
Abstract
Introduction
Together with peptidyl prolyl cis/trans isomerization (18), disulfide bond formation is often a rate-limiting step of the folding process of a protein. Therefore, cells contain dedicated pathways that catalyze disulfide bond formation to ensure fast and correct folding in vivo. The study of oxidative protein folding was initiated by Anfinsen and his colleagues in the early sixties, using ribonuclease A as a model. In a series of experiments that are now presented in some biochemistry textbooks, they discovered protein disulfide isomerase (PDI), the primary catalyst of disulfide bond formation in the endoplasmic reticulum (17). In this compartment, PDI catalyzes both disulfide bond formation and disulfide bond isomerization, ensuring the correct pairing of cysteine residues [reviewed in Ref. (19)].
The first bacterial catalyst of disulfide bond formation, disulfide bond protein A (DsbA), was identified in the periplasm of Escherichia coli by the group of Jon Beckwith (3) three decades after the discovery of PDI. In contrast to PDI, DsbA only functions as a disulfide-introducing protein and exhibits only very weak isomerase activity. In E. coli, the isomerization of non-native disulfides is ensured by another protein, DsbC.
The pathways of disulfide bond formation and isomerization in bacteria have been reviewed recently in several excellent articles, including some published in this journal (21, 24). We refer the reader to these reviews for in depth coverage of the initial experiments that led to the unraveling of the bacterial disulfide bond machineries. Here, we first briefly summarize the major achievements of the past 20 years and describe the most important properties of the oxidoreductases involved in protein disulfide formation in the E. coli periplasm. Then, we focus on the challenges ahead and on two fascinating mysteries that remain unsolved. We also highlight a new link that has been established recently between oxidative protein folding and the defense mechanisms against oxidative stress. In the last part of this review, we discuss the diversity of bacterial oxidative folding pathways.
Oxidative Protein Folding in E. coli
The proteins involved
Disulfide bond formation by the DsbA-DsbB machinery
Disulfide bonds are introduced into cell envelope proteins by the DsbA (3) (Fig. 1). In E. coli, about 300 proteins (∼40% of the envelope proteome) are known or predicted substrates of DsbA (15, 47). DsbA, like PDI and many other oxidoreductases, belongs to the thioredoxin superfamily. Proteins from that family have a canonical CXXC catalytic motif located in a highly conserved fold, which consists of four β-strands surrounded by three α-helices (37). The catalytic CXXC motif of DsbA is maintained in an oxidized state in vivo (22), which enables DsbA to react with proteins entering the periplasm to oxidize them. The oxidation reaction occurs in two successive steps. First, there is a nucleophilic attack by an active cysteine residue of the substrate on the first cysteine of the oxidized CXXC motif of DsbA. This results in the formation of a mixed-disulfide complex between DsbA and the target protein. Then, the mixed disulfide is attacked by another cysteine of the substrate, which results in the oxidation of the substrate and the reduction of DsbA (3). The increased stability of the reduced form of DsbA compared to its oxidized form endows DsbA with a strongly oxidizing redox potential compared to most structural disulfides in substrate proteins, so that DsbA readily oxidizes thiols to disulfide bonds in its substrates.
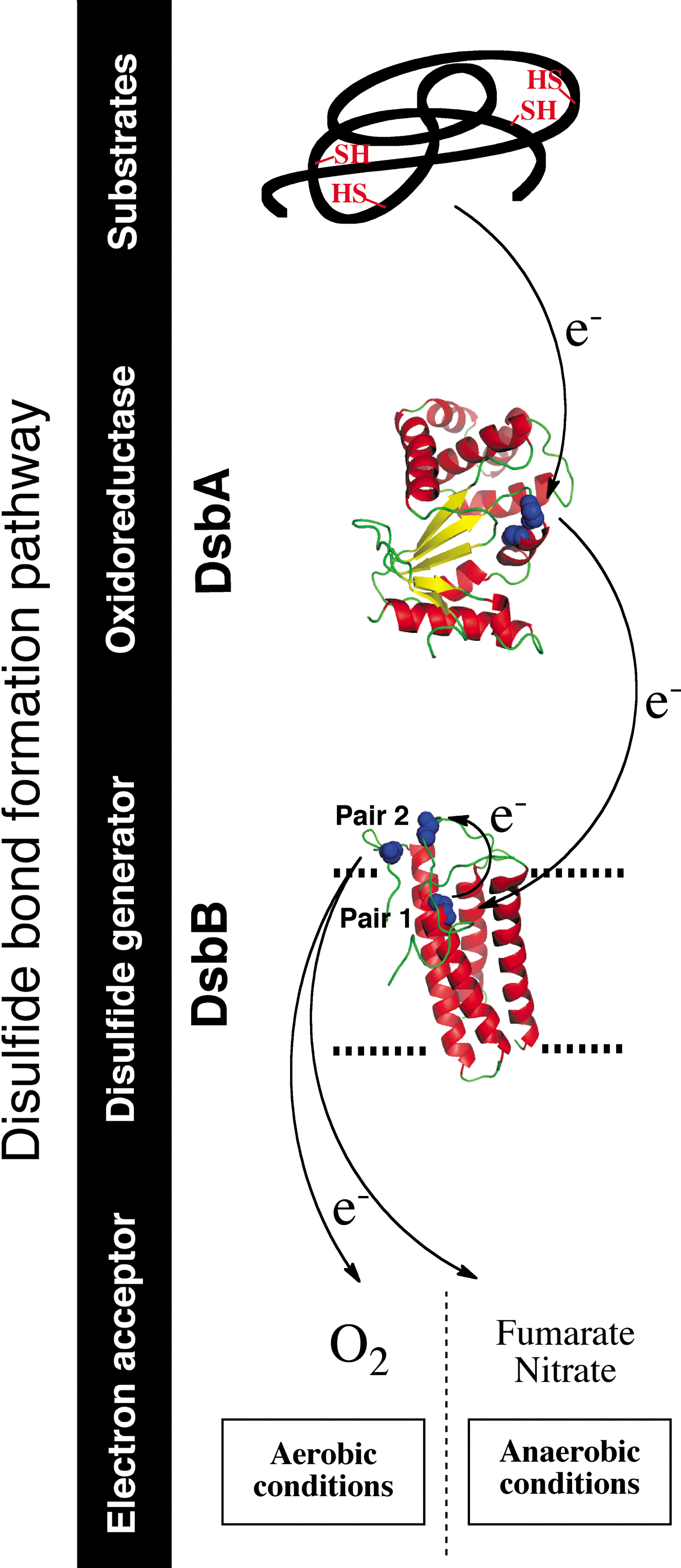
In E. coli, DsbA is recycled back to the oxidized form by the inner membrane protein DsbB (2) (Fig. 1). DsbB has four transmembrane segments and two periplasmic loops, each containing one pair of conserved cysteine residues that are maintained in an oxidized state (pairs 1 and 2). DsbB uses those cysteine residues to channel the electrons away from DsbA and to deliver them to bound quinone molecules. Thus, DsbB generates disulfides de novo with concomitant quinone reduction.
The reaction between DsbA and DsbB is initiated by the nucleophilic attack of the first cysteine residue of DsbA's CXXC motif on the oxidized cysteines of the second periplasmic loop of DsbB (pair 2), which leads to the formation of a DsbA-DsbB mixed-disulfide complex. The structure of this complex, which was solved by Inaba, Ito, and their collaborators (20), provided insightful information that significantly advanced our understanding of the mechanism used by DsbB to generate disulfides. The mixed disulfide is then transferred to DsbA, which releases the cysteine residues of pair 2 of DsbB in the reduced form. Reoxidation of these cysteines occurs by electron transfer to the cysteines of pair 1, which are finally recycled back to the oxidized state by transferring the electrons to an ubiquinone molecule. The electrons are then transferred to the electron transport chain, molecular oxygen being the terminal electron acceptor (1). The DsbA-DsbB system is also able to function anaerobically. Under these conditions, DsbB transfers the electrons to menaquinone and then to other terminal electron acceptors such as fumarate and nitrate (1) (Fig. 1).
Disulfide bond isomerization by the DsbC-DsbD system
DsbA is a powerful oxidant that preferentially introduces disulfides in a vectorial manner into proteins entering the periplasm (23), that is, between cysteine residues that are consecutive in the primary amino acid sequence. Therefore, proteins that require formation of nonconsecutive disulfides to attain their native structure are often incorrectly oxidized by DsbA. An isomerization system is present in the E. coli periplasm to catalyze the rearrangement of non-native disulfide bonds (39) (Fig. 2).
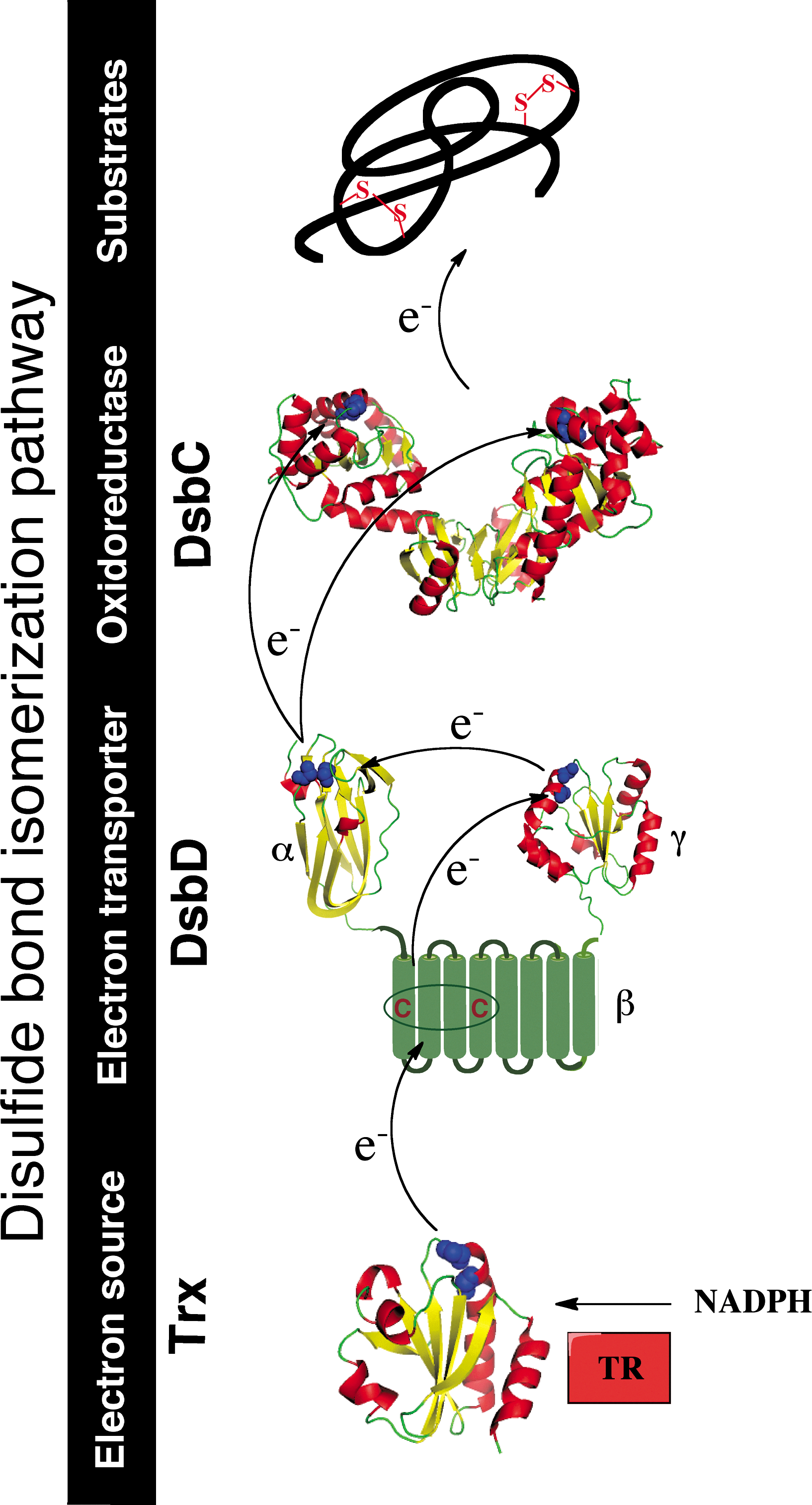
The main player of the E. coli isomerization pathway is the PDI DsbC. DsbC is a soluble homodimeric protein characterized by a V-shaped structure (36). Each monomer of DsbC presents an N-terminal dimerization domain and a C-terminal thioredoxin-like domain with a CXXC active site that is found predominantly in the reduced state in vivo (22). The dimerization of DsbC allows the formation of an uncharged cleft, which plays an important role in substrate binding. This is nicely illustrated by the fact that fusion of the N-terminal dimerization domain of DsbC to either DsbA or thioredoxin allows those proteins to function as disulfide isomerases in the periplasm [reviewed in Ref. (14)].
Upon binding of the substrate into DsbC's uncharged cleft, the N-terminal cysteine of the CXXC motif of DsbC performs a nucleophilic attack on an incorrect disulfide present in the substrate. This results in the formation of an unstable mixed-disulfide complex between DsbC and the substrate protein. This mixed disulfide is then attacked by another cysteine residue of the misfolded protein, creating a more stable disulfide in the target protein and releasing DsbC in the reduced state (here DsbC functions as an isomerase). Alternatively, the second cysteine of the CXXC motif of DsbC attacks the mixed disulfide, which results in the reduction of the substrate and the oxidation of DsbC (in that case, DsbC functions as a reductase).
The protein that recycles DsbC back to the reduced state is the inner membrane protein DsbD (39). DsbD is a 59-kDa monomeric protein composed of three distinct structural domains: an N-terminal periplasmic domain (DsbDα) with an immunoglobulin-like fold, a membrane-embedded domain with eight transmembrane segments (DsbDβ), and a C-terminal periplasmic thioredoxin-like domain (DsbDγ) (Fig. 2). Each of these domains contains a pair of conserved cysteine residues that are redox active and essential for the function of DsbD (see below) (26).
DsbD functions as an electron hub that dispatches reducing equivalents to various periplasmic oxidoreductases
The function of DsbD is to transfer reducing equivalents from the cytoplasmic thioredoxin system to the periplasm (39), by a mechanism that is still not fully understood (see below) (Fig. 2). The thioredoxin system, which involves thioredoxin and thioredoxin reductase, maintains cytoplasmic proteins reduced at the expense of nicotinamide adenine dinucleotide phosphate (NADPH) (10).
DsbC is not the only substrate of DsbD. In fact, this protein functions as an electron hub that dispatches reducing equivalents to various redox pathways present in the cell envelope (Fig. 3). For instance, DsbD provides reductants to CcmG, an oxidoreductase involved in the maturation of cytochrome c (46). Moreover, we recently found that DsbG, a homodimeric protein that is homologous to DsbC, uses electrons received from DsbD to control the global sulfenic acid content of the periplasm (13) (Fig. 3). Sulfenic acids are highly unstable modifications that form on cysteine residues exposed to reactive oxygen species (ROS). Further oxidation of sulfenic acids leads to sulfinic (−SO2H) and sulfonic (−SO3H) acids, which irreversibly modify proteins. The role of DsbG is to protect envelope proteins containing a single cysteine residue from oxidation to sulfenic acid to prevent their inactivation (13).
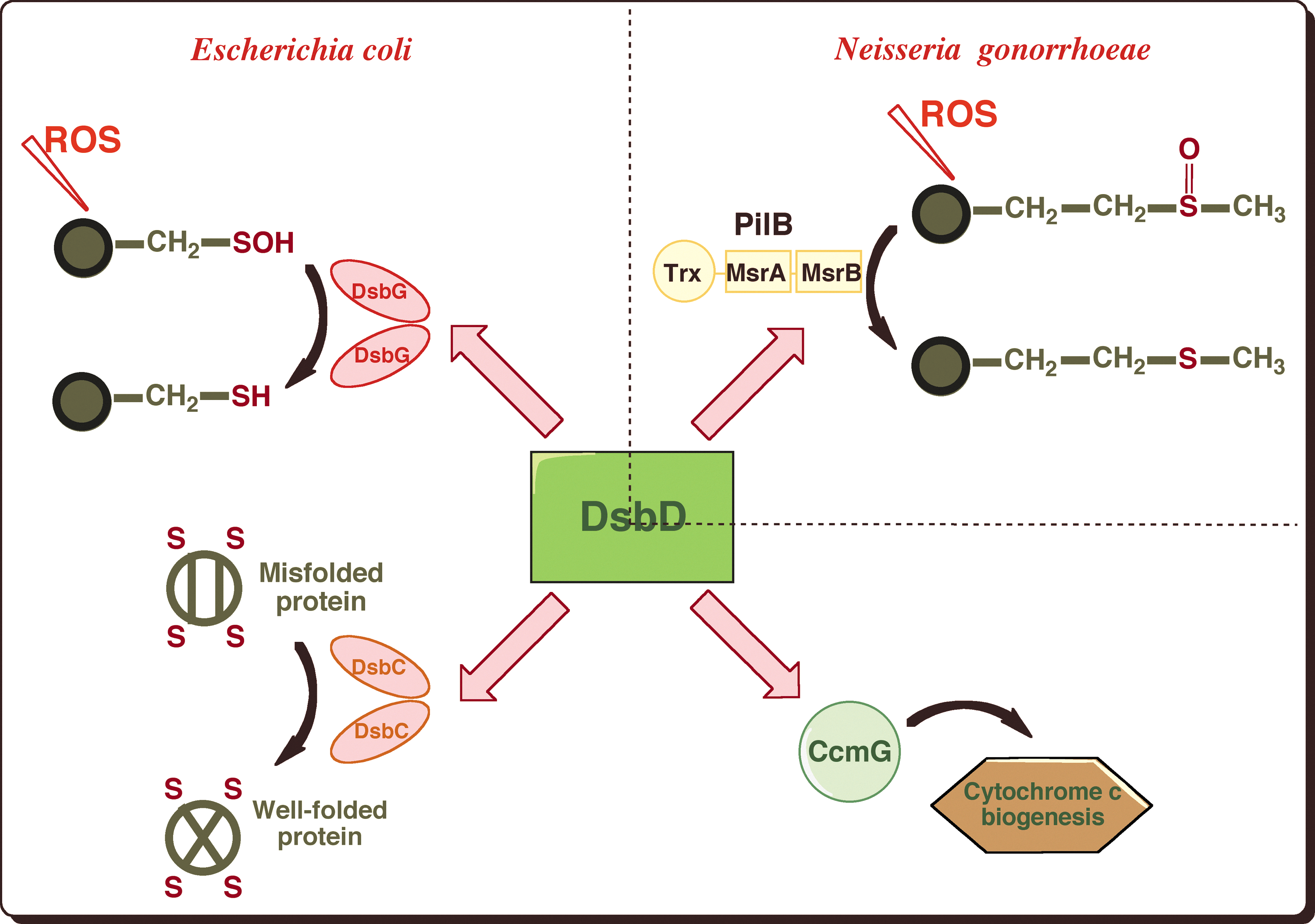
Another example of the role played by DsbD in the defense mechanisms against oxidative stress was reported in Neisseria gonorrhoeae. In this bacterium, DsbD provides electrons to the N-terminal domain of PilB (4), a multidomain protein that exhibits methionine sulfoxide reductase (Msr) activity (Fig. 3). Msrs are antioxidant enzymes that catalyze the reduction of methionine sulfoxides (MetO) back to the reduced state. MetO are formed upon exposure of methionine residues to ROS, which leads, if unrepaired, to alterations in protein conformation and loss of biological activity.
Unsolved mysteries
The work performed over the past 20 years has led to the identification and detailed characterization of most of the proteins that catalyze disulfide bond formation and isomerization in the E. coli periplasm. The oxidation and isomerization pathways have been reconstituted in vitro; the kinetic constants of the various electron transfer reactions have been determined, and the structures of most of the players involved have been solved [reviewed in Refs. (14, 24)]. However, despite these tremendous achievements, fundamental questions remain unanswered. Here, we focus on two major unsolved mysteries that require further investigation.
How does DsbD transfer reducing equivalents across the membrane?
A fascinating remaining mystery is the mechanism by which DsbD transfers reducing equivalents from the cytoplasmic thioredoxin system to periplasmic oxidoreductases such as DsbC and DsbG without using a cofactor. Elegant work by Beckwith and collaborators has revealed that the activity of DsbD depends on a cascade of thiol–disulfide exchange reactions involving the 3 pairs of cysteine residues of DsbD (26). First, thioredoxin reduces the disulfide bond involving the two cysteine residues of DsbDβ. Then, the electrons are transferred from those membrane-embedded cysteines to those of DsbDγ, DsbDα, and finally to the catalytic site of the periplasmic oxidoreductases (Fig. 2).
The reactions taking place in the periplasm, that is, the reduction of DsbDα by DsbDγ and the reaction between DsbDα and DsbC, have been thoroughly characterized [for instance, see Refs. (11, 40)]. We would like to highlight recent data reported by Redfield and coworkers, who characterized in detail the thiol–disulfide exchange reaction between the two soluble γ and α domains of DsbD (35). In particular, they demonstrated that the strength of the interaction between the two soluble subunits of DsbD depends on their respective redox states. They showed that complex formation is facilitated by a relatively high affinity (Kd of 86 μM) of the oxidized DsbDα for the reduced DsbDγ, whereas reduced DsbDα has a lowered affinity (K d of 2.4–3.5 mM) for the oxidized DsbDγ. These oxidation state-dependent interactions between both partners avoid the formation of nonefficient complexes that would prevent the transfer of reductants across the membrane.
In contrast to the abundance of details regarding the reactions taking place in the periplasm, we have a much poorer understanding of the mechanism used by DsbDβ to transfer reducing equivalents across the inner membrane. This is clearly one of the most intriguing questions that remains to be solved regarding the bacterial oxidative folding pathways. It is likely that the solution to this problem will come from solving the structure of the membranous domain of DsbD, which is still unknown. The current working model for the structure of DsbDβ is a low-resolution structure based on the work of Cho et al. (6, 7, 9). Using small thiol-modifying reagents, these authors determined the accessibility of the catalytic cysteine residues of DsbDβ as well as that of membrane-embedded residues that they substituted by cysteines. Their data suggest that DsbDβ adopts an hourglass structure in which the catalytic cysteine residues are located at the juncture of the two cavities where they are exposed to both thioredoxin in the cytoplasm and DsbDγ in the periplasm. The redox states of these cysteines do not seem to affect the conformation of the membranous domain.
How do oxidative protein-folding catalysts cooperate with periplasmic chaperones?
A second fascinating mystery is how the protein-folding catalysts DsbA and DsbC coordinate their action with the periplasmic chaperones that assist the assembly of outer membrane (OM) proteins in Gram-negative bacteria. There are two major groups of proteins present in the OM: (i) lipoproteins that are present in the periplasm, but are anchored by a lipid moiety to the inner leaflet of the OM, and (ii) β-barrel proteins that are integral membrane proteins [reviewed in Ref. (31)]. After secretion to the periplasm, OM proteins are transported across this hydrophilic compartment by dedicated chaperones that deliver them to specific OM insertion machineries: lipoproteins are escorted by LolA, whereas SurA and Skp assist the assembly of ®-barrel proteins (44) (Fig. 4). Two OM proteins, the ®-barrel LptD and the lipoprotein RcsF, have recently been shown to require the formation of nonconsecutive disulfides for activity, which raises interesting questions regarding how and where disulfides are introduced into proteins destined for the OM.

LptD has received much attention lately. It is an essential β-barrel protein that inserts lipopolysaccharides (LPS) into the OM [reviewed in Ref. (42)] and that has recently been shown to be the cellular target of a new class of peptidomimetic antibiotics (45). Although the mechanism used by LptD to insert LPS in the OM has not been elucidated yet, some key results have been obtained regarding the assembly pathway of this protein (Fig. 4). First, LptD has been shown to interact preferentially with the chaperone SurA (48): the chaperone is thought to bind to unfolded LptD upon entry into the periplasm and to deliver the protein to the Bam assembly complex for insertion in the OM (27, 50). Second, LptD forms a very stable complex with the lipoprotein LptE in the OM (5), which is required for LptD assembly. Third, LptD contains two pairs of cysteine residues that have recently been shown to form two nonconsecutive disulfides (connecting C1 to C3 and C2 to C4) bridging the N-terminal periplasmic soluble domain of the protein to the C-terminal domain that likely adopts a membrane-embedded β-barrel conformation (41). Interestingly, disulfide bond formation does not appear to be required for LptD folding and stability. However, formation of at least one of the nonconsecutive disulfides is required for function (41). Both DsbA and DsbC have been trapped in complex with LptD (12, 25), indicating that these oxidoreductases play a role in the assembly pathway of the protein. However, it is not known when oxidation of LptD takes place. Does DsbA introduce the disulfides into LptD upon entry of the protein in the periplasm, as would be suggested by the current model for disulfide bond formation? Or, does SurA interact with LptD first, the disulfides being introduced upon or after assembly of LptD in the OM, and what is the role of DsbC? The fact that the formation of the LptD/LptE complex in the OM is required for proper oxidation of LptD (5) suggests that the oxidation of this protein is only completed after LptD reaches the OM. Clearly, further investigation is required to firmly establish the oxidative folding pathway of this protein and to determine how DsbA and DsbC assist the assembly of ®-barrel proteins.
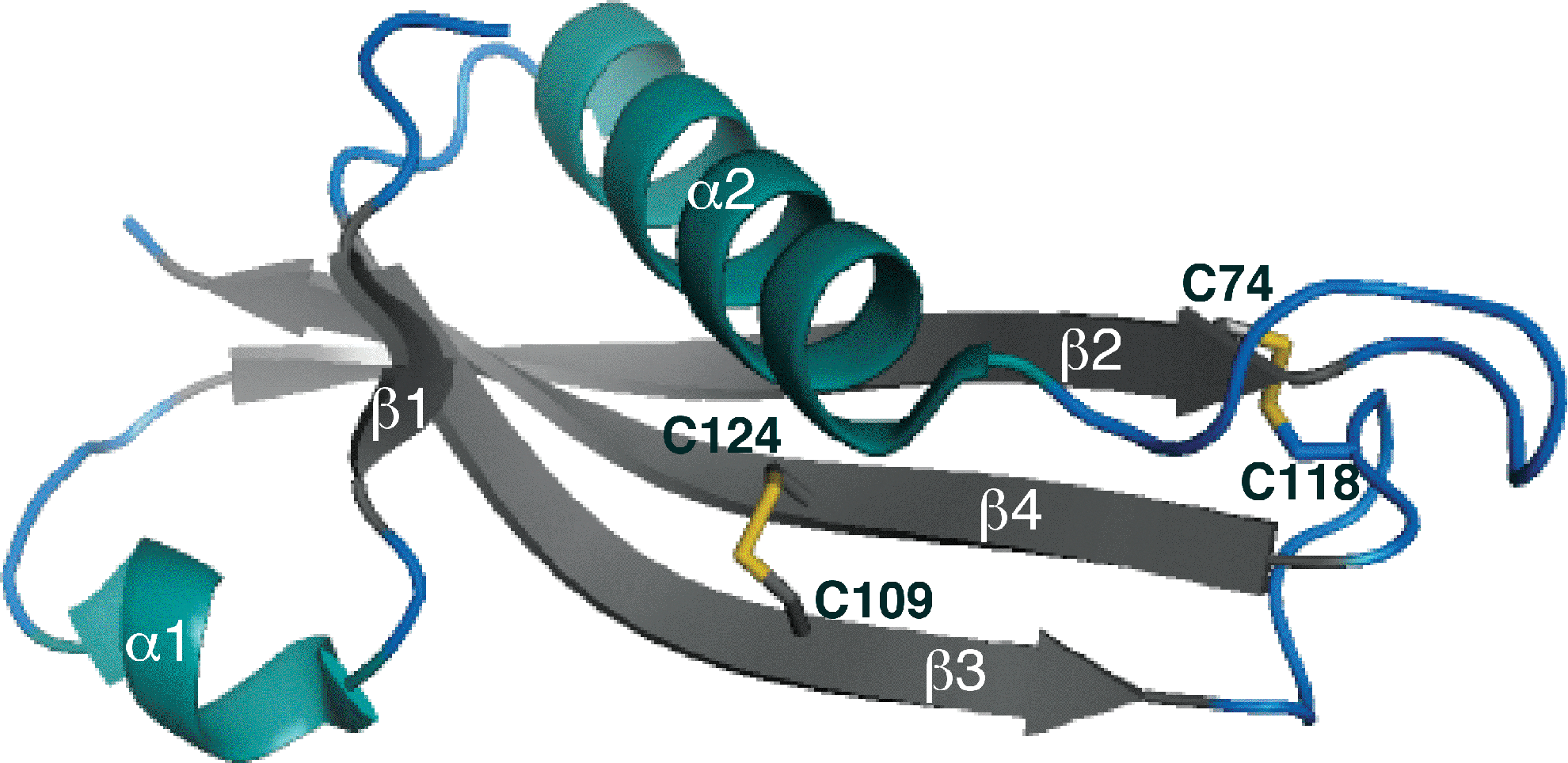
Similar questions can be raised regarding the small OM lipoprotein RcsF (14 kDa). RcsF functions as a sensor that activates a signaling cascade controlling the transcription of genes involved in cell division, motility, and biofilm formation in response to various envelope stresses [reviewed in Ref. (34)]. We recently solved the structure of RcsF, which revealed that the protein has two nonconsecutive disulfide bonds (C1–C3 and C2–C4) essential for folding and activity (Fig. 5) (30). The formation of these disulfides depends on DsbA and DsbC (25, 30). Interestingly, one of the nonconsecutive disulfides bridges, two cysteine residues that are on two adjacent β-strands, forms a so-called cross-strand disulfide (CSD) (49). CSD usually plays a functional role, suggesting that the activity of RcsF might be redox regulated. Here also, further work is required to elucidate how DsbA, DsbC, and LolA, the chaperone dedicated to lipoproteins, cooperate in the assembly process of RcsF (Fig. 4).
A Variety of Disulfide Bond-Forming Systems Among Gram-Negative Bacteria
The E. coli Dsb system is generally considered as the paradigm of oxidative protein-folding pathways in bacteria. However, bioinformatic analysis of whole-genome sequencing data, completed by molecular studies of disulfide bond machineries in bacteria other than E. coli, has revealed that there is a significant diversity of bacterial disulfide bond-forming systems [reviewed in Ref. (43)].
First, many bacterial genomes encode a repertoire of thiol–disulfide oxidoreductases that is significantly expanded compared to the E. coli K12 system. For instance, the genome of Neisseria meningitidis, a causative agent of epidemic meningitis, encodes three DsbA proteins, two of which are anchored in the inner membrane by a lipid moiety, while the third one is soluble like E. coli DsbA. Neisserial DsbAs differ in terms of redox properties, surface characteristics, and substrate specificity (29). In other bacteria, additional redox pairs that are responsible for the oxidation of specific substrates are present in addition to the classical DsbA-DsbB and DsbC-DsbD pathways. For instance, in Salmonella enterica serovar Typhimurium, DsbL and DsbI, which are homologous to DsbA and DsbB, respectively, specifically oxidize a periplasmic aryl sulfate sulfotransferase, an enzyme that detoxifies phenolic substances and antibiotics (33). Alternatively, other disulfide bond systems diverge from the E. coli machinery by the fact that they lack an isomerization pathway, such as in Wolbachia pipientis, an insect parasite (28).
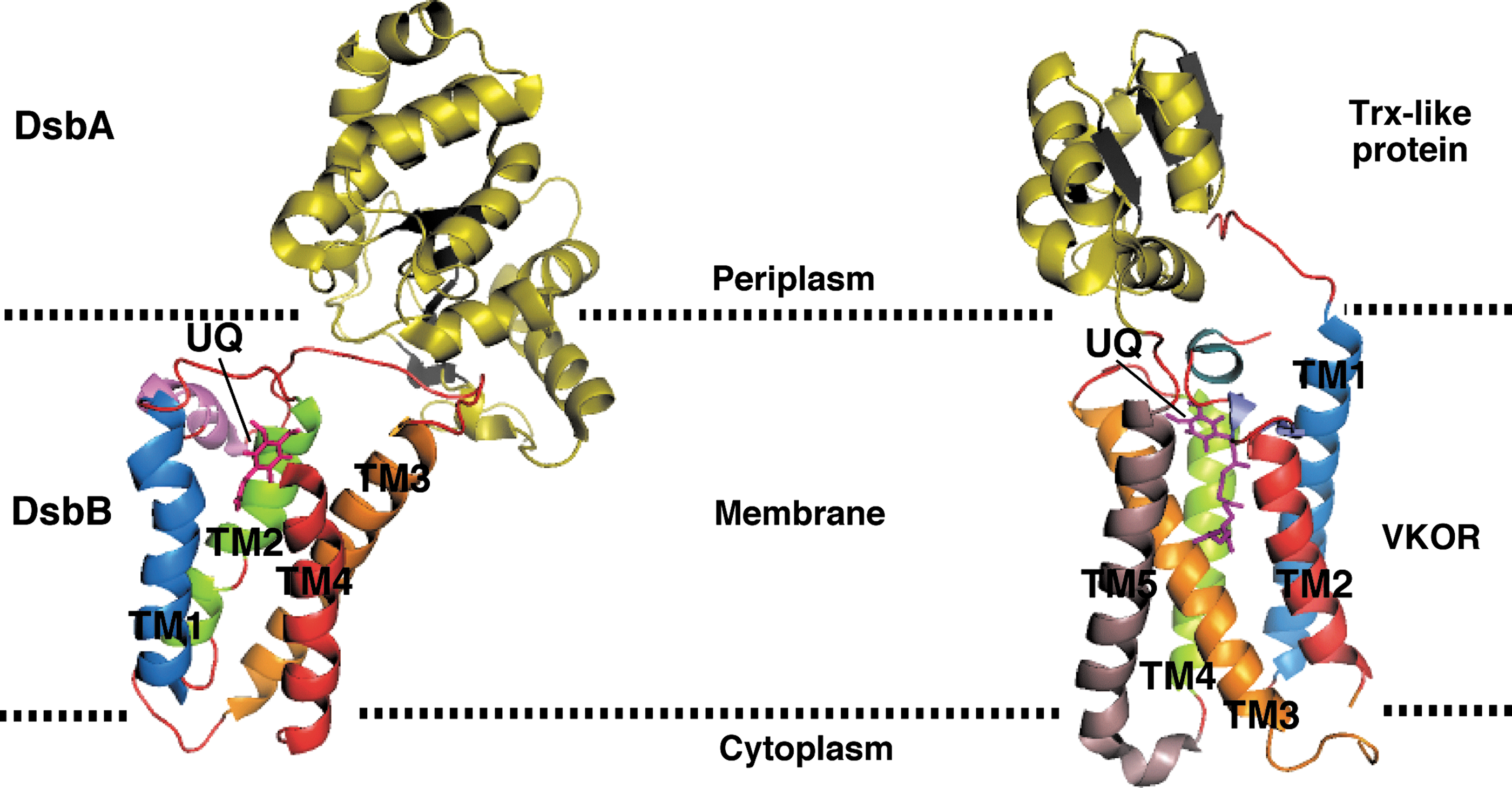
One of the most interesting findings in this area was reported recently by Dutton et al. (15, 16). The observation that some bacteria, including cyanobacteria and certain actinobacteria, ɛ-, and ™-proteobacteria, possess a DsbA homolog, but lack a homolog of DsbB, has led them to the identification of a protein with a DsbB-like activity. This protein is the bacterial homolog of the eukaryotic vitamin K epoxide reductase (VKOR), an enzyme that catalyzes the reduction of vitamin K epoxide, a quinone derivative, to vitamin K. Vitamin K is required as an essential cofactor for enzyme-catalyzed carboxylation reactions responsible for blood coagulation (38). Interestingly, the VKOR homolog from Mycobacterium tuberculosis (Mtb VKOR) can efficiently restore disulfide bond formation in E. coli dsbB mutants in a DsbA-dependent manner (15). These results suggest that Mtb VKOR and E. coli DsbB function in an analogous way in vivo, and that VKOR can substitute for DsbB in bacteria that do not encode the dsbB gene. It is noteworthy that when expressed in E. coli, Mtb VKOR is inhibited by warfarin, an anticoagulant that blocks the human VKOR function, whereas DsbB activity is unaffected by this drug (16).
Recently, the structure of a natural fusion protein from Synechococcus sp. has been solved, in which a VKOR homolog and its DsbA-like redox partner are found in complex (32) (Fig. 6). The structure revealed that VKOR and DsbB share three-dimensional structure similarities, which was unexpected because of the absence of sequence homology between these two proteins. The structural similarities suggest that DsbB and VKOR generate disulfide bonds via a similar mechanism of action.
We also would like to highlight a recent work that revealed the diversity of the proteins from the DsbD superfamily. Using bioinformatic programs to seek homologs of E. coli DsbD, Cho et al. detected a new class of reductive proteins whose prototype is Salmonella typhimurium ScsB (8). Although the ScsB class has domains comparable to those of DsbD, the N-terminal periplasmic domain of ScsB is quite different from DsbDα, suggesting that ScsB proteins act on a different array of substrate proteins from those already known for DsbD. Indeed, Caulobacter crescentus ScsB was found to transfer reducing equivalents to ScsC, a novel bacterial disulfide isomerase, and to TlpA, a thioredoxin-like protein that in turn reduces a peroxidase PprX. These results reveal that the range of electron transfer reactions involving DsbD-like proteins is much broader than has been understood. Furthermore, they suggest that the unraveling of the redox pathways at work in bacteria other than E. coli will likely lead to the identification of novel types of DsbD substrates.
Conclusions
We have a good knowledge of the pathways of disulfide bond formation in E. coli: the proteins involved have been identified, and their major characteristics have been determined. Understanding the details of the mechanism employed by DsbD to transport reducing equivalents across the membrane probably remains the most important problem to be solved to complete the molecular characterization of the E. coli Dsb proteins.
The unraveling of the E. coli disulfide bond formation machinery has allowed discovery of the basic principles of the bacterial oxidative folding pathways in general. It is now established that in bacteria, disulfides are generated by membrane proteins such as DsbB or VKOR that connect oxidative protein folding to the electron transport chain. The generated disulfides are then donated to folding proteins by oxidoreductases from the thioredoxin superfamily. In some bacteria, such as E. coli, an isomerization pathway exists next to the oxidation pathway. This reducing pathway uses electrons derived from the cytoplasmic pool of NADPH to correct wrongly formed disulfides.
One of the challenges of the coming years will be to put the knowledge of the disulfide formation machineries into the global cellular context. For instance, it will be interesting to determine how the oxidative protein folding catalysts cooperate with the other proteins involved in protein assembly. Another important challenge will be to characterize in details the oxidative folding pathways at work in bacteria other than E. coli, including pathogens. As many virulence factors are stabilized by disulfide bonds, this could lead to the design of new antibiotics.
Footnotes
Acknowledgments
We would like to thank Lloyd Ruddock and Robert Freedman for the invitation. We thank Pauline Leverrier, Seung-Hyun Cho, Alexandra Gennaris, and Carole Linster for critical reading of the manuscript. JFC is Chercheur Qualifié of the FRS-FNRS. KD is a research fellow of the FRIA. This work was supported by the European Research Council (FP7/2007–2013) ERC independent researcher starting grant 282335–Sulfenic and by grants from the FRS-FNRS to JFC. We apologize to authors whose work was not cited directly due to reference limitations.