant chromatin remodeling at the core promoter after transcription binding.
Epigenetic alterations within the intronic enhancer also constitute a mechanism for epigenetic repression of SOD2. Our group was the first which reported that decreased levels of SOD2 protein and activity correlated with CpG methylation in the intronic enhancer in a fibroblast model (20, 21). Changes in histone modifications at the intronic enhancer also appear to correlate with SOD2 expression in breast cancer. Breast cancer cell lines with low SOD2 expression have decreased levels of H3K4me2 at the intronic enhancer, compared with nontumorigenic breast epithelial cell lines with higher SOD2 expression (17). These changes were associated with decreased binding of transcription factors SP1, AP-1, and NFκB to their respective binding sites, resulting in aberrant transcriptional regulation of SOD2.
Upstream elements
In addition to the regulatory capacity provided by the intronic enhancer and the core promoter, SOD2 has numerous upstream regulatory elements that also mediate canonical transcription factor interactions and can be altered through epigenetic means (Fig. 3). These regions include NFκB, CREB, and FOXO3a binding locations, and are important for the regulation of SOD2 in distinct times of cellular differentiation. For example, the FOXO3a binding site plays a critical role in the regulation of SOD2 during cellular quiescence, while the NFκB site plays a cytokine-responsive role during active growth (27). We have identified numerous epigenetic marks present at these upstream elements that regulate their effectiveness, including DNA methylation and H3 and H4 pan-acetylation (18).
Epigenetic Mechanisms and SOD2 Biology
Although a great deal is known about the structure and regulation of the SOD2 gene, a specific mechanism that explains its repression in many cancer types remains elusive. It has been reported that the regulatory regions of SOD2 are rarely mutated or deleted in human cancer (42), which strongly suggests that epigenetic mechanisms are playing a role in stemming SOD2 transcription. In addition, the biphasic nature of SOD2 expression during cancer progression, that is, decreased early in carcinogenesis and increased later in cells at invasive lesions, indicates a plasticity that hints of epigenetic regulation. The inherent complexity of epigenetic regulation makes it difficult to pin down specifically what may be taking place at the SOD2 locus and in what order it occurs, but we do know that lasting epigenetic derangements contribute to SOD2 down-regulation in cancer cells. What has become clearer is the nature of the epigenetic marks themselves, and how post-translational modifications to histones and covalent modifications of DNA are significantly more plastic than previously thought. Until recently, histone acetylation was the only well-characterized reversible histone mark, with known acetyltransferases and histone demethylase families. In contrast, histone methylation was once presumed to be a static mark that required the replacement of histones en bloc. Recent studies have identified entire new families of histone lysine demethylases that can selectively remove methyl groups from histone lysines in a stepwise fashion (33). Similarly, the ten-eleven-translocation (TET) family of nuclear proteins appears to be capable of actively demethylating DNA through stepwise oxidation of 5-methylcytosine, which has significant ramifications for our understanding of DNA methylation (24, 44). What makes these and other epigenetic mechanisms particularly interesting with regard to SOD2 biology is their inherent sensitivity to redox biology and metabolism—systems that rely heavily on SOD2 activity for appropriate cellular function.
Figure 6a broadly summarizes the mechanisms at play in governing histone modifications that demonstrably play a role in SOD2 transcriptional regulation. Multiple histone acetyltransferase families catalyze the transfer of the acetyl group from acetyl-CoA to a histone lysine, releasing coenzyme A as a byproduct. Acetylated histone lysines can, in turn, be deacetylated by four classes of HDACs: classes I, II, and IV share a similar mechanism that requires a catalytic metal (FeII or ZnII), while class III HDACs (sirtuins) consume NAD+ as a cofactor and produce nicotinamide and the enigmatic metabolite O-acetyl-ADP-ribose in removing acetyl groups. Similarly, multiple families of histone methyltransferases selectively recognize and methylate histone lysine residues, and two known mechanisms for histone lysine demethylation. Histone methyltransferases require S-adenosyl methionine (SAM) as a methyl donor, and produce the metabolite S-adenosyl homocysteine (SAH) as a byproduct. The histone demethylation reactions are catalyzed by the lysine-specific demethylase-type demethylases, which require flavin adenine dinucleotide as a cofactor, and the jumonji-C domain-containing demethylases, which belong to the broader 2-oxoglutarate-dependent dioxygenase superfamily. These proteins utilize 2-oxoglutarate and O2 in the presence of FeII, leading to hydroxylation of the lysine methyl group and subsequent oxidative demethylation. Critically, these proteins produce succinate and CO2 as reaction byproducts, and the buildup of succinate leads to a competitive inhibition of demethylase function both in vivo in yeast and in vitro with recombinant protein (41).

Figure 6b highlights the basic mechanisms governing the placement and modification of methyl groups on CpG dinucleotides in DNA. As with histone methyltransferases, DNA methyltransferases require SAM as a cofactor and produce SAH as a byproduct. The TET family proteins that can oxidatively demethylate DNA in vitro are mechanistically similar to the jmjC histone demethylases: They belong to the 2-oxoglutarate-dependent dioxygenase superfamily as well, and catalyze the hydroxylation of 5-methylcytosine to 5-hydroxymethylcytosine. While the significance of this modified base pair has yet to be explicitly determined, evidence suggests that it functions similar to hydroxymethyllysine intermediates in histone demethylation, in that it leads to the oxidative removal of methyl groups from the parent molecule (24, 44), validating a prediction in our earlier review (16).
Notably, in each of these reaction mechanisms, there are either critical metabolic intermediates that can be altered by SOD2 activity or mechanistic elements which can be directly damaged by O2 •−. This holds true for both the enzymes that establish and remove epigenetic marks, and is broadly summarized in Figure 7. Next, we will briefly discuss how alterations in SOD2 activity can alter histone methylation, histone acetylation, and DNA methylation.
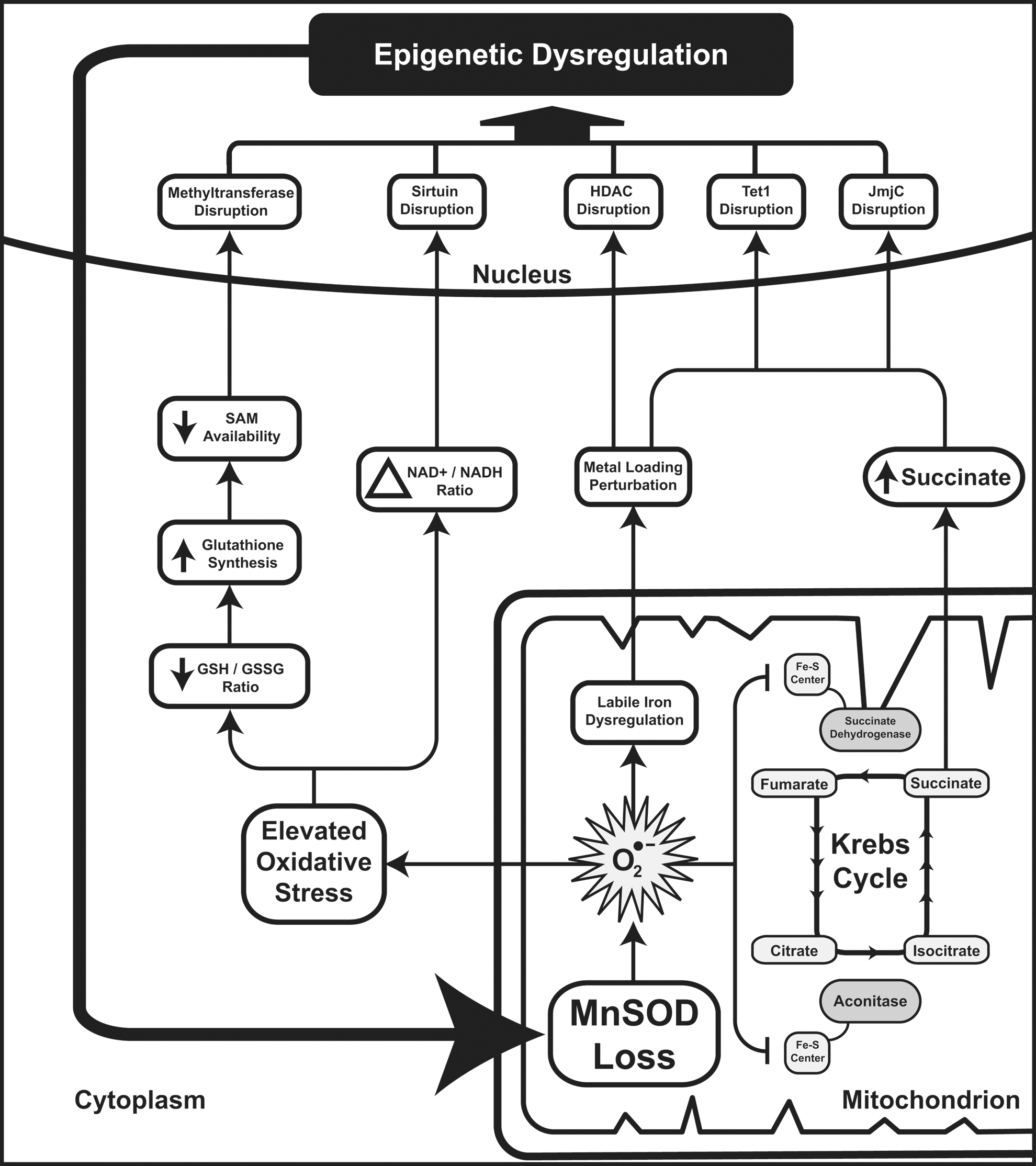
SOD2 and metabolic intermediates
SOD2 resides in the mitochondrial matrix, where it removes the O2 •− produced as an unfortunate consequence of oxidative respiration. Though O2 •− in and of itself is not the most harmful species (see Fig. 1), it is capable of interfering with proteins that depend on FeII for either structure or catalysis, particularly proteins containing Fe-S centers and some which contain heme prosthetic groups. HDAC classes I, II, and IV rely on ZnII canonically, though evidence suggests that FeII could be considered a more powerful agent for catalysis (11). This suggests that perturbations in Fe biology could have direct effects on HDAC functionality, leading to aberrant histone acetylation. Moreover, the Krebs cycle contains several Fe-S proteins, notably aconitase and succinate dehydrogenase (SDH), the latter of which also serves as complex II of the oxidative phosphorylation cascade. Indeed, reduction in these enzymatic activities may be considered a downstream marker of loss of SOD2 activity, because both aconitase and SDH are exquisitely sensitive to perturbation by O2 •− (32). Ultimately, the loss of these activities contributes to an abnormal flux of metabolic intermediates through the Krebs cycle. Loss of aconitase does not likely contribute significantly to perturbing Krebs cycle intermediate pools, as multiple anaplerotic pathways lead to the production of 2-oxoglutarate downstream of aconitase. However, the loss of SDH activity results in significant alterations in mitochondrial biochemistry. This is best characterized in SDH-deficient models, in which succinate measurably builds up in the setting of aberrant SDH subunit B expression in yeast (41). Over the years, many groups have explored the contribution of SDH activity to carcinogenesis, focusing primarily on the role of SDH in regulating the activity of the hypoxia inducible factor prolyl hydroxylases domain (PHD), which regulates the post-translational modification of HIF under normoxic conditions to promote its degradation (26). PHD enzymes belong to the same 2-oxoglutarate-dependent dioxygenase family as the TET and jmjC demethylase proteins, and are canonical “metabolic sensors” that govern HIF biology. Multiple groups have shown that PHD enzymes are perturbed by increased succinate and oxidative stress secondary to SDH activity loss, leading to a pseudo-hypoxic state in which HIF is stabilized in the absence of true hypoxia (1, 14, 40). In 2009, Cervera et al. demonstrated that these findings were extendable to the jmjC histone demethylases, showing that the abrogation of SDH activity through chemical or genetic means was sufficient to alter histone methylation patterns (3). To our knowledge, while this sort of product-level inhibition has not been demonstrated for TET proteins, we speculate that 5-hydroxymethlycytosine metabolism would be similarly perturbed by succinate buildup secondary to SDH loss, given the similarity in enzymatic function. All said, this leads to a plausible situation in which the loss of SOD2 activity would inhibit SDH activity, leading to a buildup of succinate and the subsequent inhibition of 2-oxoglutarate-dependent dioxygenases (Fig. 7, right side). This, in turn, would promote nuclear epigenetic aberrancies of both DNA and histone methylation, which could further limit SOD2 gene transcription.
The loss of SOD2 activity can also contribute to a number of additional cellular processes that can alter epigenetic regulation. Increased levels of O2 •− in the mitochondrial matrix would contribute to enhanced cellular oxidative stress by promoting the formation of downstream ROS. This, in turn, would likely alter the redox buffering capacity of the cell, affecting NAD+/NADH ratios as well as GSH/GSSG ratios. Alteration in the NAD+/NADH ratio directly alters sirtuin biology, leading to aberrant nuclear HDAC function (as well as numerous other cytosolic and mitochondrial deacetylase functions performed by sirtuins) (49). Similarly, a reduction in the GSH/GSSG ratio would be expected to indirectly affect the function of both histone and DNA methyltransferases by altering the pool of available cellular thiols. GSH biosynthesis requires cysteine, which is a downstream product of homocysteine metabolism. During times of oxidative stress, when GSSG cannot be efficiently recycled to GSH, cells export GSSG and synthesize new GSH, drawing vital thiols away from the SAM-SAH axis and decreasing the levels of SAM available for methyltransferase use (Fig. 7, left side).
When examining the potential downstream effects of SOD2 activity loss, it is apparent that multiple epigenetic sequelae are possible. In fact, unpublished data from recent work in our group demonstrates that the knockout of SOD2 in certain cell compartments produces significant global changes in histone biology. However, due to space constraints, we have not thoroughly discussed specific mechanisms for many of the epigenetic aberrations that could follow loss of SOD2 activity. For a significantly more detailed and comprehensive review of redox epigenetics, please refer our 2011 review on the topic (5).
SOD2: Spinning the Epigenetic Flywheel in Cancer
When Oberley and Buettner first postulated the free radical theory of cancer, they viewed SOD2 as a novel kind of tumor suppressor gene, one that did not necessarily perform canonical roles in signaling cascades or DNA damage repair pathways, but instead one which functioned more as the oil in an engine, keeping cellular metabolic machinery running smoothly and without damage. A loss of SOD2 activity, therefore, would cause damage to the metabolic core of the cell, leading to the metabolic and genetic derangements observed in cancer. The advent of Cre-LoxP recombination technology in murine models has enabled multiple groups, including our own, to test whether or not the absence of SOD2 expression is sufficient to suppress the development of tumors. To date, SOD2 has been knocked out in muscle (29), breast, nerve (31), kidney (36), liver (unpublished observations) (23, 45), T-cells (2), and bone marrow (manuscript in preparation). Although organ-specific disruptions in biology of varying severity and increases in cellular O2 •− were noted in each of these models, cancer was not among the noted pathologic outcomes in any of these models. Though this suggests that SOD2 does not function as a canonical tumor suppressor, these findings do not rule out the possibility that the loss of SOD2 activity contributes to carcinogenesis, at least as a tumor promoter, especially since the knockout strains described above were on the C57B/6 genetic background which is notoriously insensitive to carcinogenesis and discreet initiation regimens were not employed in these studies. Indeed, some groups have already demonstrated that total-animal SOD2 heterozygote models behave differently after initiating stimuli such as 7,12-dimethylbenz[α]anthracene and 12-O-tetradecanoylphorbol-13-acetate, producing tumor cells with differences in both apoptotic fraction and growth rates (50).
In this review, we have highlighted the epigenetic processes that are associated with SOD2 transcriptional biology, and discussed some of the ways in which SOD2 activity itself can interfere with the same epigenetic processes. As research into the redox basis of epigenetic modifications matures, we anticipate that more and more linkages between SOD2 function and epigenetic regulation will become apparent. This sets up a vicious cycle of deregulation—an epigenetic flywheel—that can continually turn from the moment SOD2 activity is altered in cancer cells. The initial loss of SOD2 activity promotes aberrant epigenetic events, which, in turn, promotes the alteration of SOD2 transcription and further modification of SOD2 activity. While we have focused primarily on the epigenetic regulation of SOD2, it is critical to note that these epigenetic changes occur throughout the genome. Broad modification of epigenetic events through loss of SOD2 activity has the potential to contribute significantly to the epigenetic plasticity that has increasingly been associated with human cancer. In the context of the epigenetic progenitor model of cancer, we can insert the SOD2-epigenetic regulatory axis framework as a driving mechanistic force that promotes the increasing epigenetic instability during carcinogenesis (Fig. 8). This combination of the free radical theory of cancer with the epigenetic progenitor model opens many tantalizing new avenues of research with regard to the role of SOD2 in carcinogenesis. Moreover, the advent of next-generation sequencing and systems biology-based approaches enables researchers to comprehensively examine the regulatory networks at play after the modification of SOD2 activity. This is especially important given the myriad downstream effects possible from the loss of this critical mitochondrial antioxidant.

To conclude, cancer metabolism, initially considered one of the primary driving forces for malignancy, largely took a backseat to genetic and other discrete models for carcinogenesis as our collective knowledge of molecular biology expanded. In recent years, interest in metabolic function in cancer cells has grown significantly, as molecular mechanisms for the metabolic phenomena described by Warburg have been established. SOD2 is a tantalizing target for future research, because as outlined earlier, it may functionally bridge the gap between metabolic and epigenetic events in tumor cells, driving tumorigenesis in a novel way. Further research using systems-based approaches to map the effects of SOD2 deregulation in tumor cells will shed more light on this exciting field and hopefully lead to well-reasoned interventions that can stem cancer development from the outset.
Footnotes
Acknowledgments
The authors thank Larry Oberley for his visionary insight and inspiration in the field of free radical cancer biology and the role of superoxide in cancer. Studies leading to the concepts underlying this work were supported in part by NIH R01 CA073612 and R01 CA115438. ARC received salary support form NIH F30 AA019856.