
Abstract
Introduction
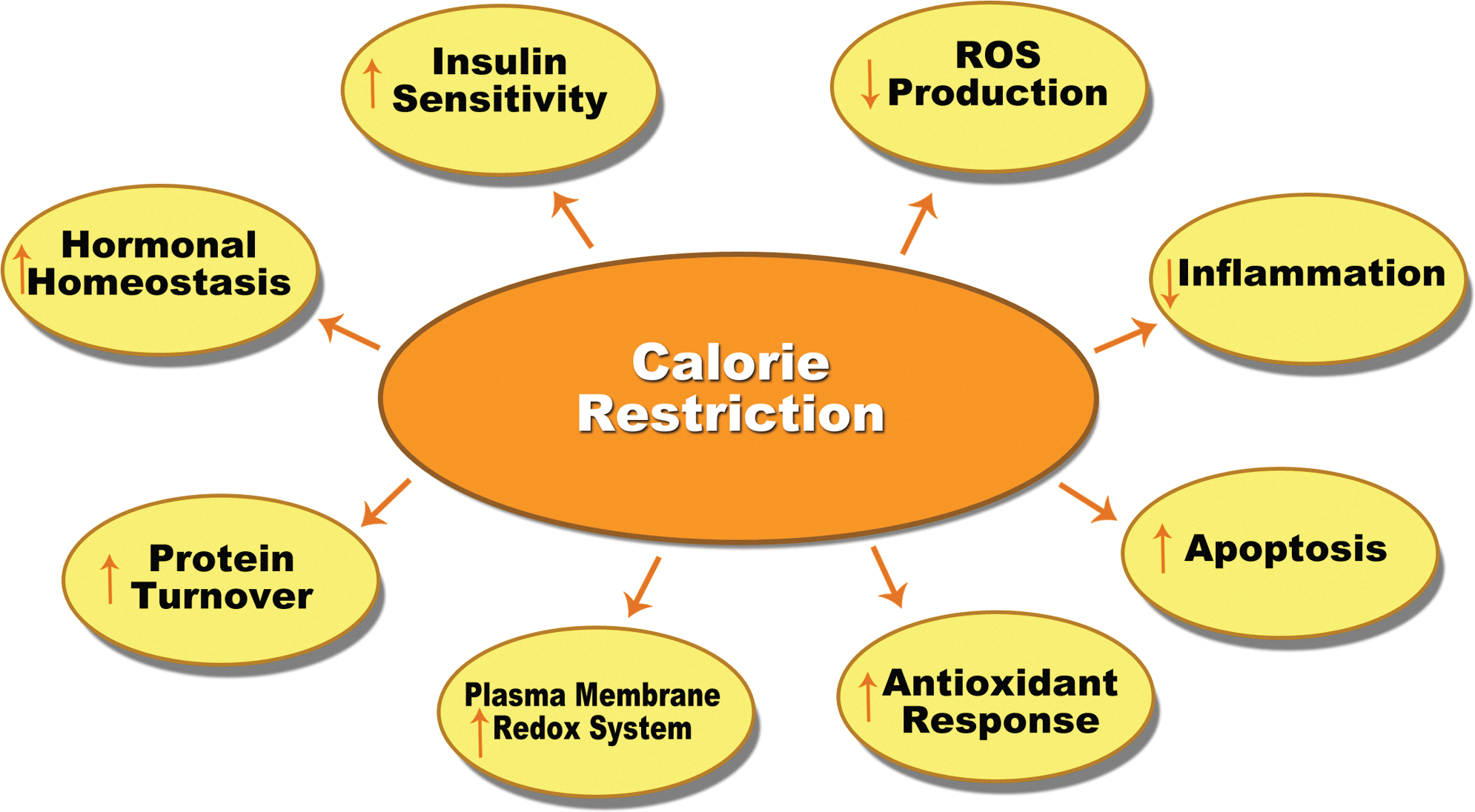
It is well known that CR decreases reactive oxygen species (ROS) production, enhances the plasma membrane redox system, decreases inflammation, and improves insulin signaling pathways (Fig. 1). Most of these benefits are thought to be associated with better mitochondrial function. In this venue, CR has been shown to produce very efficient mitochondrial performance, which is equivalent to a maintained ATP production while reducing ROS generation in the complexes of the electron transport chain (ETC) of the mitochondria. Thereby, this improvement on mitochondrial efficiency under CR is able to maintain cellular metabolism with a lower oxidative stress damage accumulation, resulting in a decreased aging rate at cellular and organismic levels.
Since adherence to a CR diet is quite difficult for humans, investigators have recently pursued compounds that could mimic CR, providing benefits to health and lifespan. In this review, we focus on the changes in molecular mechanisms produced by CR and CR-mimetics in the mitochondria that promote health benefits.
CR Prevents the Decline of Oxidative Capacity with Aging
Mitochondria are responsible for production of ∼90% of intracellular ATP. Mitochondrial respiration plays a major role in preserving longevity, as it has been demonstrated that increments in respiratory activity enhance cell and organism longevity (6). It is known that impaired mitochondrial functioning occurs with aging and leads to deterioration in the structure and function of tissues, implicating this process in the development of several age-related diseases (Fig. 2) (76). The causes of declining oxidative capacity could be based on decreased mitochondrial content and/or function. There are several contradictory reports concerning changes of mitochondrial content with aging, but the impairment of mitochondrial function is a well-established fact (76).
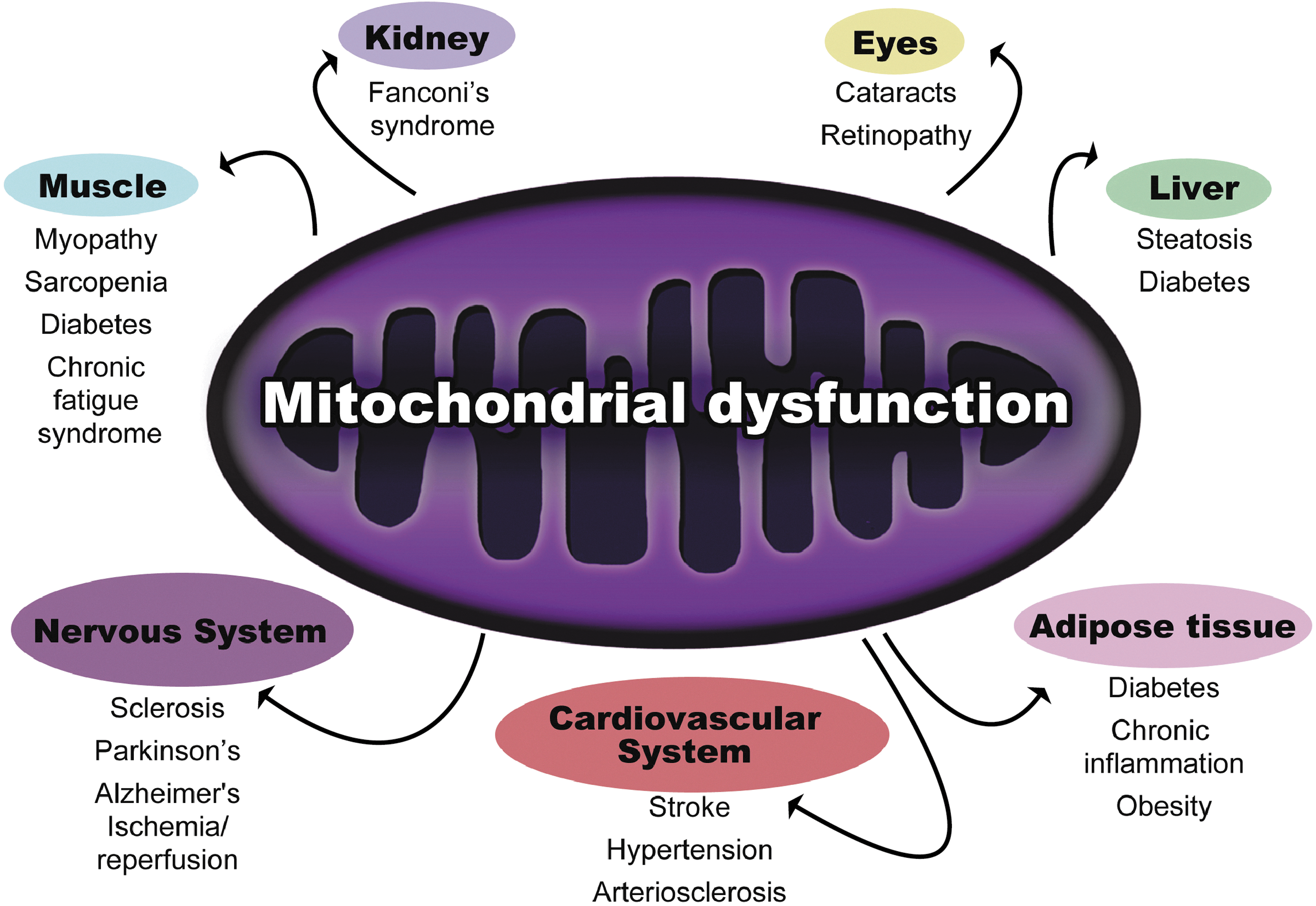
The proposed causes of mitochondrial dysfunction involve the synthesis of nonfunctional complexes of the ETC, damaged mitochondrial DNA (mtDNA), and/or the accumulation of nonfunctional proteins by ROS-induced modifications (76, 88). For instance, it has been demonstrated that aged tissues exhibit dysfunctional complex IV of the ETC. This alteration mainly affects the muscle because it requires more energy than most other tissues and is also one of the tissues most affected by aging (25). It has been recently demonstrated that the decline in oxidative capacity is the primary contributing factor to age-related muscle atrophy (88). Moreover, sarcopenia, the decline of muscle mass associated with aging, makes individuals more sedentary and is often accompanied by an increase in fat mass that compromises the quality of life and survival of the patients (63). Aged muscle tissue is characterized by a decline in the number and size of fibers, predominately type II fibers (54). CR has been shown to completely prevent the decline in mass-specific skeletal muscle aerobic performance between young and old rats (42). The protection of aerobic function was due to maintenance of skeletal muscle oxidative capacity with aging and enhanced mitochondrial function in CR animals. Accordingly, the metabolic rate when normalized to body weight does not decline with age in CR animals but does decrease in AL counterparts. It has been demonstrated that even a short-term CR period improves mitochondrial function in as little as two weeks (9). Mitochondrial oxidative capacity is a strong predictor of insulin sensitivity in skeletal muscle (12). The maintenance of skeletal muscle oxidative capacity by CR has been demonstrated even after accounting for the extension of life span. Baker et al. elegantly determined that CR completely prevented the 40% decline in plantaris and gastrocnemius muscle oxidative capacity observed in AL counterparts. Surprisingly, no differences were found in citrate synthase protein levels (5). In agreement with that, human studies in healthy overweight but nonobese individuals have shown that key mitochondrial enzymes of the tricarboxylic acid cycle, beta-oxidation, and ETC are unchanged. These data imply that CR improves the quality of individual mitochondria and ultimately results in the maintenance of healthy mitochondria with high bioenergenetic capacity and reduced propensity toward oxidant production. Improved mitochondrial function does not solely occur in the muscle tissue. For example, within the context of the liver and the white adipose tissue (WAT), increased mitochondrial biogenesis contributes to an improved metabolic profile, increasing the fatty acid oxidation and decreasing circulating triglycerides to maintain insulin sensitivity in peripheral tissues (8, 71). These events result in reduced fat accumulation, which is known to promote lifespan extension.
CR Induces Mitochondrial Biosynthesis Through PGC-1α Activation
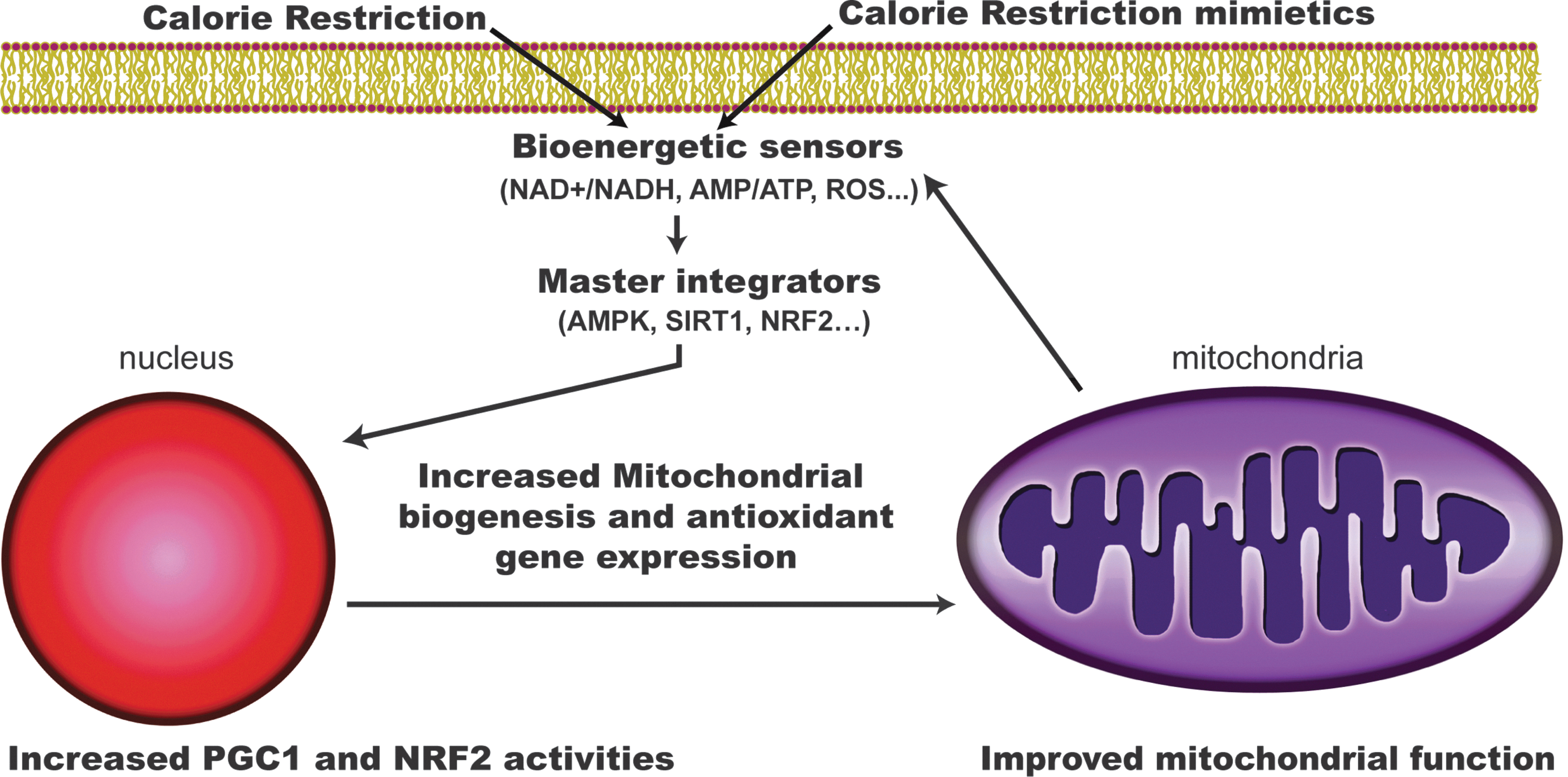
One of the hallmarks of CR is the increase of functional respiratory units (mitochondrial biogenesis) and the promotion of changes in dynamics and composition of this organelle (Fig. 3) (17, 27, 58). Mitochondrial respiration and content actually increases in yeast and nematodes under CR (46, 57). There are some reports that show increased amounts of mtDNA, ATP concentration, and oxygen consumption in WAT and many other tissues of CR mice when compared with AL mice. Additionally, it has been reported that CR may alter mitochondrial membrane fatty acid composition, allowing for increased mitochondrial function (27).

In mammals, the regulation of mitochondrial biogenesis is complex, and it is not known whether the effects of CR on mitochondrial biogenesis are tissue-specific. Mitochondrial biogenesis is a highly regulated process that coordinates the activity of the ∼1000 genes involved in mitochondrial function (Fig. 4). This process requires coordination of the nuclear and mitochondrial genomes. Under CR, transcriptional coactivator peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC-1α) is expressed and activated, leading to an increase of mitochondrial mass. PGC-1α gene expression has been shown to be maintained with aging in CR models (5). In addition to PGC-1α there are other master regulators that are expressed under CR conditions like peroxisome proliferator activated receptor (PPAR) family and liver X receptor, which control fatty acid metabolism. PGC-1α physiological importance has been demonstrated since repression of this protein by a mutant form of the huntingtin protein leads to mitochondrial dysfunction, whereas its overexpression rescues cells from the deleterious effect of huntingtin (20). PGC-1α specifically modulates the activity of several transcription factors and coactivators involved in mitochondrial respiration and biogenesis such as nuclear respiratory factor 1 (NRF-1), NRF-2, PPARγ, steroid receptor coactivator-1, and mitochondrial transcriptor factor A. NRF-1 and NRF-2 coordinate the expression of nuclear and mitochondrial genes that encode most of the subunits of mitochondrial complexes (91). In the same sense, PGC-1α activates the shift of substrate utilization from carbohydrates to fatty acids through coregulation of PPARγ (75). Although it has been reported that CR enhances mitochondrial activities, the mechanism remains controversial as reports conflict about the extent to which CR changes expression of genes involved in nutrient sensing, mitochondrial biogenesis, and other key mitochondrial enzymes involved in the Krebs cycle, β-oxidation, and ETC activities in humans (17). The differences found in the literature might be based on the species used in the studies, the different ages of the animals, the tissues analyzed, and/or the way that the data are presented (e.g., activity/g of wet tissue weight versus activity/g of protein extracted). In any case, we consider the quantification of mitochondrial number by electron microscopy the best option to determine changes in mitochondrial number (58).
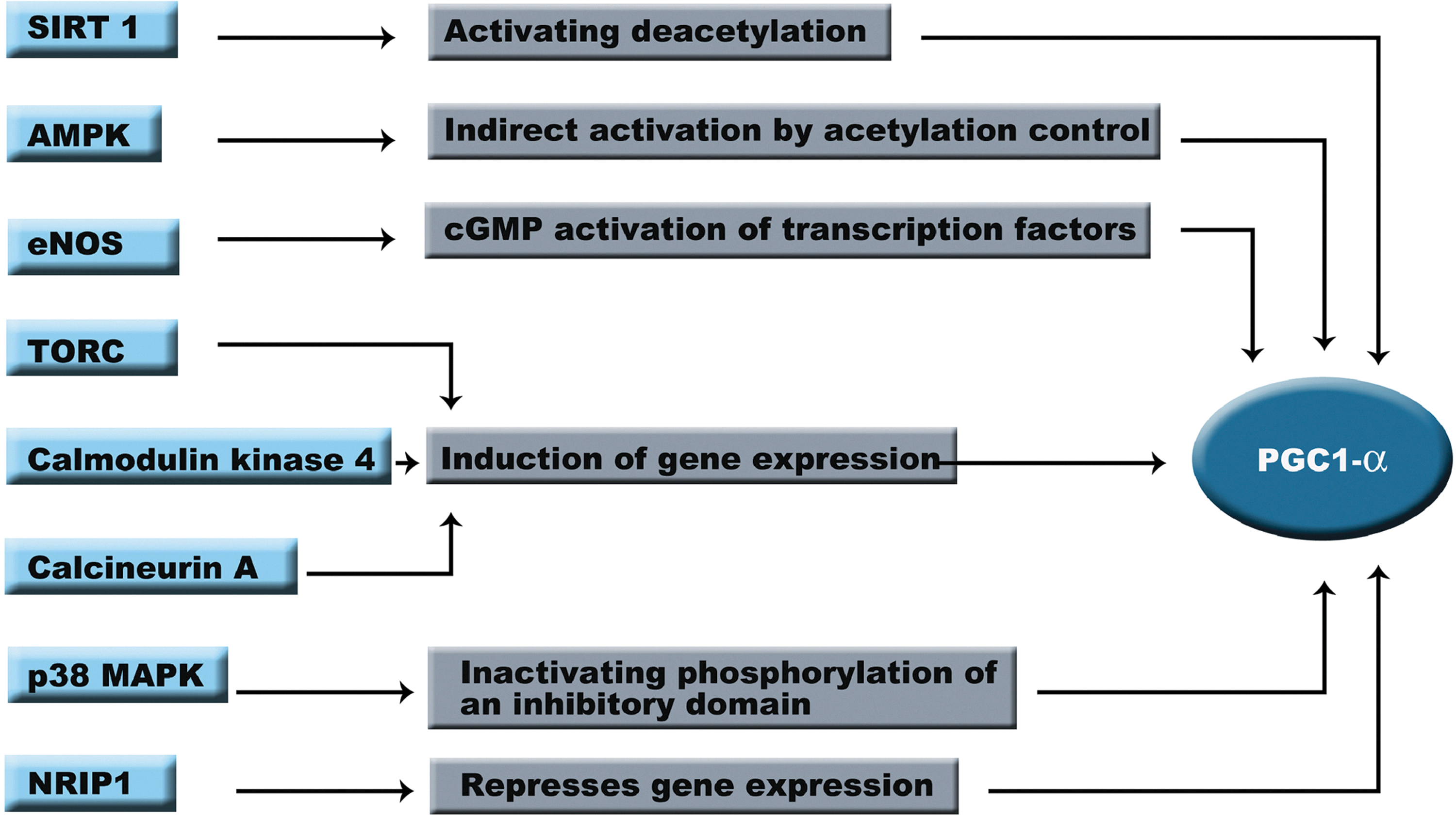
Regulation of PGC-1α Activity
CR activates several regulatory pathways that lead to the activation of PGC-1α and increases mitochondrial biogenesis. During the years a new field has developed to understand the regulation of PGC-1α and its downstream target activities. In this section, we will describe the present knowledge about molecular pathways that regulate PGC-1α (Fig. 5).
Sirtuin 1
Sirtuin 1 (SIRT1) is a NAD+-dependent histone deacetylase that is highly expressed under CR in most of the animal models. For instance, it has been reported that SIRT1 expression is induced under CR and exercise in humans (17). SIRT1 belongs to the sirtuin family. This family consists of seven enzymes that share a conserved deacetylation catalytic domain but differ in cellular localization, tissue distribution, and substrate preference (23). Increasing evidence implicates mammalian sirtuins as regulators of CR-induced physiological responses. Although there are controversial reports, additional copies of SIRT1 orthologues have been shown to extend lifespan in yeast, worms, and flies (13). In mammals, SIRT1 performs an important role in epigenetic silencing and genomic stability, regulating energy metabolism and endocrine signaling. This protein is also expressed under stress conditions and low-nutritional status. SIRT1 overexpression or activation confers a metabolic state that resembles CR; however, it does not extend lifespan in mammals (10). The lack of lifespan extension on Sirt1 transgenic mice could be due to the mild overexpression obtained in Sir1 transgenic mice, between two- to four-fold increased mRNA expression, which could not be high enough to produce an increase in longevity. Another possibility could be that Sirt1 might not protect against all the age-related diseases and could be beneficial only in protecting against a subset of these diseases, such as some types of cancer and diabetes (43).
It has been proposed that SIRT1 acts as a master regulator of the cellular metabolism via the redox status. Under CR there is a molecular adaptation that shifts the NAD+/NADH ratio toward higher NAD+ levels inducing SIRT1 activity (30). SIRT1-mediated effects result from deacetylation of lysine residues on substrate proteins. To date, several proteins such as nuclear factor-κB, forkhead box class O3, p53, PGC-1α, and endothelial nitric oxide synthase (eNOS) have been identified as targets of SIRT1 (28). Increased SIRT1 activity triggered by elevated NAD+ levels has been shown to increase the transcriptional activity of PGC-1α (75). Accordingly, decreased acetylation status of PGC-1α has also been demonstrated to increase mitochondrial PGC-1α-dependent expression. These data suggest a direct link between SIRT1 and mitochondrial bioenergetics. Studies with mice lacking SIRT1 have shown that PGC-1α mRNA is reduced while PGC-1α is acetylated in skeletal muscle, suggesting that SIRT1 regulates PGC-1α by both transcriptional and post-translational mechanisms (34). Further, the addition of a SIRT1 inhibitor, nicotinamide, decreases the expression of downstream targets of PGC-1α in muscle cells (34).
SIRT1 appears to be involved in the regulation of the antioxidant defenses within the mitochondria since SIRT1 overexpression attenuates the high fat-induced hepatic steatosis by induction of antioxidant proteins, such as mitochondrial superoxide dismutase and NRF-1, while SIRT1 deficiency enhances liver steatosis. Moreover, the regulatory role of SIRT1 on cellular respiration has been demonstrated by studies with SIRT1-silenced cells treated with serum from CR mice.
AMP-activated protein kinase
Another master integrator of external signals involved in PGC-1α regulation is the AMP-activated protein kinase (AMPK). This kinase coordinates the vast majority of the responses to the energy status of the cell and is activated by an increase in AMP/ATP ratio. Impaired regulation of AMPK activity is associated with several aging-associated processes, such as obesity and insulin resistance. AMPK expression is also induced under CR conditions (17, 74). However, it is not clear if CR directly affects AMPK function, but short term CR increases phosphorylation and thereby the activity of AMPK in young and old animals (35). Interestingly, chronic AMPK inactivation is associated with age-related complications (74) and constitutively, activated AMPK leads to increased lifespan in nematodes (21). In mammalian studies, it has been shown that AMPK phosphorylates PGC-1α and thereby controls glucose uptake, fatty acid oxidation, and mitochondrial biogenesis in primary muscle cells. Starvation conditions activate AMPK that initiates a signaling process that increases PGC-1α and PPARδ activities, leading to an increase of the mitochondrial content.
Endothelial nitric oxide synthase
Nitric oxide (NO) is a free radical involved in several physiological processes. NO production is essential for mitochondrial biogenesis and is increased under CR conditions. This mechanism also works through the activation of PGC-1α (71). NO is synthesized by eNOS, which is sensitive to nutritional status. AKT modulates eNOS activity through phosphorylation, leading to an increase of the NO levels (31). The consequence of eNOS expression and 3,5-cyclic guanosine monophosphate (cGMP) production is the activation of PGC-1α via cGMP-mediated upregulation of transcriptional factors. Increases in cGMP levels have also been related to the expression of the deacetylase SIRT1 (71). eNOS gene expression is increased in humans under CR and CR plus exercise. Interestingly, NO can induce mitochondrial biogenesis in human skeletal muscle without increasing SIRT1 protein expression in humans (17). These data suggest that the effect of NO in mitochondrial biosynthesis might not be related to SIRT1 activity. Decreased levels of NO in eNOS null-mutant mice attenuated the caloric restriction-induced expression of PGC-1α and TFAM mRNA and the increase of mitochondrial content induced by CR.
Other activators
There are several reports that include other families of coactivators in PGC-1α activation. One of them is the transducer of regulated CREB (TORC) family (90). TORCs proteins induce PGC-1α gene transcription in response to an increase of cAMP through cAMP response element-binding (CREB). Increased expression of TORC1, TORC2, and TORC3 in primary muscle cells induced PGC-1α expression and its downstream targets leading to increased cellular respiration (90). This is a PGC-1α dependent mechanism, since the lack of this gene abolished the increment of mitochondrial content. A different regulation process depends on an increase in muscle cytosolic calcium levels induced by exercise. Calcium stimulates calmodulin kinase IV, which promotes PGC-1α expression through CREBP. Calcineurin A also enhances PGC-1α expression in cardiac muscle through activation of myocyte enhancer factor-2 (22).
The inhibition of PGC-1α
PGC-1α also contains a negative regulatory domain. The p160 myb binding protein (p160MBP) represses PGC-1α by binding it to this regulatory region. This interaction is further regulated by p38 mitogen-activated protein kinase (MAPK), which phosphorylates the inhibitory domain of PGC-1α, thereby disrupting p160MBP-binding and inducing mitochondrial biogenesis (26). Another protein that inactivates PGC-1α is the nuclear receptor-interacting protein 1 (NRIP1). This protein seems to function primarily as a scaffold protein that links nuclear receptors to chromatin remodeling enzymes repressing PGC-1α activity. NRIP1 is a coregulator that shares a number of features with the PGC-1 proteins and has been shown to repress PGC-1α activity (89).
CR Promotes Mitochondrial Efficiency
Preservation of mitochondrial structural and functional integrity by CR is believed to result from the attenuation of oxidative damage, which is considered one of the major factors contributing to slowing the aging process and preventing tumor formation. ROS are produced by mitochondrial oxidative phosphorylation and extracellular oxidants. When ROS levels are elevated, they can modify cellular molecules resulting in lipid peroxidation, DNA strand break, and protein carbonylation. All these alterations impair cellular functions and increase the chance of cell death (Fig. 1). The accumulation of oxidative damage eventually impairs the function of the organs, which has been identified in many age-related diseases. We and others have shown that serum from CR animals decreases membrane potential and oxygen consumption in human cells lines and primary culture cells, thereby decreasing the electron flow (58). Therefore, CR prevents the electron leakage from mitochondrial complexes to form superoxide, decreasing the accumulation of oxidative damage. Interestingly, the ATP levels and the ATP/ADP ratio do not change in CR. Thus, these results agree with the hypothesis that CR produces very efficient mitochondrial electron transport based on low-potential mitochondria that sustain reduced oxygen consumption while maintaining cellular ATP levels and reducing ROS production.
Results from different laboratories support that, besides the decreased ROS production, CR also upregulates the expression of genes involved in ROS scavenging (51, 59, 60, 69). One of the most important transcription factors involved in the upregulation of the antioxidant defenses is the nuclear factor erythoid-derived 2-like 2 (NFE2L2). For instance, under CR, SKN-1 (NFE2L2 orthologue) is upregulated in the ASIs neurons in Caenorhabditis elegans. Studies in our lab have focused on its antioxidant activity and its implications in cancer and aging. CR was not effective against chemically induced tumorigenesis in the NFE2L2 knockout (KO) mice. AL NFE2L2 KO and CR NFE2L2 KO mice reached total tumor incidence at the age of 30 weeks, while 40% CR wt mice did not show any papillomas until 42 weeks of treatment (73). These data suggest that the anticarcinogenic effect of CR solely depends on the activity of NFE2L2 due to the lack of activation of the antioxidant defenses. Moreover, several reports have shown that rodents overexpressing antioxidant enzymes, such as mitochondrial catalase and copper-zinc superoxide dismutase in fruit fly, increase lifespan through increased antioxidant capacity.
There is extensive evidence that mitochondrial dysfunction and loss of mitochondria are due in large part to mutations in mtDNA since the vast majority of the ROS production is localized within the mitochondria. Interestingly, there is a body of literature showing that uncoupled mitochondria could give some health and lifespan benefits since several organisms have been shown to live longer (81). Supporting this hypothesis, studies with mice lacking uncoupling protein 2 (UCP2) presented shortened lifespan and increased oxidative damage (2). On the other hand, mice overexpressing UCP1 displayed increased lifespan, consumed more oxygen, increased body temperature, lowered body weight, and reduced oxidative damage accumulation (32, 49). The explanation of the increased longevity found in these animals could be that uncoupled mitochondria decreases mitochondrial potential and thereby ROS production. These events might produce the induction of mitochondrial biogenesis, as suggested by the increased Sirt1 activity and decreased acetylation of PGC-1α, which in turn, would be able to maintain ATP production (32).
The mitochondrial theory of aging proposes that mutations of mtDNA induced by ROS are the primary cause of cellular energy decline with aging. Accordingly, complex I is particularly affected since seven of its subunits are encoded by mtDNA (33). Moreover, the lack of protective histones and the low abundance of introns increase the probability to mutate coding sequences. Mutations in mtDNA have been found to accumulate and colocalize with impaired oxidative phosphorylation in muscle fibers (88). Mitochondria presenting mutated mtDNA have been shown to produce lower activities in the ETC complexes that are codified by mtDNA in several research models and humans. On the other hand, several studies have refuted the involvement of mtDNA mutations as a major contributing factor in the decline of the skeletal muscle oxidative capacity (82). These reports suggest that the different amount of mtDNA mutations in muscles under AL and CR are not associated with a decreased oxidative capacity. Further, different oxidative capacities in several muscles from very old CR and AL rats do not correlate with increased mtDNA mutations (5).
CR Promotes Apoptosis
Another cellular consequence of reduced energy intake in CR models is the induction of pro-apoptotic gene expression. It is known that aside from their role as energy suppliers, mitochondria are also involved in the regulation of the apoptotic program through caspase-dependent and caspase-independent mechanisms (62, 87). Apoptosis activation depends on the release of mitochondrial enzymes such as apoptosis inducing factor (AIF) and endonuclease G (EndoG), which induce DNA-fragmentations (87). When apoptosis is induced, cytochrome c is released into the cytoplasm and promotes the oligomerization of the apoptosis protease activating factor-1, which finally activates the caspase pathway. In addition, some reports have shown that AIF and EndoG can also translocate to the nucleus and start a caspase-independent apoptosis mechanism.
CR Enhances Protein Turnover
CR promotes autophagy in many species, leading to the selective elimination of damaged proteins and whole organelles. The autophagic response persists in old CR individuals while becoming dysfunctional in AL counterparts (61). Autophagy is the main cellular mechanism attributed to the degradation of dysfunctional and damaged mitochondria. This process is critical for cellular survival since the progressive accumulation of damaged proteins in cells lacking autophagy results in cell death and loss of tissue function (29). Although the autophagic activity in liver declines with age, there must be a tissue-specific regulation of this process, since macroautophagy is maintained in heart and skeletal muscle in old rodents. Macroautophagy plays an important role promoting the health benefits of CR because the inhibition of this process suppressed the lifespan extension promoted by CR in worms. Under CR, autophagy is regulated by at least two signaling pathways. The first one involves the activation of the phosphoinositide 3-kinase class III, which is bound to Beclin 1. The second one acts through the mitochondrial target of rapamycin (19, 83). Experimental evidence indicates that protective effects of CR on mitochondrial function rely on the ability to reduce the incidence of mitochondrial abnormalities through maintenance of proper autophagic responses. Additionally, CR may induce a shift toward an enhanced turnover of mitochondrial proteins and increased mitochondrial biogenesis by PGC-1α. This hypothesis would explain the lack of differences in citrate synthase protein content between AL and CR old animals (17).
CR Mimetics
Due to the difficulty for humans to adhere to CR similar to that performed under laboratory conditions, many investigators have sought compounds that could mimic CR and produce physiological and anti-aging benefits similar to CR. Epidemiological studies have clearly documented that the action of some phytochemicals resembles, at least partially, the CR phenotype (Table 1) (15, 16).
One of the most known CR mimetics proposed is resveratrol. This is a naturally occurring polyphenol that has been shown to increase the activity of PGC-1α, leading to an increased mitochondrial biogenesis in liver, muscle, and brain (7, 53). SRT1720 is an analog of resveratrol that acts through the same enzymatic mechanism, binding to an allosteric site in the enzyme-substrate complex and lowers the Km of SIRT1 (80). It has been described as 1000 times more potent than resveratrol. We and others have shown that resveratrol and other polyphenols such as SRT1720 and SRT501 activate SIRT1 directly or indirectly in a variety of models leading to increased deacetylation of PGC-1α when administered in the diet (86). In fact, resveratrol is completely unable to modulate PGC-1α function in SIRT1−/− MEFs (53). SRT501 and SRT1720 generate a gene expression profile similar to CR (80). An analysis of the gene expression shift induced by CR compared to SRT1720 and SRT501 depicts that the gene expression profile is significantly shifted in the same direction, including the mitochondrial biogenesis network among others. Further, this analysis also showed that the PPAR family and estrogen-related receptor are induced by SRT501. These genes are known to be activated when mitochondrial biogenesis is required. Correspondingly, the data show that NRIP1, an inhibitor of PGC-1α, is reduced. These data suggest that both act via the same mechanism in vivo.
Another CR mimetic candidate is Metformin. This CR mimetic has been used since 1960 in the treatment of diabetes due its ability to reduce blood glucose levels thereby reducing the risk of glucotoxic effects such as glycosylation and oxidative damage. This compound induces a gene expression profile that closely aligns to long-term CR (24). Metformin supplementation in diet has also been shown to extend lifespan in several laboratory models, including mammals (3, 72). Metformin also induces the expression of glycolytic genes, which is known to occur in CR as well. It has been proposed that metformin compromises ATP production in the mitochondria. The mechanism of action would be based in a mild inhibition of the mitochondrial complex I. Interestingly, although the increase of oxidative damage due to the inhibition of mitochondrial complexes is a well-established fact, metformin seems to reduce ROS generation and oxidative damage accumulation (1, 37, 40, 84). Supporting these findings, an increase of the expression of antioxidant proteins has been reported in cells and animals treated with metformin (45, 72). A slightly inhibited complex I activity under metformin treatment may lead to lower ATP production without affecting, or even decreasing, oxidative damage. In this context, the decrease of ATP production renders the increase of the AMP/ATP ratio, which is known to activate AMPK. Although AMPK activation is not responsible for all the benefits achieved by CR, it is likely that AMPK activation due to metformin treatment can mimic certain aspects of CR (14).
The regulation of AMPK, SIRT1, and PGC-1α pathways using CR mimetics would induce mitochondrial biogenesis and antioxidant gene expression. The post-translational regulation on PGC-1α by CR mimetics may confer the gene expression changes that induce a switch in the source of energy utilized from glucose to lipids in the mitochondria leading to the improvement of the mitochondrial function and thereby, protecting against metabolic stresses.
Concluding Remarks
During the last decades, researchers have been trying to determine the mechanisms whereby CR delays aging and prevents age-related diseases. Understanding the metabolic reprogramming in the mitochondria induced by CR would offer potential to develop mechanism-based interventions to promote longevity and healthy aging. The next step in future investigations would be developing a mechanism to stimulate the regulatory components of mitochondrial biogenesis, mainly PGC-1α, SIRT1, AMPK, and their downstream targets to shift the metabolic balance. To develop a preventive strategy to treat the age-related complications in humans, we propose the use of nontoxic CR mimetics as a potential therapeutic target. A plausible aging prevention strategy could be based on an approach that includes moderate CR, physical activity, and CR mimetic supplementation, which seem to be the best way to maintain mitochondrial activity.
Footnotes
Acknowledgment
This work has been supported by the Intramural Research Program of the National Institute on Aging, National Institutes of Health. We thank Jessica Curtis for editing the article.