
Abstract
Introduction
Bipolar disorder (BD) is a mood disorder, which occurs with a prevalence of ∼2% and, similar to SZ, develops progressively often undetected during the premorbid phase with symptom onset during late adolescence. The disorder is characterized by alternating episodes of mania and depression, which can vary in frequency, duration, and severity. Symptoms can include affective disturbances (irritability, depression, and suicidality), behavioral changes (psychomotor agitation), psychotic symptoms (delusions and hallucinations), as well as cognitive impairments (attentional and judgemental deficits).
SZ and BD are categorized as two distinct disorders in diagnostic manuals, such as the current diagnostic and statistical manual of mental disorders (DSM-IV). However, in the clinic, it is often difficult to distinguish both disorders. It is rather common that patients with SZ have affective symptoms, such as depression or mania, while bipolar patients express psychotic symptoms, such as delusions and hallucinations. This symptomatic overlap could be one of the reason for the delay of months and sometimes years between the occurrence of first psychotic or manic symptoms and the diagnosis of SZ or BD (97). In support of the clinical observations, research is now accumulating evidence for commonalities between both disorders. The typical onset of both disorders is during adolescence or young adulthood, with an earlier onset in men than in women. Moreover, there is a substantial comorbidity index between the spectrum of SZ and BD (69). Both diseases are proposed to be neurodevelopmental disorders with aberrant brain connectivity occuring long before symptoms appear (74, 96). It is becoming evident that SZ and BD share genetic, pathological, and epidemiological characteristics. Both SZ and BD are considered complex diseases of polygenic and environmental origin. A large number of vulnerability genes are common to both diseases (i.e., disrupted-in-schizophrenia 1 [DISC1], brain-derived neurotrophic factor [BDNF], catechol-O-methyltransferase [COMT], and neuregulin 1 [NRG1]) (20, 79, 102). Shared neuropathologies include abnormal myelination (67, 78, 117), impairment of GABAergic interneurons (73, 94), dysregulated HPA axis (68, 122), mitochondrial dysfunction (24), and signs of pro-inflammatory processes (68, 86).
In spite of all these etio-pathological similarities, some clinical manifestations are more characteristic of one or the other disorder. The literature strongly suggests that patients with SZ display relatively global cognitive impairments (83). Some of these impairments may result from early developmental problems, since impairments in cognition, language, and motor performance and also in social, emotional, and behavioral capabilities are already present in children, who will later develop SZ. Interestingly, premorbid neuropsychological deficits are found in a substantial proportion of children who later develop SZ, especially in the SZ with family history subgroup, but less so in BP, suggesting especially impaired neurodevelopment underlying cognition in pre-SZ children (104). In patients with BD, cognitive performance is generally better than in SZ patients (105). Furthermore, the course of cognitive deficits differ between the two diseases. In SZ patients, cognitive impairments precede the onset of the illness and worsen during the prodromal stage and the first years following diagnosis. By contrast, cognitive deficits in bipolar disease appear mostly by the first episode and become more severe as symptoms worsen (72). On the other hand, affective disturbances, hyperactivity, and alterations of the sleep–wake cycle are more frequent and severe in BD patients, although they are also present to some degree in SZ.
The pathophysiology of both disorders is highly diffuse and heterogeneous among patients, emphasizing the importance of the identification of common final pathways. Our group and others [see reviews (33, 128)] have suggested that increased levels of oxidative stress and impaired antioxidant systems, including a deficit in glutathione (GSH), may represent one of these common final pathways upon which risk factors converge and from which established pathophysiological impairments originate. Here we review behavioral, morphological, electrophysiological, and neurochemical evidence from experimental rodent models supporting the concept that redox dysregulation due to GSH deficit may play an important role in both disorders. As presented below, these effects are quite different from those implicating oxidative stress in neurodegenerative diseases.
GSH Deficit and Redox Dysregulation in SZ and BD
The tripeptide GSH (γ-glutamyl-cysteine-glycine) is the main intracellular nonprotein antioxidant and redox regulator (see Fig. 1 for the GSH synthesis pathway and redox cycle). GSH is crucial for the protection against cellular oxidative damage caused by reactive oxygen species (ROS) and reactive nitrogen species (RNS) and for the neutralization of toxins and reactive metabolites. It is further required for the maintenance of the thiol redox status, which modulates redox-sensitive processes, such as cell cycle regulation and cell differentiation, receptor activation, signal transduction, and the binding of transcription factors to DNA. A deficit in GSH can result in oxidative stress and subsequent oxidation of lipids, proteins, and DNA. Therefore, a redox dysregulation due to GSH deficit could alter many biological processes resulting in abnormal brain development and function.
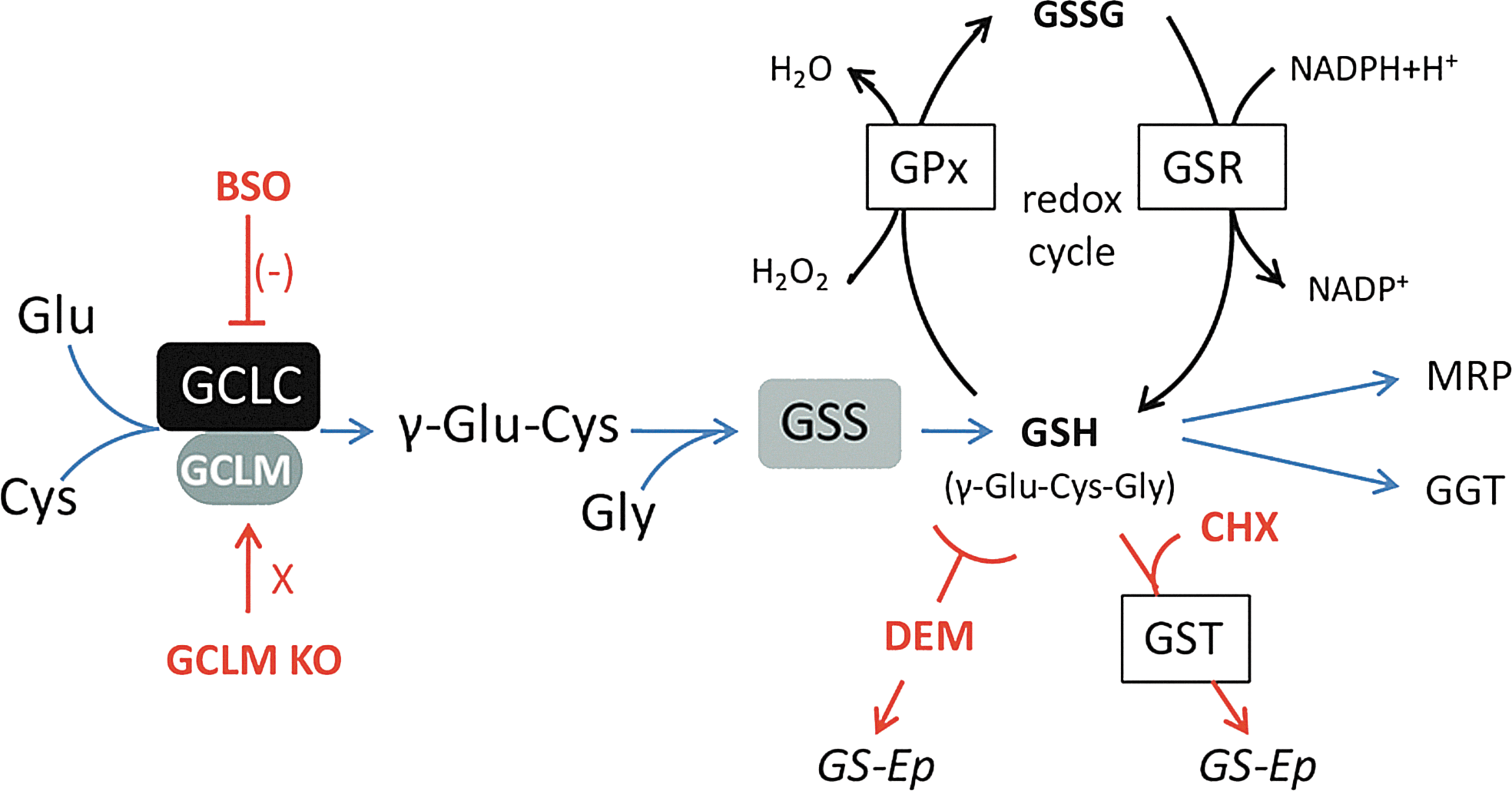
What is the evidence for a dysfunctional GSH system in SZ and BD? In SZ patients, GSH levels are decreased in the cerebrospinal fluid. Moreover, magnetic resonance spectroscopy (MRS) studies have shown decreased GSH levels in vivo in the prefrontal cortex (34) as well as in the postmortem prefrontal cortex (42) and the striatum (127). Interestingly, lower levels of GSH in the frontal cortex of SZ patients correlate with a greater negative symptom severity (82). Importantly, variants of the genes for both modulatory (GCLM) and catalytic (GCLC) subunits of the key GSH-synthesizing enzyme glutamate cysteine ligase (GCL) are significantly associated with SZ in a Swiss and Danish cohort (50, 118). Accordingly, cultured skin fibroblasts of Swiss individuals expressing high-risk GAG-trinucleotide repeat (TNR) polymorphisms in the GCLC gene have decreased the GCLC protein expression, GCL activity, and GSH content under oxidative stress condition compared to fibroblasts with low-risk GCLC genotypes (50). Two studies on Japanese cohorts did not find an association of GCLM or GCLC with SZ (52, 82). However, in the study by Hanzawa et al. (52), the six genotypes related to the GAG-TNR polymorphisms of the GCLC gene have only been tagged with various single-nucleotide polymorphisms (SNPs). In a Danish subset of the SGENE cohort, no SNP haplotype has been found to be a perfect surrogate for any of the repeat lengths (see Table 1). Thus, a standard methodology (50) should be used to correctly genotype these TNR as they cannot be derived from available SNP data from a genome-wide association study (GWAS). Polymorphisms and copy number variations of genes coding for glutathione S-transferases (GST), which catalyse the reaction of GSH with electrophilic and hydrophobic compounds, have also been found associated with SZ (48, 100). Postmortem studies on brains of SZ patients have, moreover, reported decreased activity of enzymes associated with the GSH system, such as glutathione peroxidase and reductase (GPx and GSR, respectively) (127).
In a Danish sample genotyped on the Illumina HumanHap610-Quad platform (56), three markers show strong LD to the GCLC GAG-TNR polymorphism, which was genotyped using standard procedure (50). A haplotype analysis of the roughly 650 samples typed for both the SNP array and the GCLC repeat using the NEMO algorithm (49) shows that there are five main haplotypes (>5%) in the LD region based on the TNR and two of the three SNPs; rs4712036 and rs7738142 (adding the third SNP, rs510088, further breaks down the haplotype structure with respect to the TNR polymorphism). Allele C of rs4712036 is mostly found together with nine GAG repeats, and allele T of rs7738142 is almost exclusively found together with seven GAG repeats. None of the three SNP haplotypes, however, are perfect surrogates for any of the respective GCLC GAG-TNR alleles, and the only allele with a predictive value is C of rs4712036, which predicts the nine repeats GAG-TNR allele with ∼80% specificity (four out of five chromosomes with C-rs4712036 have the nine repeats allele) and ∼80% sensitivity (four out of five chromosomes with the nine repeats allele have C-rs4712036 as well). There is a slight excess of high repeat number (8–9) in patients compared to controls.
GCL, glutamate cysteine ligase; GCLC, GCL catalytic subunit; LD, linkage disequilibrium; SNP, single-nucleotide polymorphism; TNR, trinucleotide repeat.
There is also converging evidence for increased oxidative stress/damage and redox imbalance in BD [see reviews (3, 11)]. Similar to SZ, GSH levels are decreased in the postmortem prefrontal cortex of BD patients (42). Moreover, lipid peroxidation levels are elevated in the BD anterior cingulate cortex (ACC) (123). To our knowledge, only one study has so far looked at oxidative pathway genes in BD patients. This study reports no association of GCLC or GCLM, but finds haplotypes in the superoxide dismutase 2 (SOD2) gene associated with BD (39).
Finally, as proof of concept, clinical trials have shown that the GSH precursor and antioxidant N-acetylcysteine (NAC), given as adjunct to antipsychotic medication, has positive effects in both SZ and BD. NAC improves negative symptoms and reduces side effects in chronic SZ patients (10). Moreover, NAC improves mismatch negativity (70), a preattentional, auditory-related, N-methyl-D-aspartate (NMDA)-dependent evoked potential (120) typically impaired in SZ (60, 106). In BD patients, adjunctive NAC treatment causes a prominent reduction in depressive symptoms, as well as improvement in general functioning and quality of life (11). One study in a small sample of bipolar II patients has shown that adjunctive NAC treatment is associated with significantly more full remissions of depressive and manic symptoms compared to placebo (77).
Together, these results provide evidence for a dysregulated redox system and increased levels of oxidative stress that could contribute to the pathophysiology of both SZ and BD (see Fig. 2).

Disease-Relevant Consequences of a GSH Deficit in Animal Models
To study the consequences of GSH dysregulation in experimental models, we are using a reverse-translational approach, moving from studies in patients to understanding how a given risk gene alters brain development and function in rodents and how it relates to specific disease phenotypes. Gene variants of both GCLM and GCLC are associated with greater risk for SZ (50, 118). Modification or deletion of one of these two genes in a mouse line would, therefore, provide a model with high construct validity. Deletion of GCLC in mice is lethal at early embryonic state due to a total loss of GCL function (27). By contrast, mice knockout (KO) for GCLM (responsible for the regulation, but not catalytic function of GCL) (126) are viable and display a permanent decrease in brain GSH levels of 70% to 80% (36, 111). Thus, GCLM-KO mice represent a good model to identify and study the anomalies induced by a GSH deficit as observed in patients suffering from either SZ or BD.
Another approach to study the effects of GSH deficit that we and others have used is to deplete GSH pharmacologically. L-buthionine-(S,R)-sulfoximine (BSO) is the most specific GSH-depleting pharmacological agent, which selectively inhibits the GCL enzyme activity resulting in a significant, but reversible GSH decrease. The electrophile compounds, diethylmaleate (DEM) and 2-cyclohexene-1-one (CHX), represent another class of pharmacological agents that induce GSH deficit. Both conjugate to GSH directly or via GST. In the process of detoxifying these compounds, GSH is rapidly depleted. These pharmacological models can be used to explore the impact of transient rather than permanent GSH deficits. The fact that GSH levels rapidly normalize following treatment cessation, allows investigating the acute and long-term effects of transient GSH deficit during defined developmental periods, thus indicating which periods are particularly vulnerable to GSH deficit/oxidative stress.
To model human disease conditions more closely, some studies have introduced additional manipulations. Some have used rats of the osteogenic-disorder Shionogi (ODS) strain. In normal rats or mice, a GSH deficit results in a compensatory increase of ascorbic acid synthesis. ODS rats, similar to humans, cannot synthesize ascorbic acid to partially compensate for a GSH deficit; these rats can only rely on a daily uptake of ascorbic acid that is contained in food. In other studies, dopamine (DA) itself or the DA reuptake inhibitor GBR12909 (GBR) have been administered with the idea to partially mimic environmental stress, which stimulates the stress axis and increases the DA release (84). This is in keeping with the conception of SZ and BD as complex disorders resulting from the interplay of endogenous vulnerabilities and environmental stressors. Importantly, DA metabolism generates ROS and reactive quinones (54) that are neutralized by GSH. Thus, GBR or DA administration increases the oxidative burden similar to an exogenous insult, particularly when there is an innate deficit in GSH.
Clearly, it is impossible to entirely model SZ and BD in mice or rats. Positive symptoms, such as hallucinations and delusions, in particular, cannot be modeled in the rodent at all. However, laboratory mice and rats can be used to study a number of specific behavioral and cognitive domains that are relevant to both SZ and BD, including learning and memory processes, social, stress- and emotion-related behaviors. In addition, these models allow to explore morphological, physiological, and biochemical alterations likely to underly the behavioral deficits. Such models are also highly valuable to test potential therapeutic molecules.
Behavioral studies
Does a permanent GSH deficit induce behavioral or cognitive anomalies that resemble SZ or BD symptomatology? Permanent GSH deficit leaves the appearance and general status of GCLM-KO mice relatively unaltered compared to nonmutant wild-type (WT) mice except for a small, but significant decrease in body weight (126), an increased risk for cardiovascular problems (63), and reduced KO female fertility (90). Closer investigation reveals behavioral changes. In particular, GCLM-KO mice respond to stress with increased locomotion and exploration, and display more risky behaviors compared to WT mice. When placed for 30 min on an elevated platform (a 8-×8-cm platform positioned 1.20 m above the ground), pubertal (P28–42) KO mice under such a stressful situation actively jump off the platform, while WT mice crouch, freeze, or move very carefully on such a platform (unpublished observations). Young adult (P60–80) KO mice show hyperlocomotion in a bright open field arena, while their activity is normal in the home cage. In an elevated plus maze, adult GCLM-KO mice enter the open/unprotected arms more frequently than WT controls; however, the number of total arm entries is comparable to WT. In addition, young adult KO mice swim for a longer time than WT during the initial few minutes of a forced swim test when the cold water stress is strongest (66). These data suggest that mice with low GSH levels from gestation onward display, already at young age, altered behavior in unfamiliar and stressful situations. In novel and slightly aversive contexts, KO mice show increased locomotor activity and engage in more risky exploratory behavior than WT animals. At fully adult age (P120 and older), KO mice display, however, normal activity levels in the open field (25, unpublished observations) suggesting that open field hyperlocomotion is a transient phenotype that is only present in young animals. Fully adult KO mice spend more time in the aversive light compartment of the light–dark transition box and engage in more frequent light–dark transitions as compared to WT (111). Finally, at 120 days and older, KO mice also display less freezing (i.e., immobile posture representing fear) during the tone–shock conditioning period and during the retention test of a fear conditioning paradigm (111). Common to the behavior of KO mice at all ages is an altered response to novel and slightly stressful situations. KO mice show a stronger exploratory drive (elevated platforms, open field, light–dark box) and more risk-seeking behavior (elevated plus maze, light–dark box) than WT mice. These behavioral manifestations could be a reflection of reduced anxiety and/or an altered stress response system. Increased locomotion and exploratory drive may, in part, be similar to acute agitation and motor restlessness, a positive symptom observed in psychotic SZ and BD patients (1, 5).
It is well established that both SZ and BD patients have an altered stress response system. Patients of both disorders display increased stress reactivity (35, 51) and trait arousability (32), associated with increased plasma cortisol levels at resting state (31, 41, 57, 89, 101). Moreover, when pharmacologically challenged with a synthetic cortisol (dexamethasone [DEX]), medicated and nonmedicated SZ and BD patients display an attenuated hormonal negative feedback response (88, 124) indicating that the stress response system does not sufficiently downregulate cortisol to resting levels following a stressor exposure. Interestingly, unlike WT mice, GCLM-KO, which have been exposed to repeated mild stress during puberty (P35 to 42) also show a permanent increase in plasma glucocorticosterone (CORT; rodent cortisol equivalent) levels at rest. Furthermore, upon challenge with DEX, naive KO mice show an attenuated negative feedback response similar to that observed in patients (unpublished observation). Together, these data suggest that permanent GSH deficit in KO mice induces emotion- and stress-related behavioral and hormonal changes similar to those observed in SZ and BD patients.
Other disease-relevant behavioral domains are also affected by permanent GSH deficit. Compared to WT, GCLM-KO mice display a slight impaired sensory motor gating in the form of prepulse inhibition (PPI) of the acoustic startle reflex. PPI refers to the weakening of the startle response when the startle stimulus is shortly preceded by a weaker prepulse (PP) sound. PPI is a preattentional gating mechanism that is hypothesized to prevent sensory overload and cognitive fragmentation. KO mice show a PPI impairment only when a low PP intensity is used (69 dB) (66). This selective PPI impairment at a low PP intensity has been observed by our group in separate cohorts of fully adult animals, including one cohort of animals that had been exposed to repeated mild stress during the peri-pubertal period (P28–41). Others have not observed a PPI deficit in GCLM-KO mice (25). This discrepancy may be due to the fact that Cole et al. (25) used a higher PP intensity at which GCLM-KO mice show also close-to-normal PPI in our own study (66). In SZ, reduced PPI has been associated with thought disorder, distractibility, and a lower overall level of function (121). PPI deficits occur not only in SZ (43) and BD (46, 44) patients, but also in their unaffected siblings (44) and first degree relatives (17) suggesting that PPI deficits are at least, in part, genetically mediated.
GCLM-KO mice also display an increased locomotor response to a low-dose amphetamine injection (66). It has been shown that both, SZ and manic symptoms can be induced or exaggerated by amphetamine and cocaine (58, 65). Moreover, it has been demonstrated that the psychoactive drug L-dopa can switch BD patients from a depressive to a manic state (47).
So far, we have discussed behavioral phenotypes that are observed in patients of both disorders. A recent study has compared the behavior of SZ and manic BD patients in a human open field/novel object exploration paradigm (95). Locomotion in an unfamiliar room is elevated in SZ patients and remains high throughout the test period. Manic patients show initially an even stronger hyperlocomotion than SZ patients, but in contrast to SZ patients display a steady decline (habituation) of activity over time. In addition to hyperlocomotion, manic, but not SZ patients show highly increased object exploration, which also habituates over time. Intriguingly, the exploratory behavior of KO mice very closely mimics that of manic patients: when exposed to three unfamiliar objects within a novel environment, GCLM-KO mice show hyperactivity and extensive object interactions. Both, activity and exploration, habituate to the WT level following repeated exposures (111). This is consistent with hyperlocomotion in the open field observed in young adult KO mice, which also habituates. This suggests that this particular subphenotype of GCLM-KO mice resembles the general phenomenon of increased locomotor and exploratory activity in novel contexts that is expressed by patients of both disorders, but more closely mimics the distinct temporal pattern of exploratory behavior of manic BD patients.
Besides these altered behavioral manifestations, we and others have observed that a number of cognitive capacities remain largely intact in GCLM-KO mice, particularly in the domain of spatial cognition. Respectively, KO mice display normal short-term memory of previously visited arms in the Y-maze and in the baited T-maze, normal learning and memory of a hidden platform position in the water maze. In addition, KO mice have an intact short-term memory of object positions (memory for place), while they show impairment in a novel object recognition task (memory for object) (25, 66, 111). This strongly suggests that the circuitry involved in spatial aspects of cognition is relatively resistant to permanent GSH deficit. However, this contrasts with the spatial learning and memory impairments observed following a transient GSH deficit induced pharmacologically (see next chapter and also Tables 2 and 3 for comparison).
This table summarizes the behavioural and cognitive domains for which the phenotype of GCLM-KO mice has been assessed. Tests and the respective outcomes are listed for the investigated age. Note that spatial learning and memory are intact in GCLM-KO mice.
FC, fear conditioning; GCLM, GCL modulatory subunit; KO, knockout.
This table summarizes the behavioural and cognitive effects of transient GSH deficit during early postnatal development or adult age with or without additional hyperdopaminergia. Note that the phenotype changes depending on the age tested. Note also that spatial learning and memory are disturbed following early postnatal but also adult GSH deficit.
AMPH, amphetamine; BSO, L-buthionine-(S,R)-sulfoximine; CHX, 2-cyclohexene-1-one; DA, dopamine; DEM, diethylmaleate; GBR, GBR12909; GSH, glutathione; NAC, N-acetylcysteine; ODS, osteogenic-disorder Shionogi; PCP, phencyclidine; WT, wildtype.
Altogether, behavioral anomalies identified in GCLM-KO mice are consistent with symptoms of both SZ and BP. These mice show an enhanced response to psychostimulant, slight impaired PPI, and social behavior (66). One remarkable phenotype is an increased exploratory activity under novel and stressful contexts leading to more risky behaviors. This suggests reduced anxiety and/or reduced behavioral inhibition that could resemble mania manifestations. By contrast, these mice do not show despair behavior and have intact spatial learning and memory. As we shall see below, severe transient stresses leading to strong and acute redox dysregulation might be more critical in inducing cognitive deficits than a chronic redox dysregulation as in GCLM-KO mice.
How does transient GSH deficit during early postnatal life or adulthood affect behavior and cognition? BSO-induced GSH deficit from postnatal day (P) 5 to 16 does not affect the general physical status of the animals (21, 22). The main effect of early BSO treatment in pubertal (P26 to 34) ODS rats, which cannot synthesize ascorbic acid, is an impaired spatial working memory performance in the water maze. BSO-treated ODS rats persistently require more time than controls to find a hidden platform. This deficit is observed 10 days after cessation of BSO treatment when GSH levels have just returned to the physiological level, but not at the adult age (16). This suggests that early GSH deficit delays the maturation of the circuitry necessary for the successful performance in the water maze (99). At a young adult age (P65 to 90), ODS rats treated with BSO during postnatal early life show impaired memory for familiar objects (22). This impairment is more pronounced in rats that received simultaneous postnatal GBR-injections to induce hyperdopaminergia, which results in increased oxidative burden via DA catabolism (21). Five-month-old WT or ODS rats exposed to early BSO treatment display also significant deficits in place discrimination when performance depends on the integration of multiple distributed olfactory and visual cues (16). Interestingly, deficits in multisensory integration are also well described in SZ patients (29, 30, 125) and correlate with a greater negative symptom severity and the presence of multisensory (as opposed to uni-sensory) hallucinations (125). Equivalent studies with bipolar patients are lacking. Together, these results show that the transient GSH deficit during early postnatal life induces short- and long-term cognitive alterations. Further studies are required to investigate the consequences of GSH deficit at different developmental time points to specify vulnerable periods. It is plausible that a redox dysregulation occurring at different times of development could differently alter brain development and function resulting in distinct behavioral phenotypes.
Finally, several studies demonstrate that a transient GSH deficit in the adult age can interfere with normal behavior and cognition. Acute intracerebroventricular injections of BSO in adult mice impair spatial short-term memory performance in the Y-maze (80), decrease the locomotor response to mid-range doses of amphetamine (59), and impair memory of familiar objects (80). In mice or rats, systemic administration of CHX or DEM results in spatial learning and memory deficits in the Y- and the water maze, respectively (23, 26, 28). When adult BSO treatment is combined with hyperdopaminergia, it impairs locomotor coordination and also spatial learning in the water maze (107, 108).
Apart from cognitive function, behavioral domains, such as stress and emotional reactivity, social capacity and PPI have not been investigated in BSO-treated rats or mice making it difficult to fully compare genetic and pharmacological models. However, deficit in object recognition/memory are observed in both GCLM-KO mice (111) and mice and rats treated with BSO either during early postnatal development or during adulthood (22, 80). This strongly suggests that the capacity and the circuitry needed to encode and/or remember object-specific information is, particularly, vulnerable to oxidative stress/redox dysregulation. Indeed, object memory deficits in GSH-deficient rodents translate well to object recognition impairments observed in a large percentage of SZ (18, 40, 71) and BD patients (45).
However, while rats or mice with a transient GSH deficit either during early development or adulthood have difficulties in spatial information processing and storage (Table 3), KO mice display intact spatial learning and memory (Table 2). These apparent contradictory data may be due to different factors. In contrast with animals with a transient GSH deficit, GCLM-KO mice might be able to maintain or acquire normal spatial cognitive functions via compensatory mechanisms that cannot fully operate in mice or rats following an acute GSH deficit. On the other hand, a strong oxidative stress induced by BSO+GBR treatment during early development might impact more severely the brain structures implicated in spatial cognitive function than the chronic redox dysregulation in GCLM-KO mice. It, thus, can be speculated that the emergence of cognitive deficits associated with a deficit in GSH might be related to the timing and severity of additional insults. Further investigations on the impact of additional stress in GCLM-KO mice should allow clarifying this point and determine whether differences in cognitive impairment between BP and SZ could be related to such environmental factors.
Morphological and electrophysiological studies
The brains of both SZ and BD patients are characterized by prominent GABA dysfunction [see reviews (7, 73)]. Among GABA inhibitory interneurons, those that express the calcium-binding protein parvalbumin (PV) seem to be particularly affected in both diseases. Accordingly, a decrease in glutamate decarboxylase 67 (GAD-67) occurs primarily in PV-positive interneurons (53). Cell count studies on postmortem tissue have revealed reduced numbers of PV-immunoreactive interneurons in prefrontal areas, including the ACC and in the hippocampus (9, 8, 130). Proper functioning of PV interneurons is crucial for the tight control of the firing rate of pyramidal neurons and for the initiation of neuronal network synchronization in the gamma range. It is, therefore, likely that the observed deficits in gamma oscillations and the associated cognitive impairments in SZ and BD are closely related to PV interneuron deficits (93, 119).
Could permament GSH deficit be one causal factor contributing to GABA deficits? To answer this question, we have concentrated on the mouse hippocampus and the ACC because structural and functional anomalies in these two structures are consistently observed in both SZ and BD (12, 37, 91, 103). We observe PV impairment in both structures. In GCLM-KO mice, the integrity and function of PV interneurons is selectively impaired in the ventral, but not the dorsal hippocampus (VH and DH, respectively; see Fig. 3) (111). Based on cytoarchitechtonical and functional differences, the hippocampus can be divided along the rostrocaudal axis into the ventral, intermediate, and dorsal portions (see Fig. 3). The DH receives highly processed sensory information from sensory cortical areas (87). DH lesions, accordingly, result in spatial learning and memory deficits (4). The VH has connections to the prefrontal cortex, amygdala, and nucleus accumbens (98) and is implicated in the regulation of the stress response and emotional and affective states (81). In GCLM-KO mice, PV immunoreactivity is decreased in the adult dentate gyrus and the CA3 area of the VH, but not DH. The PV impairment in VH starts to appear at P40 with a concomitant rise in oxidative stress markers and is associated with reduced gamma oscillations at the adult age. The region selective impairment of the VH, but not the DH indicates that interneurons in the VH are more susceptible to oxidative stress or alternatively that the oxidative burden in VH is higher compared to the DH. This might be attributed, among others, to rich catecholamine innervation of the VH, which is associated with increased oxidant production (111). The morphological and functional impairments selectively of the VH, but not the DH are well in line with the behavioral/cognitive phenotype. Respectively, GCLM-KO mice display altered emotion- and stress-related behaviors (111), as well as novelty- and amphetamine-induced hyperlocomotion and PPI deficit (66), which are associated with VH function. In contrast, KO mice show intact DH-associated spatial learning and memory.
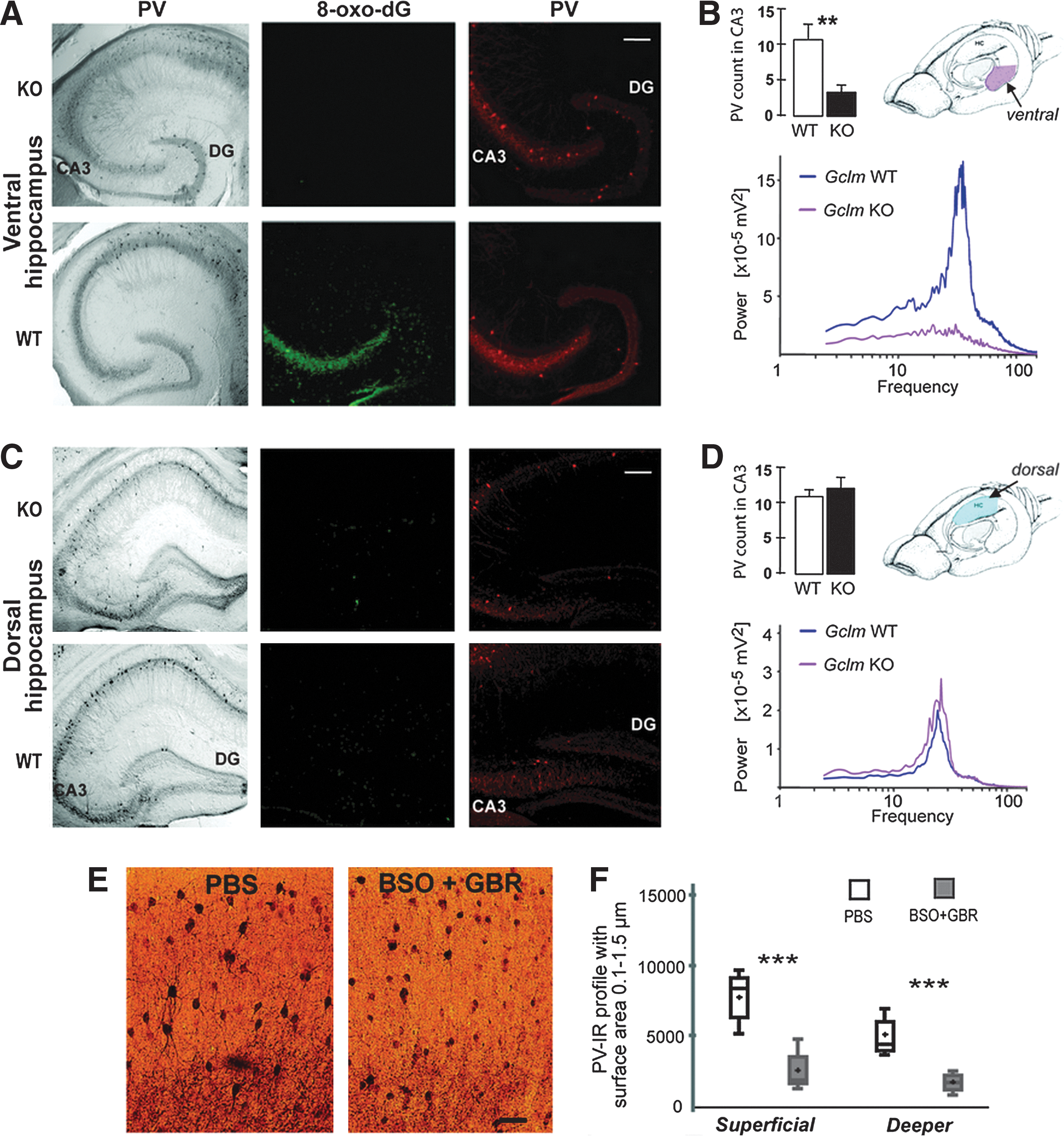
Ongoing experiments also indicate signs for PV impairments in the ACC of GCLM-KO mice (112). Together, these data indicate that a chronic GSH deficit impairs PV interneurons in brain structures known to be particularly affected in both SZ and BP.
Could a transient GSH deficit during development be one causal factor contributing to GABA deficits? BSO-induced GSH deficit from postnatal day (P) 5 to 16 affects PV-positive interneurons in the ACC of ODS rats as demonstrated by a reduced number of PV-positive processes radiating from the somata (15). Additional GBR treatment augments this effect. Postnatal BSO+GBR treatment leads, moreover, to a drastic decrease in the number of small PV-positive profiles in superficial and deeper layers of the ACC, corresponding to PV-positive synaptic boutons and dendritic and/or axonal arborization (see Fig. 3). These morphological anomalies of PV interneurons induced by a transient postnatal GSH deficit are permanent; that is, they are observable long after GSH levels have returned to the normal physiological level. Thus, at adult age P90, BSO− and BSO+GBR-treated rats have significantly fewer numbers of PV-immunoreactive interneurons than phosphate-buffered saline (PBS)-treated rats (unpublished observations).
Together, data from GCLM-KO mice and BSO-treated rats (15, 111) suggest that a GSH deficit affects selectively PV interneurons, while other subpopulations of GABAergic interneurons (e.g., expressing calbindin and calretinin) remain unaltered, showing that PV cells seem to be particularly sensitive to GSH deficit and oxidative stress. Further evidence comes from the ketamine rodent model of SZ. Ketamine-induced activation of the enzyme, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, results in the production of superoxide radicals, which are directly associated with PV interneuron dysfunction and loss of PV expression (6, 109). Together, these observations strongly suggest that PV impairment in SZ and BD patients can be, in part, mediated by a GSH deficit/redox dysregulation.
In addition, BSO+GBR treatment (from day 5 to 24) affects the morphology of ACC pyramidal neurons. Specifically, apical dendrites have more numerous and expanded branching. In addition, the density of dendritic spines on apical and basal dendrites is reduced in ACC layer III (unpublished observation). Together, these data show that transient early postnatal GSH deficit, particularly in combination with GBR-induced elevated extracellular DA levels, affects the prefrontal circuitry. Similarly to what we have observed in the behavioral studies, some of the effects are still observable at adult age, suggesting that perinatal insults leading to increased oxidative stress can have long-lasting detrimental effects in vulnerable individuals with a reduced antioxidant defense capacity.
Could a GSH deficit affect glutamatergic and dopaminergic systems? Many proteins possess thiol residues that are redox-sensitive through which the redox conditions can modulate the function of these proteins. Several proteins implicated in synaptic transmission and plasticity are redox-sensitive, including NMDA receptors (64, 115), GABAA receptors (2), and L-type voltage-gated calcium channels (19). Therefore, redox dysregulation due to a GSH deficit may alter aspects of synaptic transmission and plasticity, which could directly be related to psychiatric disorders. Our group has shown that NMDA receptor-mediated field excitatory postsynaptic potentials are weaker in hippocampal slices of BSO-treated rats compared to control slices, while basal neurotransmission mediated by AMPA receptors is not altered by GSH deficit (114). This reduced NMDA receptor function under GSH deficit is consistent with the NMDA receptor hypofunction theory of SZ.
An abnormally stimulated locomotor activity following an amphetamine injection in GCLM-KO mice is also in line with DA dysregulation following GSH deficit. A detailed examination of the DA system in GCLM-KO mice is, however, still pending. Nevertheless, a BSO-induced GSH deficit results in an elevation of DA metabolites in the frontal cortex, nucleus accumbens, and hippocampus (59). Moreover, we have observed that a transient GSH deficit induced by BSO alters DA modulation of calcium signaling in neuronal cultures. In neurons with normal GSH content, DA enhances calcium responses to NMDA. In GSH-deficient neurons, this modulation is reversed (113). This is due to an alteration of DA modulation of L-type voltage-gated calcium channels. The alteration of DA modulation of these calcium channels is mediated by the enhanced function of redox-sensitive ryanodine receptors (113). Interestingly, there is converging evidence for a strong genetic association between CACNA1C, the gene encoding for the α-1C subunit of the L-type voltage-gated calcium channel, with BP and SZ (75, 92). Healthy human subjects carrying the risk-associated SNP display altered activation of the hippocampus, ACC, amygdala, and prefrontal cortex (13, 38).
Together, morphological and electrophysiological data show that the effects induced by GSH deficit and associated oxidative stress are consistent with alterations observed in SZ and BD.
Neurochemical studies
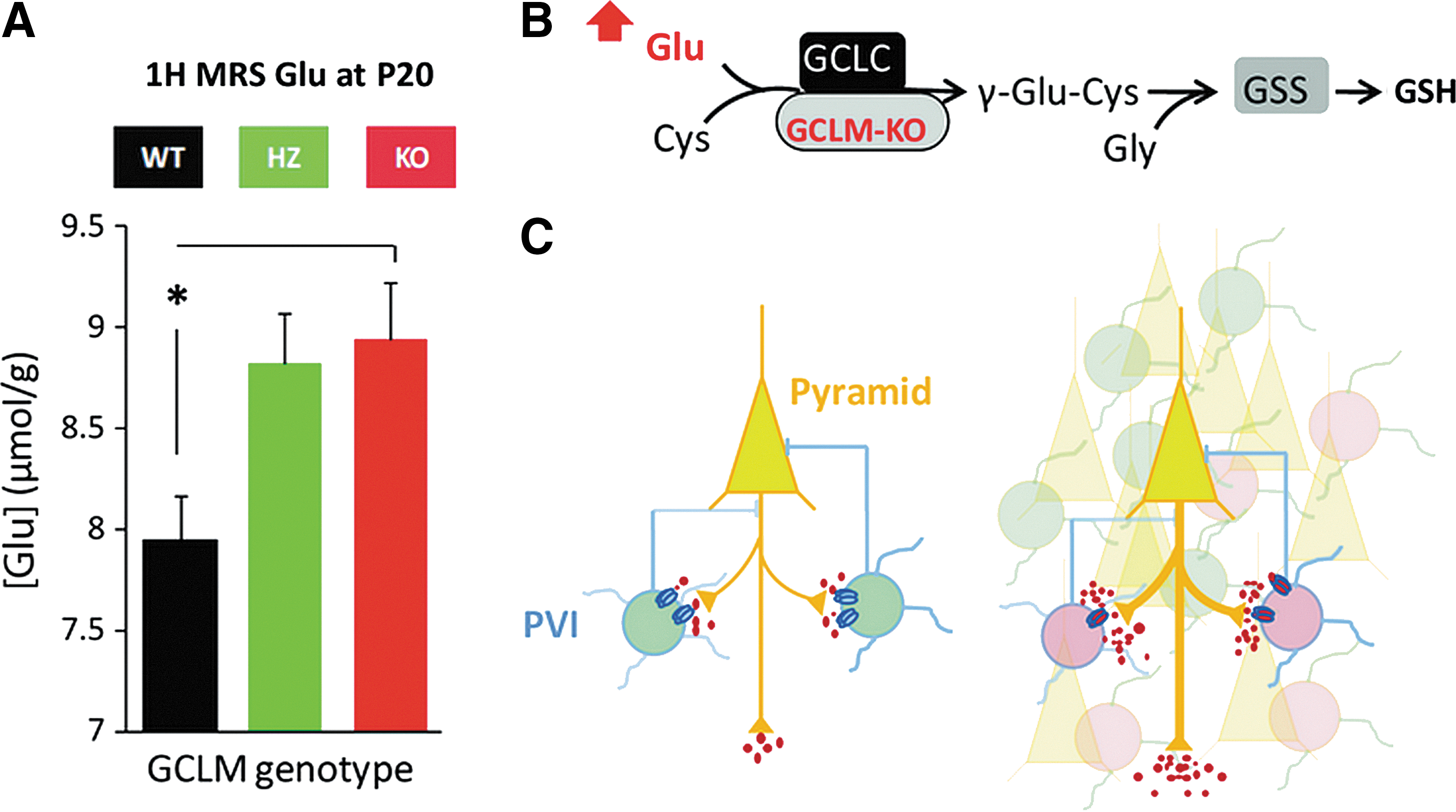
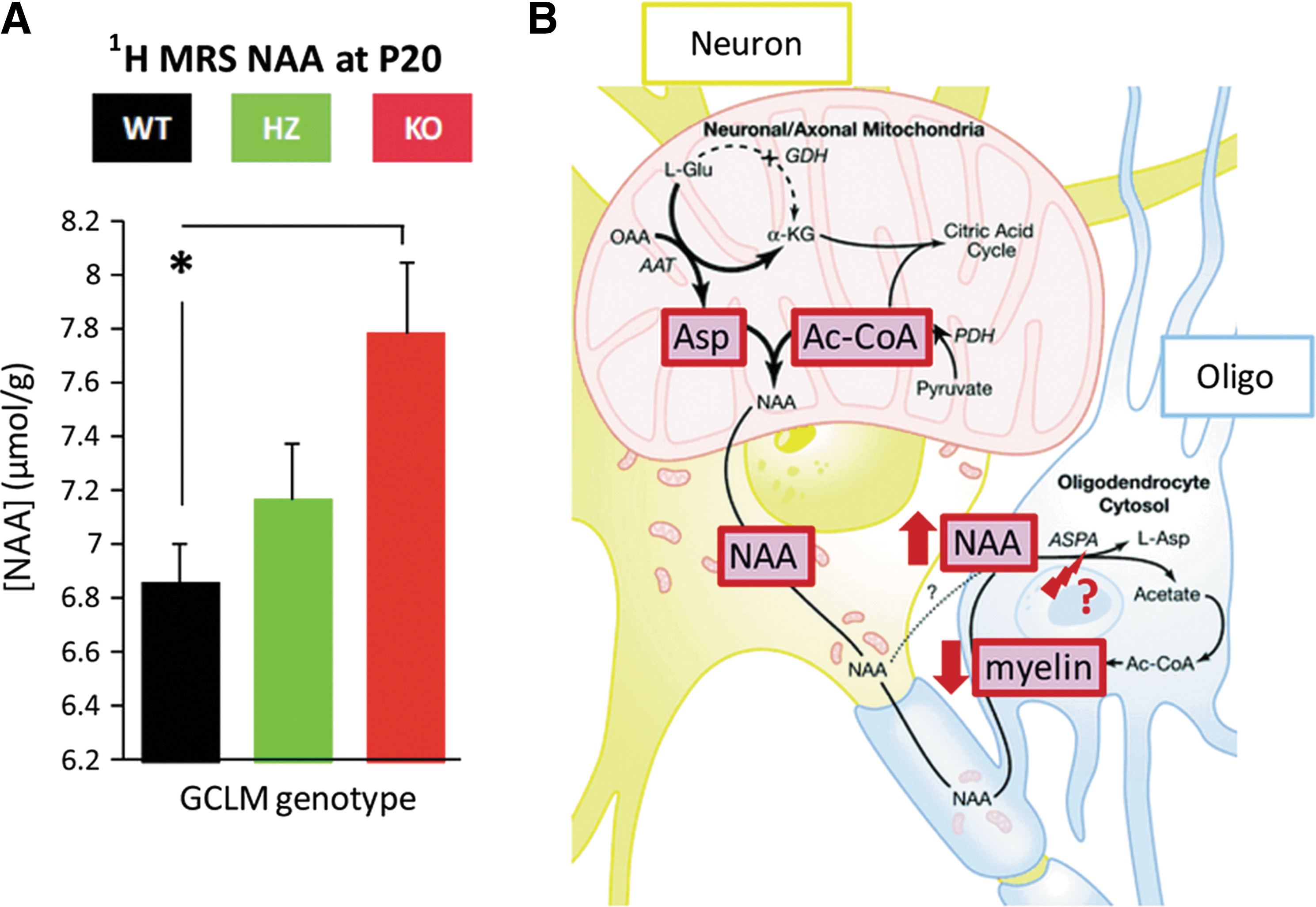
Using high-resolution in vivo MRS technology, we have quantified the brain neurochemical profile (composed of 18 metabolites) of GCLM-KO and WT mice throughout development (P10, 20, 30, 60, and 90) using a longitudinal study design. Given that KO mice display morphological and functional anomalies in the ACC, we have determined the profile in a voxel of the anterior cortex. Though the hippocampus is of similar interest, we have not quantified the hippocampal neurochemical profile due to technical constrains posed by the small brains/hippocampi of early postnatal mice. Compared to WT, the anterior cortex of KO mice shows elevated concentrations of glutamate (Glu), glutamine (Gln), Gln/Glu, and N-acetylaspartate (NAA) at prepubertal ages P20 and P30 (36). Notably, the increases in Glu, Gln, and Gln/Glu, are similar to alterations observed in early stage SZ (14, 116) and in adult BD patients (129). As illustrated in Figure 4, cortical concentration changes in Glu are consistent with the NMDA receptor hypofunction theory of SZ (62), as well as with PV interneuron impairment. Altered NAA levels are also observed in both SZ and BD. However, in contrast to our findings in KO mice, NAA levels tend to be decreased in patients compared to control subjects (110). Thus, our findings are in agreement with a general NAA pathology in both disorders, but do not mimic the directional change. As shown in Figure 5, increased NAA levels in GCLM-KO mice could be a result of insufficient deacetlylation by oligodendrocytes, which may cause deficits in myelination. This is in line with our recent finding that myelin basic protein expression is decreased in ACC of pubertal GCLM-KO mice as compared to WT (unpublished observation). Interestingly, myelination anomalies are also observed in SZ and BD (85).
Chronic treatment with NAC from gestation onward normalizes most of the neurochemical alterations, including the concentrations of Gln and Glu (36). Thus, NAC exerts protective effects on the neurochemical profile in GSH-deficient mice. Together, these data show that GSH deficit in the KO mouse model induces many neurochemical alterations in the anterior cortex that resemble those observed in SZ and BD patients. The data also highlight the prepubertal period as a sensitive time for redox-related neurochemical changes and demonstrate beneficial effects of early NAC treatment.
Concluding Remarks
Redox dysregulation appears to be a common final pathway on which genetic and environmental factors converge during brain development to induce structural and functional alterations, which may contribute to psychiatric disorders, such as SZ and possibly BD. The phenotypes of animal models deficient in GSH synthesis are consistent with what is observed in SZ and BD at behavioral, morphological, electrophysiological, and neurochemical levels. Cognitive and affective deficits are likely to be underlined by developmental alterations of the circuitry involving PV-positive inhibitory interneurons in hippocampus and anterior cortex. The efficacy of the GSH precursor and antioxidant NAC to normalize neurochemical anomalies in the mouse model of GSH deficit and to improve clinical symptoms in patients suggests that such relatively safe compounds could be used as a preventive treatment in risk individuals.
Footnotes
Acknowledgments
This work was supported by the Swiss National Science Foundation (# 310030-135736 to Kim Q Do), the “Loterie Romande,” and the Alamaya Foundation.