
Abstract
Introduction
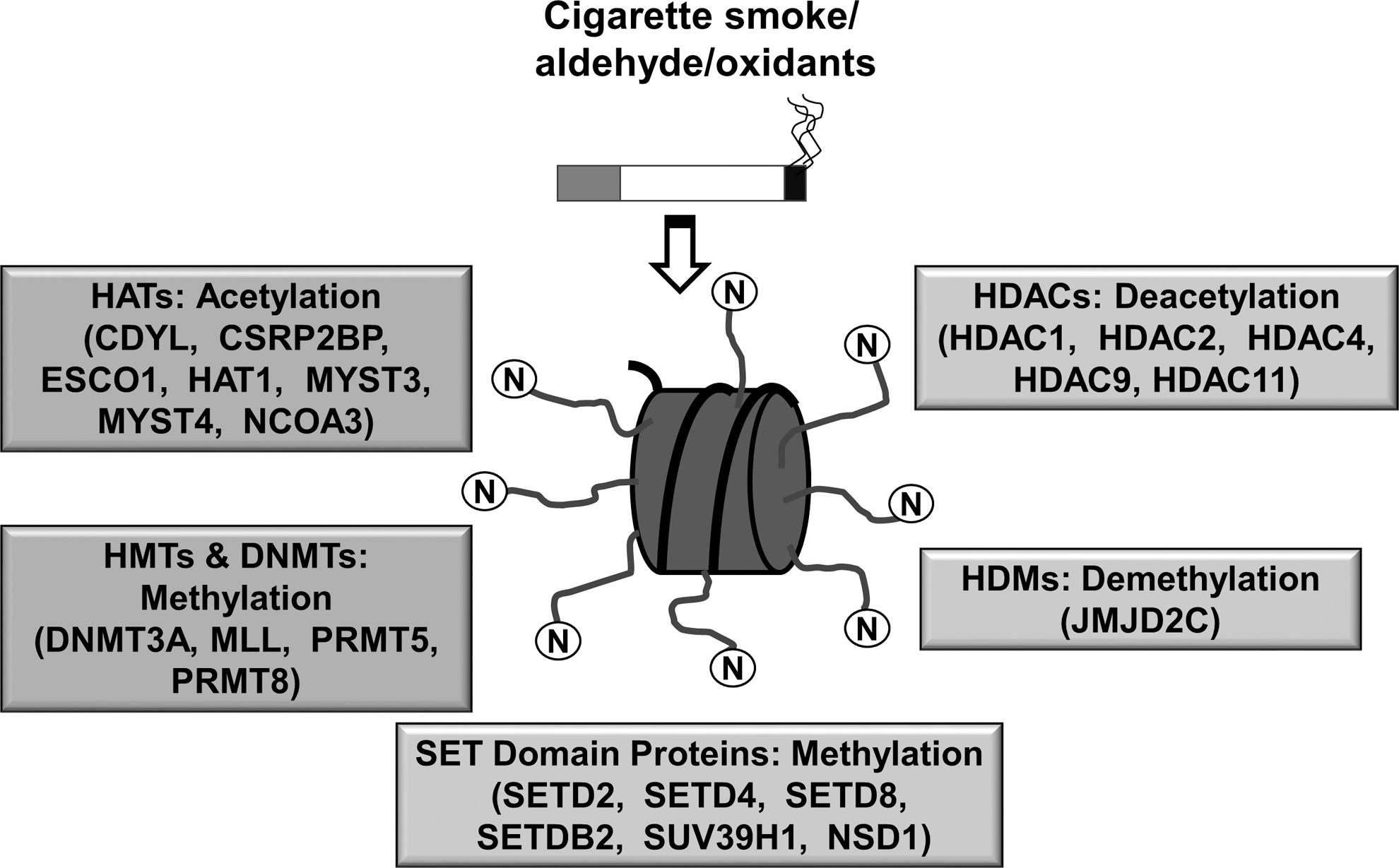
DNA is packed into chromatin by histones to form a tightly coiled structure in the nucleus. Chromatin remodeling by histone modifications plays a key role in the regulation of cellular processes, including gene transcription/repression, DNA repair, differentiation, and proliferation. Post-translational histone modifications predominantly occur via acetylation/deacetylation and methylation/demethylation reactions (41, 127) (Fig. 1).
Histone acetyltransferases (HATs, including p300- CREB-binding protein [CBP]-associated factor, p300/CBP, and GCN5) and histone deacetylases (HDACs e.g., HDAC2 and SIRTUIN1 [SIRT1]) maintain the balance between histone acetylation and deacetylation. HATs affect the binding of DNA sequence-specific transcription factors and, subsequently, recruit coactivators or corepressors on gene-specific regions to form either coactivator or corepressor complexes (166). It is well known that transcriptional coactivators possess intrinsic HAT (CBP/p300) and HDAC activities, suggesting that histone acetylation and deacetylation play a causal role in regulating gene transcription (72, 170, 171). It is generally accepted that increase in histone acetylation leads to increased gene transcription, while histone hypoacetylation is linked to decreased gene transcription (72, 170, 171). Histone acetylation by HATs and deacetylation by HDACs are also linked to cell-cycle progression, proliferation, senescence, DNA repair, and recombination events, as well as proinflammatory gene transcription, which can be affected by oxidative stress and redox signaling (15, 69, 146).
Methylation of histones occurs either on lysine (K) or arginine (R) residues in a reaction that is catalyzed by specific histone methyltransferases (HMTs) (11, 192). HMTs utilize S-adenosylmethionine as a cofactor during transmethylation and produce S-adenosylhomocysteine as a by-product. Methylation of specific lysine residues results in the formation of binding sites/interacting domains, which allow recruitment of other regulator proteins. HMTs are deregulated in several chronic lung diseases, thereby affecting the global methylation status. Histone demethylases (HDMs) catalyze the removal of methyl groups from lysine or arginine residue of histones. These include lysine-specific demethylase 1 and Jumonji C domain family proteins involved in the regulation of gene expression (149). These enzymes demethylate specific histone residues, thereby forming transcriptional repression complexes (133). HMTs and HDMs are subject to redox regulation such that post-translational modifications of histone proteins by oxidants and environmental stresses can trigger the transcription of genes that are involved in chronic inflammatory events. This review is focused on the role of ROS-mediated effects on chromatin modification enzymes and epigenetic chromatin modifications (histone acetylation/deacetylation and histone methylation/demethylation) in inflammatory response, steroid resistance, and cellular senescence that occurs in chronic inflammatory lung diseases associated with cigarette/tobacco smoking.
Cigarette Smoke and Oxidative Stress
ROS can affect the lung by directly oxidizing proteins, DNA, and lipids or induce changes indirectly through the generation of secondary metabolic reactive species. ROS can fragment the extracellular matrix, and cause mucus secretion, apoptosis, and alter cell proliferation (113). Relevant ROS generating enzyme systems in Chronic Obstructive Pulmonary Disease (COPD) include xanthine–xanthine oxidase, NADPH oxidase, and heme peroxidases (108, 117). CS, which contains 1015 free radicals and 4700 different chemical compounds per puff, is the major source of inhaled environmentally derived ROS. It has been estimated that 15%–20% of smokers develop COPD. The Global Initiative for Chronic Obstructive Lung Disease defines COPD as a syndrome of chronic, irreversible obstruction of lung airflow characterized by progressive decline in forced expiratory volume in one second. The inflammatory immune features of the COPD phenotype includes, activation of epithelial cells, and resident macrophages, as well as the recruitment and activation of neutrophils, monocytes and B and T lymphocytes into the lung. Inflammatory cells are activated in response to cytokines/chemokines/chemoattractants after recruitment into the interstitium, thereby generating various ROS. ROS cause lipid peroxidation (oxidation of membrane phospholipids) and the generation of malondialdehyde, 4-hydroxy-2-nonenal, acrolein, and F2-isoprostanes (114, 120).
Oxidative Stress and Kinases Signaling in Chromatin Remodeling
It is well known that ROS can activate signal transducing molecules through the effects on oxidation-prone cysteine-rich domains, thereby activating gene transcription (6, 118). These include the mitogen-activated protein kinase family, extracellular signal-regulated kinase, c-Jun N-terminal kinase (JNK), p38 kinase, and phosphoinositide 3-kinase (PI3K)/Akt. Based on the redox status of the cells, members of the MAPK family are activated, leading to a multifaceted transactivation of redox-sensitive transcription factors (ATF-2, CBP) (6, 147). Furthermore, activation of upstream kinases, such as nuclear factor-κB (NF-κB)-inducing kinase, mitogen- and stress-activated kinase 1, and IκB kinase-α, results in downstream chromatin remodeling events that alter the function of various gene promoters (27, 28, 138, 179, 181). This epigenetic mechanism modulates a series of specific proinflammatory gene transcription events that modulate apoptosis, autophagy, senescence, proliferation, transformation, and differentiation (53). CS induces activation of protein kinase C (PKC), mediates regulation of intracellular signaling pathways (174). PKCζ-knockout mice exposed to CS or lipopolysaccharide showed a reduction in lung inflammatory response due to chromatin modifications, suggesting that PKCζ is an important modifier of lung inflammatory response (Fig. 2) (186). Hence, activation of kinases by ROS and aldehyde/lipid peroxidation products can lead to a proinflammatory response by chromatin remodeling.

The role of PI3K and p38 signaling pathways have been shown in pathogenesis of COPD and lung cancer (34, 37, 51). Inhibition of PI3K/p38 lowers tobacco/CS-induced lung inflammatory response (34). Interestingly, PI3K inhibition significantly increased the steroid efficacy to inhibit an inflammatory response under oxidative stress (14, 82, 151). Therefore, in addition to p38 MAPK and PKCζ, oxidants/CS-mediated activation of the PI3K signaling pathway may be a target for intervention in COPD. These oxidant- and redox-sensitive pathways (PI3K/p38) activate various kinases, such as NF-κB-inducing kinase, mitogen- and stress-activated kinase 1, and IκB kinase-α, resulting in histone acetylation and transcriptional activation of proinflammatory genes (e.g., IL-6, IL-8, and COX-2) (27, 138, 179) (Fig. 2).
Chromatin Remodeling: Histone Acetylation, Deacetylation, Methylation, and Demethylation
Histone acetyltransferases
HATs transfer acetyl groups from acetyl-CoA to the N-terminal lysine residues of histones (H3 and H4), thereby uncoiling DNA resulting in the transcription factor binding to promoters followed by transcriptional gene activation (56, 58, 119, 144). HATs and HDACs play opposing roles in regulating acetylation of core nucleosomal histones. HATs are classified into five distinct families. These include the CBP/p300 HATs, Gcn5-related acetyltransferases, the general transcription factor HATs (TFIID subunit TAF250), and the nuclear hormone-related HATs SRC1 and ACTR (SRC3) (144, 153). Each of these HAT families has diverse roles in regulating the chromatin assembly and structure (chromatin remodeling) although they share a common enzymatic activity (112).
CS/oxidant-mediated acetylation of histone H3 occurs in macrophages and epithelial cells, and in lungs of humans and rodents, suggesting that histone acetylation/deacetylation plays an important role in chromatin remodeling. Oxidative stress and/or glutathione depletion is associated with increased histone acetylation (119, 120). CS-mediated alterations in chromatin remodeling are shown to be involved in sustained lung inflammatory diseases, such as COPD (83, 140, 143, 179, 186). CBP and p300 are the known coactivators that possess intrinsic HAT activity. They are regulated by the JNK/p38 MAP kinase pathway, which activates redox-sensitive transcription factors, such as NF-κB and activator protein-1 (148). Therefore, histone acetylation by CBP/p300 has an impact on the activation of specific proinflammatory mediators linked with NF-κB/AP-1-mediated gene expression (22, 64, 115). Transcription factor NF-κB (RelA/p65) is acetylated at K310 residue by CBP/p300 HAT, but also causes histone acetylation by recruiting other cofactors/coactivators and chromatin remodeling complexes to activate proinflammatory gene transcription (e.g., IL-6 and IL-8) (46, 57, 75). It is shown that NF-κB-induced acetylation of histone H3, but not histone H2A, H2B, or H3, occurs in epithelial cells on specific lysine residues (Lys8 and Lys12) at NF-κB-responsive regulatory elements on proinflammatory genes (57). Oxidative stress and CS activate NF-κB, HAT, CBP/p300 leading to specific histone acetylation in macrophages and lung cells (60, 93, 178, 179, 187). Therefore, development of small molecules that target for inhibition of HATs (e.g., CBP/p300, PCAF, and GCN) may be useful for therapeutic intervention in COPD and other chronic inflammatory lung diseases associated with smoking mediated oxidative stress (8, 99).
Histone deacetylases
HDACs play a crucial role in removing the acetyl groups from histones, thus providing a regulatory role toward the epigenetic control of gene expression (17). Presence of HDACs within the gene regulatory regions leads to formation of closed chromatin complexes (heterochromatin) resulting in gene silencing. So far, eighteen mammalian HDAC enzymes are known, which are classified into four classes based on their homology to a prototypical HDAC found in yeast (32) (Fig. 3). Class I HDACs (HDACs 1, 2, 3, and 8) are ubiquitously expressed with the possible exception of HDAC3 and HDAC10, which are predominantly localized in the nucleus (145). Class II HDACs (HDACs 4, 5, 6, 7, 9, and 10) are expressed in a tissue-specific manner, and they have the ability to shuttle between the nucleus and cytoplasm (13, 50). The shuttling of class II HDACs from the nucleus is mainly regulated by nuclear export signaling 14-3-3 proteins (13). Based on the existence of tandem deacetylase domains, class II HDACs are further classified into class IIa (HDACs, 4, 5, 7, and 9) and class IIb (HDACs 6 and 10) (62). Class I and II HDACs share significant homology at the deacetylase domain, but differ in their N-terminal sequence (Fig. 3).

The class III HDACs or sirtuins are named based on their homology to the yeast Sir2 gene, which is a highly conserved gene family. In humans, sirtuins comprise seven members, SIRT 1–7 (86). Among these, SIRT1, SIRT2, SIRT3, and SIRT5 have an NAD+-dependent deacetylase domain (distinct from the zinc-dependent deacetylase domains of class I and II HDACs), which catalyze the deacetylation of histones and nonhistone proteins. Since sirtuins utilize NAD+ obtained from metabolic processes, it plays vital roles in linking molecular mechanisms between cellular metabolic status (NAD+/NADH levels) and several cellular processes (16). In contrast, SIRT4 and SIRT6 have an NAD+-dependent ADP ribosylation domain and catalyze protein ribosylation. The subcellular localizations of sirtuin family members are well defined. SIRT1, SIRT6, and SIRT7 are localized to the nucleus; SIRT2 to the cytoplasm and SIRT3, SIRT4, and SIRT5 are localized to the mitochondria. Class III HDACs share less homology compared to the class I and II HDACs, and are not inhibited by widely used HDAC inhibitors, such as butyrate, valproic acid, trichostatin A (TSA), or suberoylanilide hydroxamic acid (175) (Fig. 3). Instead, they are inhibited by nicotinamide, splitomicin, and sirtinol. SIRT1 is a redox-sensitive deacetylases, which requires intracellular NAD+ for its regulations (20). HDAC11 belongs to class IV HDACs, which share limited homology with class I and II HDACs (44).
HDACs are also reported to deacetylate nonhistone proteins, such as the redox-sensitive transcription factor NF-κB (RelA/p65) and forkhead box class O3 (FOXO3), thereby regulating transcription of NF-κB-dependent proinflammatory genes (132). The levels and activities of HDACs are significantly decreased in macrophages, and lung cells in vitro in response to oxidative/carbonyl stress, as well as in lungs of COPD patients via alterations in intracellular GSH/GSSG redox ratio (caused by H2O2 and CS exposures). Decreased HDAC2 activity is linked with COPD severity, asthma, and steroid resistance in asthmatics who smoke tobacco (5, 60, 93, 178, 187). Intracellular GSH maintains HDAC2 in a reduced environment, and increasing intracellular GSH inhibits HDAC2 oxidative post-translational modifications (5, 85, 178). A decrease in levels of HDAC2 in several cellular systems is due to post-translational modifications, such as oxidation/carbonylation, nitrosylation, acetylation, and phosphorylation in response to oxidants derived from CS (3, 5, 59, 93, 178). Hence, HDAC2 levels/activities and the status of oxidative post-translational modifications dictate the effects of coactivators on the chromatin status on various promoters to drive the gene activation or repression.
HDAC2 and steroid resistance in inflammation
Decreased HDAC2 occurring as a result of CS/oxidants/aldehydes is associated with activation of NF-κB RelA/p65 subunit (evident as increased levels of total and phospho-acetylated RelA/p65). RelA/p65 interacts with HDAC2, and nuclear accumulation of RelA/p65 result in transcription of proinflammatory genes when HDAC2 is post-translationally modified, ubiquitinated, and degraded (79, 178, 185, 187). HDAC2 modification is dependent on the cellular thiol (GSH/GSSG) redox status (85, 178). In vitro studies using trichostatin A (HDAC inhibitor) in different cell lines showed enhanced NF-κB-driven inflammatory gene transcription (24, 57). Hence, alteration of HDACs (particularly HDAC2) by CS/oxidative stress leads to increased acetylation of histones (histone H3 and H4) and activation of NF-κB, thus enhancing transcription of proinflammatory genes (4, 123, 178). In humans, a marked reduction of HDAC2 expression/activity in lung parenchyma, bronchial biopsies, and alveolar macrophages of patients with COPD is correlated with the severity of inflammation and the progression of the disease (60). The underlying mechanism for reduced HDAC2 levels/activities include post-translational modifications (i.e., nitrosylation, phosphorylation, and ubiquitination) leading to proteasome-dependent degradation, particularly, in response to CS (5, 43, 123), and/or oxidative/carbonyl modifications/degradation of HDAC2 (178). HDAC2 is important for the anti-inflammatory effects of glucocorticoid treatment. The levels/activity of HDAC2 is decreased in lungs of patients with COPD who are resistant to corticosteroid (60). Corticosteroid resistance is also associated with increased oxidative/carbonyl stress (aldehyde-mediated carbonylation of proteins on cysteine, histidine, and lysine residues). Corticosteroids do not block the inflammatory response when there is an increased oxidative stress, such as in severe asthmatics and patients with COPD. This inefficacy is not associated with alterations in nuclear HAT activity (60). Increasing HDAC activity, by dietary polyphenol curcumin, sulforaphane, or theophylline (HDAC2 activators), significantly enhances the steroid-induced suppression of IL-8 release in monocytes and alveolar macrophage from COPD patients; conversely, HDAC inhibitors have the opposing effects (30, 52, 85). This suggests a specific role of HDAC2 in steroid resistance. Furthermore, deacetylation of glucocorticoid receptor (GR) by HDAC2 enhanced the association of GR and RelA/p65, which resulted in the attenuation of proinflammatory gene transcription in monocytes and lung cells (61). Consequently, restoration or attenuation of decreased HDAC2 levels/activities will further enhance the glucocorticoid sensitivity by deacetylating RelA/p65 and GR. This can be achieved by reversing the post-translational modifications of HDAC2, such as decarbonylation or dephosphorylation, via inducing aldehyde dehydrogenases/reductases, thioredoxin reductase, and phosphatases, or inducing the antioxidant buffer systems by using Nrf2 activators and extracellular superoxide dismutase mimetics (2, 183). Thus, HDAC2 is a redox-sensitive protein, whose levels/activities are decreased under oxidative/carbonyl stress conditions, resulting in chromatin modifications associated with steroid resistance leading to lung inflammation.
HDAC2, DNA damage/repair, and cellular senescence
CS/oxidants can also induce cellular senescence (i.e., stress-induced premature senescence, SIPS) in alveolar epithelial cells and fibroblasts, which is independent of telomere shortening (95, 101, 102, 155 –157). Cellular senescence (irreversible cell growth arrest) impairs the repair of damaged lung tissue, perhaps, explaining why COPD progresses even after cessation of smoking. The role of cellular senescence in COPD is supported by animal studies (74, 130, 137, 184), where CS is known to cause premature senescence. The senescent cells are metabolically active and prone to secrete proinflammatory mediators (i.e., IL-6 and IL-8) and matrix metalloproteinases (i.e., MMP-1, MMP-3, and MMP-9) (40, 125). These proinflammatory mediators may in turn initiate and maintain cellular senescence as the deficiency of IL-6, IL-6R, or CXCR2 prevents premature senescence (1, 71). Indeed, the percentage of proinflammatory senescent type II cells expressing both p16 and phosphorylated NF-κB (i.e., senescent-associated secretory phenotype) was increased in lungs of patients with COPD as compared to smokers and nonsmokers (157). However, the mechanism underlying premature senescence and associated secretory senescence phenotype (secretory senescent cells) in the development of COPD remains unknown.
Persistent DNA damage causes premature senescence and senescence-associated secretory phenotype via forming persistent foci, termed as DNA segments with chromatin alterations reinforcing senescence (29, 125, 126). Further study demonstrated the requirement of the upstream signals of DNA damage response, including ataxia telangiectasia mutated and its substrates Nijmegen breakage syndrome 1 and checkpoint kinase 2 for the NF-κB-dependent inflammatory phenotype in senescent cells (36, 125, 173). The double-strand break is the most dramatic form of DNA damage, which can be repaired either by homologous recombination, classical, or alternative nonhomologous end joining. CS-mediated oxidative stress has been shown to cause DNA damage, which is increased in patients with COPD (21, 23, 33, 106, 134). The nonhomologous end joining repair proteins, such as Ku70, Ku80/Ku86, are also impaired by CS, which may, in turn, accelerate the formation of persistent DNA segments with chromatin alterations reinforcing senescence leading to sustained DNA damage and an associated secretory senescence phenotype observed in patients with COPD (21, 126, 135). Ku70, Ku80/Ku86 proteins may be regulated by acetylation/deacetylation. However, it is unclear how chromatin remodeling influences premature senescence and associated secretory senescence phenotype during DNA damage/repair by CS/oxidants in lung cells.
HDAC2, not only functions in DNA damage response to promote repair, but also regulates cellular senescence and inflammation via deacetylating histones and transcription factors (e.g., NF-κB) (89, 150, 165, 169, 193). Indeed, histone acetylation and methylation as well as NF-κB activation play an important role in DNA damage repair and genomic instability as well as premature aging (159, 190). Additionally, other histone modifiers, such as CBP/p300, regulate DNA damage and repair (103, 129, 163, 167). Cellular senescence and its associated inflammatory phenotype occur in lungs of patients with COPD (156, 157). It is evident that HDAC2 regulates cellular senescence along with its complex, such as HDAC1, in response to stress (169). However, no information is available regarding the role of HDAC2 epigenetic modifications in DNA damage/repair, as well as premature senescence and associated secretory senescence phenotype, particularly, in response to CS exposure in lung cells.
Histone modifications, including acetylation and methylation, play an important role in DNA damage repair and genomic instability as well as premature aging (159, 163, 190). It is possible that HDAC2 regulates CS-mediated DNA damage, premature senescence, and associated secretory senescence phenotype through histone deacetylation (Fig. 4). HDAC2 can interact with protein methyltransferases to form a large and multiple-protein complex(es), thereby regulating transcriptional repression or activation (161). Our preliminary studies have recently showed that CS exposure alters histone methylation (e.g., H3K4me, H3K27me, H3K79me, H3K36ac/me) in cells and mouse lung (27, 177). CS/oxidant-mediated HDAC2 reduction may alter histone methylation by regulating enzyme methyltransferases. In addition to HDACs, CBP/p300 also has shown to acetylate histone H3 and H4 at the sites of DNA double-strand break, thereby regulating DNA damage and repair (103, 163). HDAC2 interacts with CBP in response to CS exposure (3). Therefore, other coactivators/repressors, such as CBP/p300, and NF-κB along with HDAC2 participate in the CS-mediated DNA damage and premature senescence and associated secretory senescence phenotype.

SIRT1 deacetylase, FOXO3, NF-κB, and cellular senescence
Sirtuin 1 (SIRT1) belongs to class III HDACs, which possess anti-inflammatory, anti-aging/senescence, and anti-apoptotic/autophagy properties due to their ability to deacetylate both histones and nonhistone proteins, including transcription factors (e.g., NF-κB, FOXO3, p53, Ku70, and PGC-1α) (182) (Fig. 5). SIRT1 levels and activities were decreased in monocytes (MonoMac6 cells), bronchial epithelial cells (Beas-2B), mouse lungs exposed to CS, as well as in the lungs of COPD patients and smokers (54, 121, 122, 180), suggesting the involvement of SIRT1 in the pathogenesis of COPD. The mechanism for SIRT1 reduction involves post-translational modifications, such as phosphorylation, nitrosylation, and oxidation/carbonylation by ROS/carbonyls-aldehydes in response to CS/oxidative/carbonyl stress, ultimately leading to SIRT1 degradation (20, 121, 122). SIRT1 is also regulated by the intracellular redox thiol (GSH/GSSG) pool (20, 180). SIRT1 knockdown by siRNA leads to heightened NF-κB activation and inflammatory response. SIRT1 activators, such as SRT1720 and resveratrol diminished the release of proinflammatory mediators in response to oxidative stress/CS exposure (122). These findings suggest a role for SIRT1 activators (90, 91) in the therapeutic intervention of COPD. Administration of the SIRT1 activators, SRT2172 and SRT1720, attenuated CS-induced lung inflammation in mice (96). Furthermore, SIRT1-mediated protection in response to CS/oxidative stress against lung inflammatory response and injury are linked to the deacetylation of NF-κB RelA/p65 and FOXO3, and negative regulation of MMP-9 via the tissue inhibitor of metalloproteinases-1 (25, 96, 180, 184). Further studies are required to investigate the key molecular mechanisms to understand how SIRT1 can mediate protection against oxidative stress during COPD progression (184, 188).
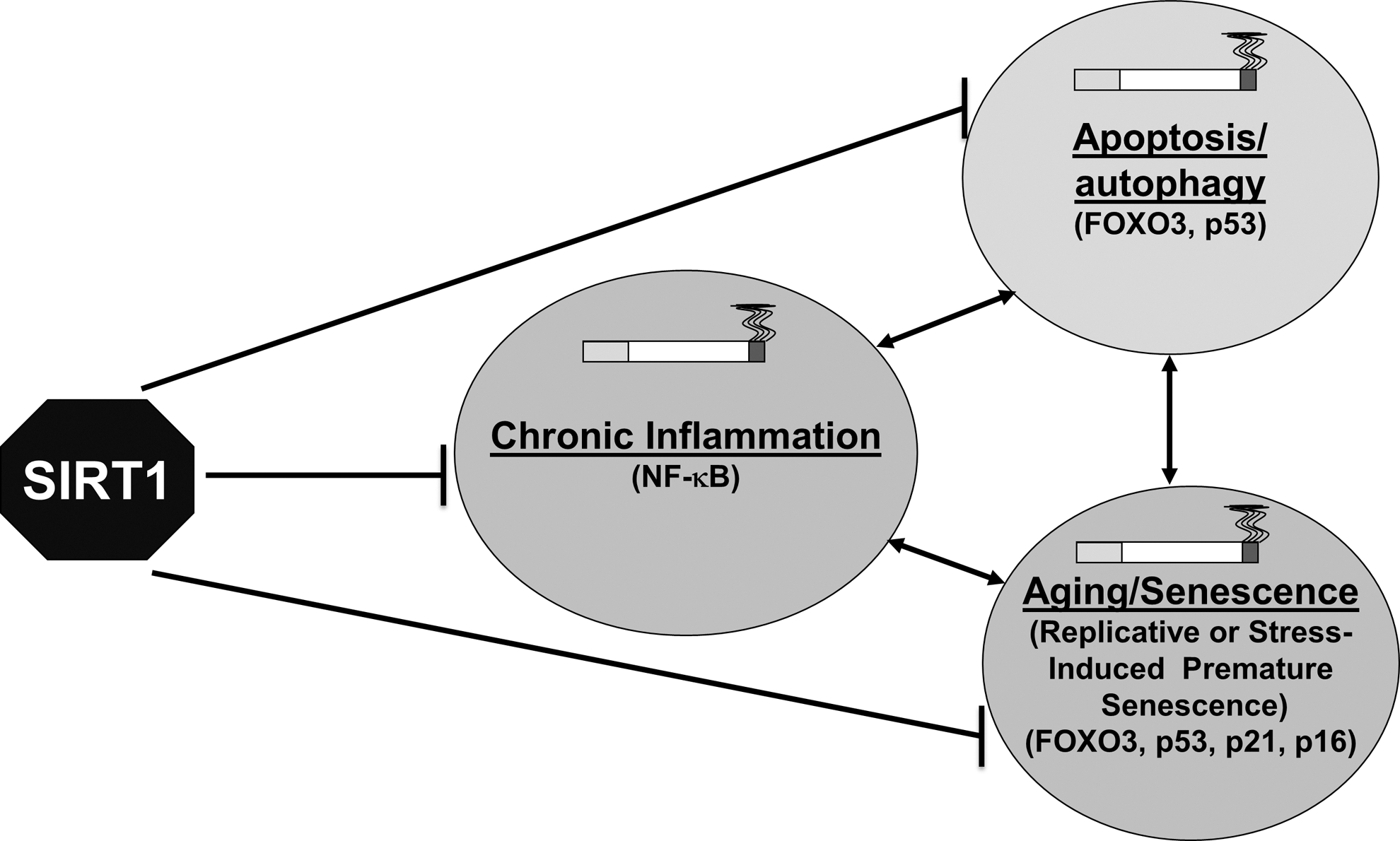
SIRT1 also deacetylates other transcription factors, such as FOXO3, p53, and NF-κB, thereby regulating CS/oxidative stress-induced cell cycle arrest, apoptosis, and cellular senescence, which play important roles in the pathogenesis of COPD (Fig. 6) (188). Transcriptional activity of FOXO3 is regulated by its phosphorylation and acetylation status. SIRT1 deacetylates FOXO3, leading to its activation and to the induction of cell cycle arrest, thereby reducing premature senescence (94, 184, 189). FOXO3 is acetylated when SIRT1 levels/activity are reduced in response to CS in mouse lung (121). Furthermore, FOXO3 has a regulatory role in lung inflammatory response and regulation of antioxidant genes, such as manganese superoxide dismutase and catalase. Targeted disruption of FOXO3 results in downregulation of these antioxidant genes in mouse lungs and increases the susceptibility for the development of COPD/emphysema (55). Yao and colleagues have shown that the SIRT1-FOXO3-mediated pathway is important in pathogenesis of COPD independent of the NF-κB pathway (116, 184). Further studies on the SIRT1-FOXO3 pathway will elucidate the underlying mechanisms in response to oxidative/carbonyl stress, and will provide the possible therapeutic interventions in treatment of COPD based on SIRT1 activation.
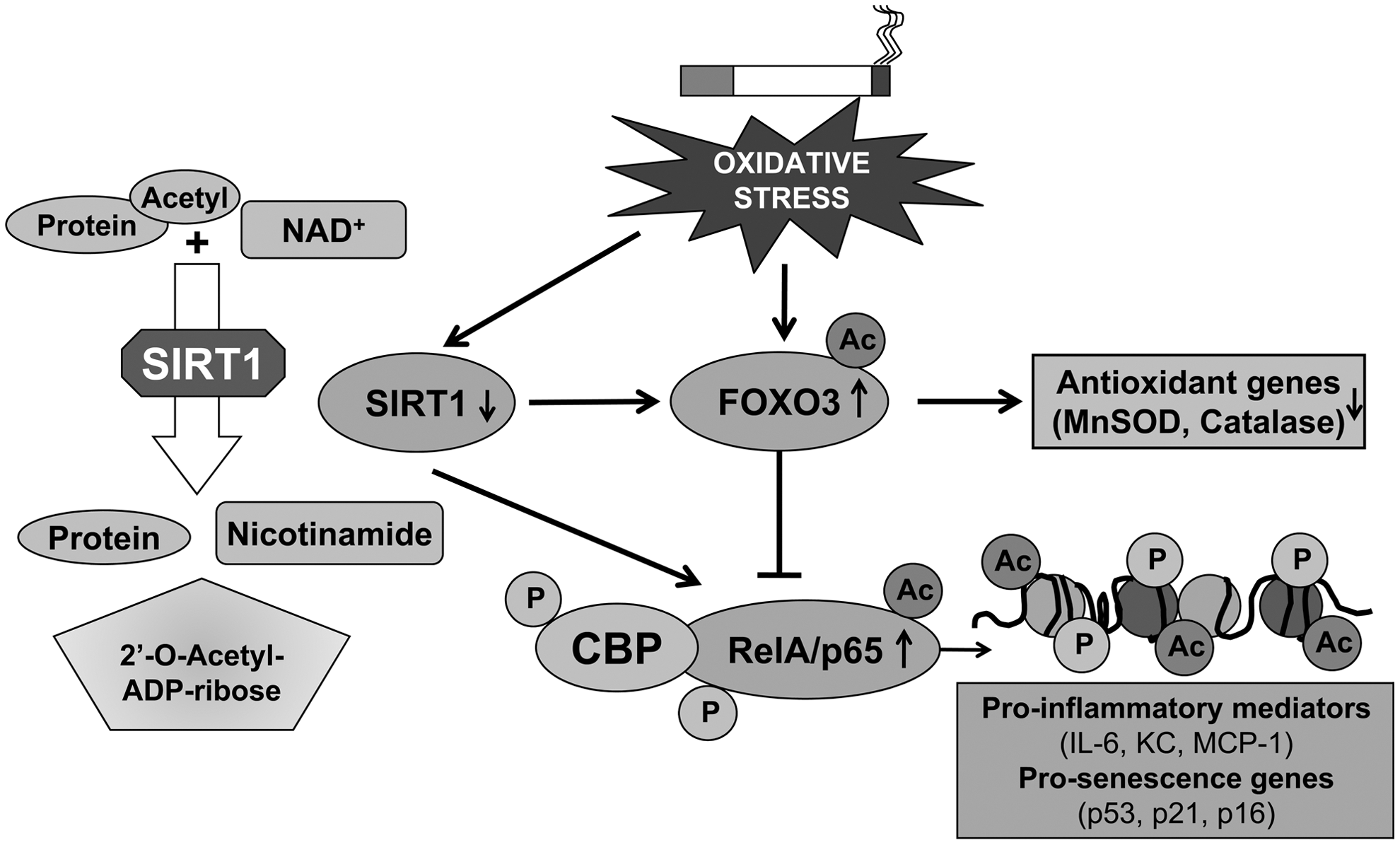
SIRT1 interacts with p53, and deacetylates lysine residue at the C-terminal regulatory domain (162). Decreasing SIRT1 levels/activity increases p53 acetylation, thereby promoting apoptosis and senescence (80, 162). Oxidative stress is shown to accelerate cellular senescence, due to increased p53 acetylation via decreasing the function of SIRT1 through NAD+ depletion (42) (104). Furthermore, blocking p53 by antisense oligonucleotides reversed the inhibitory effect of SIRT1 on cellular senescence (104). Earlier studies have shown that the nuclear levels of SIRT1 were decreased in response to CS both in vitro and in vivo in lungs (180), but still it remains unclear whether SIRT1-mediated regulation of p53 (acetylation/deacetylation) plays an important role in oxidants/CS-mediated apoptosis, autophagy, and cellular senescence/aging.
Other sirtuins have also been shown to modify cellular functions under oxidative stress. For example, a recent report demonstrated the role of SIRT6 in DNA repair under oxidative stress through the activation of poly [adenosine diphosphate-ribose] polymerase 1 (PARP1) (81). Oxidative stress in mammalian cells recruits SIRT6 to the sites of DNA double-strand breaks, where it interacts with PARP1 (redox sensitive), and mono-ADP-ribosylates PARP1 on Lys521, thus stimulating PARP1's poly-ADP-ribosylase activity, and repairing DNA double-strand break through both nonhomologous end joining and homologous recombination repair mechanisms (81). Therefore, SIRT6, in addition to SIRT1 is implicated to play crucial roles in oxidative stress-induced inflammatory response, senescence, genomic instability, DNA damage/repair, and aging (65, 81, 88, 158). Consequently, other sirtuin members possibly gain equal credence in understanding the pathogenesis of COPD and other chronic lung diseases associated with tobacco smoking.
SIRT1 is a key regulator of vascular endothelial homeostasis controlling angiogenesis, vascular tone, and endothelial dysfunction by regulating the endothelial nitric oxide synthase activity (109, 172). Endothelial dysfunction plays an important role in pathogenesis of emphysema, and CS-induced emphysematous alveolar destruction, which is almost avascular, associated with decreased expression of endothelial nitric oxide synthase (35, 38). SIRT1 deacetylates lysines at position K496 and K506 in the calmodulin-binding domain of endothelial nitric oxide synthase, leading to enhanced nitric oxide production, which is vital for endothelial-dependent vasorelaxation, cell survival, migration, and postnatal neovascularization (10, 84). NO activates the SIRT1 promoter, thereby increasing SIRT1 mRNA and protein levels (100, 105), suggesting that a positive feedback mechanism exists between SIRT1 and endothelial nitric oxide synthase (110). Therefore, the activation of SIRT1 by small molecules (such as SRT1720) may help in resetting the endothelial nitric oxide synthase activity during endothelial dysfunction in patients with COPD, where NO availability is limited (87). SIRT1 overexpression attenuated CS-induced apoptosis and inflammatory response in cultured coronary arterial endothelial cells (31). Therefore, SIRT1 acts as a possible epigenetic chromatin redox regulator in the treatment and prevention of chronic lung diseases, including COPD and comorbidities associated with endothelial dysfunction and cardiovascular problems by protecting endothelial cells from stress-induced premature senescence, autophagy, apoptosis, and inflammatory response.
Histone methyltransferase
Methylation of histones occurs either on lysine (K) or arginine (R) residues in a reaction that is catalyzed by specific HMTs (PRMT family, the SET-domain [Suppressor of variegation-Enhancer of zeste-Trithorax domain]-containing protein and the non-SET-domain proteins DOT1/DOT1L) (11,192). HMTs utilize S-adenosylmethionine as a cofactor during transmethylation and produce S-adenosylhomocysteine as a by-product. The substrate specificity and activity of HMT depends on SET domains and associated motifs (191). Methylation of specific lysine residues results in the formation of binding sites/interacting domains, which allow recruitment of other regulator proteins (coactivators/corepressors). HMTs are deregulated in several chronic lung diseases, thereby affecting the global methylation status. Histone H3K4, H3K9, and H3K27, and H4K20 are frequently and preferentially methylated as mono-, di-, or tri-methylated histone H3 and histone H4 (131). Methylation at H3K4, H3K36, and H3K79 is linked to gene activation, whereas H3K9, H3K20, and H3K27 methylation is associated with gene repression (49) (Fig. 7).

Histone modifications are regulated by specific chromatin modification enzymes, which have a residue-specific role in regulating gene expression with respect to histone modifications (9, 19, 68, 127). In vitro exposure of airway epithelial cells or immortalized bronchial epithelial cells to CS condensate/extract (oxidants) reduced H4K16ac and H4K20me3, but increased H3K27me3. This may have implications in tumor development in several cancer types associated with tobacco smoking (77). It is well known that COPD is associated with a significantly increased incidence of lung cancer and other comorbidities in susceptible chronic smokers. HDACs, including HDAC2 and SIRT1, play an important role in cancer cell epigenetic regulation and gene expression. Histone H4K16ac and H4K20me3 has been suggested as a hallmark in different kinds of human tumors and tumor-derived cell lines, thus characterized as known epigenetic marks in cancer (39). SIRT1 depletion by siRNA in mammalian cells resulted in hyperacetylation of histone H4K16 and decreased H3K9me3 and H4K20me3 (160). However, it remains to be seen whether the regulation of these enzymes and specific histone modifications via modulation by oxidative stress and/or redox status of the cells also occur in redox epigenetic chromatin regulation of proinflammatory genes in COPD and smoking-induced diseases.
The cross-talk between histone methylation sites also controls the transcriptional activation of target genes (26, 168). The positive and negative cross talk, ultimately, generates the complex pattern of gene- or locus-specific histone marks, which are linked with distinct chromatin states (euchromatin or heterochromatin), leading to transcriptional repression or activation. Cross-talk exists between various epigenetic events on histones and DNA methylation (Fig. 7). For example, the methylcytosine-binding protein recruits HDACs to methylated DNA as well as to methylated histones. These events are associated with histone deacetylation and chromatin condensation, leading to transcriptional silencing of genes (47, 98). On the contrary, inhibition of HDACs and histone acetylation/methylation leads to upregulation of proinflammatory mediators, such as the granulocyte–macrophage colony-stimulating factor by IL-1β in lung epithelial cells (63).
Although histone H3K4 tri-methylation has been reported to play a role in tumorigenesis and X-chromatin inactivation in human cells (73), little evidence is available for its role in inflammatory responses. Furthermore, hypermethylation of histone H3K4 was found to be unrelated to acute TNF-α-induced gene expression (128). In contrast, the involvement of H3K4 tri-methylation in IL-1β-induced gene expression has been reported (164). Furthermore, H3K4 tri-methylation plays a critical role in IL-1β stimulated secretory leukocyte protease inhibitor expression by modulating RNA polymerase II recruitment, and subsequent engagement of SET1 (164). Conversely, very few reports are available on cross-talk between histone acetylation and methylation in response to CS/oxidative stress-mediated lung inflammation and in pathogenesis of chronic inflammatory lung diseases. Studies on sequential events of histone acetylation/histone methylation will provide the understanding on the new epigenetics-based biomarkers and/or treatment for chronic inflammatory diseases, including COPD (Fig. 7).
Histone demethylases
HDMs catalyze the removal of methyl groups from lysine or arginine residue of histones. They are classified into two types, the lysine-specific demethylase 1 and JmjC domain family proteins involved in the regulation of gene expression (149). Lysine-specific demethylase 1 specifically demethylates histone H3K4me2, and is an important member of HDMs among other transcriptional repression complexes (133). Lysine-specific demethylase 1 utilizes oxygen as an electron acceptor to reduce methylated lysine to form lysine, formaldehyde, and hydrogen peroxide (133). JmjC demethylates mono-, di-, or trimethyl lysine residues by a different mechanism that requires cofactors, such as molecular oxygen, α-ketoglutarate, Fe2+, and ascorbate via a redox modulating process (66, 67). Aberrant expression of HDMs occurs during the course of tumor initiation and progression (76) possibly due to increased oxidative stress. However, the role of ROS/redox GSH status in modulation of HDMs in COPD and other chronic lung diseases remains unknown. Hypoxia is known to occur in tumor microenvironments, as well as in lungs of patients with COPD. The level of lysine-specific demethylase 4B is upregulated in response to hypoxia, which depends on hypoxia-inducible factor 1 alpha (176). Inhibition of H3K4 demethylase (JmjC domain-containing histone demethylation protein 1A) reduces tumor growth in vivo demonstrating its role in regulating histone methylation in hypoxia. Furthermore, induction of JmjC domain-containing histone demethylation protein 1A by hypoxia-inducible factor 1 alpha acts as an epigenetic signal amplifier to enhance hypoxic gene expression, thus facilitating tumor growth (70). In contrast, hypoxia increases global levels of H3K4me3 in alveolar A549 and bronchial Beas-2B cell lines, due to the inhibition of demethylation process, particularly, demethylase (Lysine-specific demethylase 5A). Therefore, hypoxia targets lysine-specific demethylase 5A, which induces H3K4me3 both globally and at gene-specific promoters (heme oxygenase-1 and decay-accelerating factor), thus promoting alterations in the pattern of gene expression and tumor progression (194). Understanding the molecular epigenetic mechanism that involves HDMs in COPD/lung cancer will influence treatment options and outcomes in COPD, lung cancer, and other chronic lung diseases associated with tobacco smoking (139).
DNA Methylation
DNA methylation is a nonhistone epigenetic event that plays an important role in transcriptional regulation, for example, gene silencing. Methylation occurs on cytosine residues, producing 5-meC (5-methylcytosine) (142). DNA methylation is associated with epigenetic silencing, and this effect is, in part, mediated by recruitment of HDACs through the methyl-DNA-binding motifs, including components of several HDAC-containing complexes (97). Environmental factors/agents that trigger oxidative stress, such as diet, genetic predisposition factors, and aging can gradually affect the promoter CpG methylation by recruiting methyl CpG-binding protein 2 and DNA methytransferases to various promoters along with HDACs. This leads to alterations in the expression of tumor suppressor, onocogenes, and pro- and anti-inflammatory genes. Gene-silencing occurs in several genes (e.g., GDNF, MTHFR, OPCML, TNFRSF25, TCF21, PAX8, PTPRN2, and PITX2) due to promoter-specific hypermethylation in lung cancer (7, 12). However, the relationship between gene-specific DNA methylation and smoking history/disease progression is not known (154). Georgiou et al. reported methylation of the p16 promoter in sputum of patients with COPD that significantly correlated with heavy cigarette smoking, suggesting DNA methylation is associated with CS/oxidant-mediated lung diseases (45). Array-based methylation screening from two family-based cohorts (n=1,085 and 396 subjects) revealed 349 CpG sites significantly associated with the presence and severity of COPD. Significant association was shown between SERPINA1 hypomethylation and pathogenesis of COPD with lower lung function. Similarly, Philibert et al. have recently shown that smoking altered DNA methylation in alveolar macrophages correlates with vascular endothelial growth factor 1 gene expression (107). This implicates that DNA methylation/hypomethylation that is caused by oxidative stress in specific genes may be an epigenetic event in pathogenesis of COPD (111).
CS affects DNA methylation on several genes on their CpG island directly or indirectly. CS downregulates DNA methyltransferase 3B in alveolar A549 epithelial cells resulting in demethylation of the CpG island in the oncogene synuclein-gamma and leading to high expression of this prometastatic oncogene in A549 cells (78). Sood et al. identified the promoter methylation from sputum samples of patients demonstrating wood smoke (biomass fuel burning) exposure associated with the COPD phenotype (decline in lung function, airflow obstruction, and chronic bronchitis), and smokers with aberrant p16 and GATA binding protein 4 methylation (136). It would be interesting to conduct studies on identifying epigenetic changes in DNA isolated from chronic CS or biomass/wood smoke exposed mouse lungs to identify the genes that are specifically modified by CS and/or biomass/wood smoke exposure that can be used as better biomarkers in smoke-mediated chronic lung diseases. Suzuki et al. demonstrated methylation of IL-12Rb2 and Wnt inhibitory factor-1 in COPD patients compared to the non-COPD group, suggesting the molecular epigenetic events that influence COPD-related nonsmall cell lung cancer 141). Another report described the association of prenatal exposure to tobacco smoke with significant changes in two types of DNA methylation (global methylation and promoter CpG island methylation) (18). Furthermore, children exposed to second-hand tobacco smoke had a lower methylation profile of their AluYb8 repetitive elements compared to unexposed children. Genetic susceptibility plays a vital role in smoking related effects based on differences in gene variants (methylation of LINE1 with the common GSTM1 null genotype and CpG-specific methylation with a common GSTP1 haplotype) among children involved in detoxification of tobacco smoke. This supports that epigenetic effects of in utero exposures may be due to alterations in DNA methylation patterns (18). Coordinated changes in DNA methylation, particularly, at the aryl hydrocarbon receptor repressor was observed in lymphoblasts and alveolar macrophages from smokers suggesting the role of epigenetic effects mediated by CS on carcinogenesis and other related comorbidities (92). The DNA methylation status on susceptibility genes may have implications in smokers, who are at high risk for COPD development. These studies suggest that DNA methylation may play an important role in the regulation of proinflammatory response. The role of various histone modifications versus promoter DNA methylation, particularly, in response to oxidative stress in inflammatory conditions, and in pathogenesis of COPD and other smoking related airway diseases is an emerging field of further research.
Translational Impact of Chromatin Remodeling in COPD and Smoking Related Diseases
COPD is predominantly a tobacco smoke-triggered disease characterized by premature lung aging and steroid resistance, which is, in part, due to CS-mediated chromatin modifications. The translational impact of chromatin remodeling in the pathogenesis of COPD and other smoking related diseases is as follows: (i) CS-mediated ROS production activates various kinases leading to histone acetylation and chromatin remodeling on proinflammatory genes. Inhibition of HATs by small molecule inhibitors would inhibit lung inflammation. (ii) HDAC2 reduction is associated with steroid resistance in the inflammatory response by CS-mediated oxidative stress. Activation of HDAC2 by theophylline and curcumin would reverse steroid resistance, and hence, provide protection against lung inflammatory response. (iii) SIRT1 undergoes oxidative/carbonyl post-translational modifications leading to degradation in response to CS. SIRT1 regulates several biological processes via FOXO3 and NF-κB-dependent mechanisms, which are involved in lung inflammaging. Activation of SIRT1 by pharmacological agents would protect lung cells against premature senescence and inflammaging. (iv) HDAC2-dependent epigenetic mechanism may be associated with DNA damage-induced premature senescence and associated secretory senescence phenotype in response to oxidative stress imposed by smoke. (v) CS/oxidative stress causes DNA damage, which is increased in patients with COPD/emphysema. Persistent DNA damage can lead to premature senescence with increased proinflammatory mediators release by secretory senescent cells via forming persistent DNA damage foci, termed as DNA segments with chromatin alterations reinforcing senescence (distinct nuclear structures that sustain damage-induced senescence growth arrest and inflammatory cytokine secretion). (vi) Chromatin remodeling, including histone modifications (e.g., histone acetylation/deacetylation and methylation/demethylation) are shown to regulate cellular senescence. Histone acetylation and NF-κB-dependent proinflammatory cytokines are increased in CS-exposed rodent lungs and in lungs of patients with COPD. Histone modifications are associated with DNA damage/repair and epigenomic instability as well as premature lung aging, which has implications in pathogenesis of COPD. The chromatin modification enzymes responsible for histone methylation/demethylation and acetylation/deacetylation (e.g., HDACs) also regulate cellular senescence and aging. Overall, reversing the CS-induced aberrant chromatin modifications on proinflammatory and prosenescent genes would have effects on attenuating the premature lung aging in the progression of COPD.
Conclusions and Future Directions
Oxidative stress by CS/oxidants is critical for the chronic lung inflammatory response via activation of stress kinases, redox-sensitive transcription factors, and modulation of epigenetic chromatin modifications, which result in gene transcription. Recent studies have focused on identifying genes that undergo epigenetic modifications; it is increasingly important to better understand the molecular mechanism that controls the overall epigenetic state of a cell, particularly, in response to environmental stresses. Another key area that will gain more importance is to understand whether these histone modifications enzymes, such as HATs/HDACs and HMTs/HDMs share any common targets, so as to design a common therapeutic agent. Recent studies have highlighted the pharmacological significance of HDACs and SIRT1 in regulation of lung inflammation. Identification of specific substrates/molecules targeted by HDACs and SIRT1 may have considerable therapeutic implications against chronic inflammatory lung diseases. Histone acetylation and deacetylation, as well as histone/DNA methylation play an important role in oxidative stress-induced chronic inflammatory diseases, such as asthma, COPD, and other smoking induced diseases. HDAC2 is required for glucocorticoids to function as anti-inflammatory agents. The level/activity of HDAC2 is reduced in inflammatory cells and lungs of patients with severe asthma and COPD (114, 187). Activation of HDAC2 or reversal of oxidative post-translational modifications of HDAC2 is another avenue to devise epigenetic-based therapy in treatment of severe asthma and COPD. Thus, understanding the molecular epigenetic redox regulation mechanisms will enable the development of newer epigenetic-based therapeutic strategies in the treatment of chronic inflammatory lung diseases.
Cross-talk exists between various epigenetic markers on histones and DNA methylation. Future studies will help to understand the extent to which these post-translational modifications, which are regulated by ROS in inflammation and steroid resistance, are linked to chronic inflammatory lung diseases associated with cigarette smoking. Studies on ROS and redox epigenetic regulation will further unravel the mechanisms and identify possible epigenetics-based therapies for chronic inflammatory diseases, particularly, COPD and smoking induced diseases.
Footnotes
Acknowledgments
This study was supported by NIH 1R01HL085613, 1R01HL097751, 1R01HL092842, and NIEHS Environmental Health Science Center grant P30-ES01247.