
Abstract
Introduction
The formation of red blood cells (RBCs) begins with the pluripotent stem cells of the bone marrow (48, 81). These stem cells undergo proliferation and differentiation into progenitor committed cells and then into progressively maturing precursors and finally mature RBCs, white cells, and platelets. The earliest progenitor cell committed to the erythroid is the BFU-E, which forms large bursts of erythroblast colonies. Further differentiation results in the first morphologically recognizable erythroid precursors, the proerythroblasts, which eventually develop into reticulocytes. The reticulocytes eject the nucleus before being released into the circulation, but retain the mitochondria, ribosomes, and ribosomal RNA necessary for the synthesis of hemoglobin (Hb) and other proteins. It has been estimated that ∼20%–30% of the total Hb is synthesized in reticulocytes. The reticulocytes in the circulation have a half-life of ∼30 h during which time they transform to mature RBCs with complete elimination of traces of DNA, mitochondria, endoplasmic reticulum, and ribosomes.
Recent proteomics studies indicate that Hb accounts for 95 to 97% of the cytosolic protein of RBCs. Most of the other cytosolic proteins are involved in neutralizing oxidants (peroxiredoxin2 [PRDX2], catalase, superoxide dismutase (SOD) and glutathione peroxidase [GPx]), and in the metabolism of glucose and synthesis of ATP (both anaerobic glycolysis and the hexose monophosphate shunt) (14, 74). The RBC membrane consists of a lipid layer with phospholipids and cholesterol. There are glycol proteins and glycol lipids on the exterior surface. The inner surface is secured through transmembrane proteins to a cytoskeleton consisting of an elastic network of skeletal protein that enables the RBC to deform and flow through the narrow capillaries of the microcirculation to deliver oxygen to the tissues. The membrane also contains proteins that provide channels for the uptake and release of ions and low molecular weight hydrophilic compounds necessary for maintaining ionic homeostasis.
The RBC is, thus, especially designed for the delivery of oxygen to the tissues by the reversible binding of oxygen to the very large pool of Hb contained in the cell. The other properties of RBC are necessary to facilitate this process. The synthesis of ATP facilitates ionic homeostasis, which retains an excess surface area, and the cytoskeleton provides the elastic properties. Both the biconcave shape with excess surface area and elasticity are necessary for the cell to deform and deliver oxygen to the tissues (Fig. 1). The extensive antioxidant system is designed to neutralize the harmful reactive oxygen species (ROS) generated as a result of the constant exposure to variable oxygen tensions.

To optimize the amount of oxygen the cell can carry, by increasing the Hb concentration, and the ability to transport this oxygen, by increasing the cellular deformability, the mature RBC has ejected all the nonessential components of the RBC that prevent this optimization (29, 52).
The Removal of RBCs from the Circulation
The mature RBC without a nucleus to regulate regeneration also lacks many of the features of other cells that are responsible for their survival and proper functioning. They have no mitochondria required for efficient oxidative metabolism, no DNA or ribosomes necessary for protein synthesis to replace damaged proteins, and the de novo synthesis of lipids is also metabolically precluded. The RBC continuously undergoing normoxic and hypoxic cycling is constantly exposed to oxidative insults during its 120-day lifespan that results in continuous biochemical, physical, and immunological changes. These changes impair the ability of the RBC to transport oxygen and eventually trigger removal from circulation by the reticuloendothelial system. The reticuloendothelial system involves the mononuclear phagocytic cells primarily in the spleen, and also in the liver and lymph nodes. The processes responsible for the actual triggering of the removal have been extensively studied (5, 6, 13, 88).
Membrane microvesiculation is a regulated process that accelerates in older cells (104). It is responsible for the increase in cell density coupled with a decrease in deformability and flexibility (10, 101). These changes limit the ability of the RBC to maintain the highly deformable biconcave shape necessary to pass through narrow pores, thus contributing to their removal from circulation (Fig. 1). It has, however, been shown that the vesicles formed contain elevated levels of phosphatidylserine (PS), IgG, and breakdown products of band 3 that have exposed antigenic sites, which have been shown to trigger the removal of RBCs from circulation (see below). Based on this finding, it was suggested that vesiculation may actually be a self-protective mechanism that increases the RBC lifespan (103, 104).
The RBC membrane band 3 is the dominant integral transmembrane protein. It has several crucial functions: (i) the maintenance of anion homeostasis, (ii) providing a link between the membrane and the cytoskeleton responsible for maintaining the cell shape, and (iii) providing for the interaction of a number of cytosolic proteins with the membrane via the amino terminal region that protrudes into the cytosol. This region of band 3 competitively binds both Hb, and a number of glycolytic enzymes (60). The changes in Hb binding to band 3 as a function of the Hb oxygenation, therefore, couple Hb oxygenation to glycolysis and ATP production.
Damage to band 3 has been linked to RBC aging including the exposure of senescent specific neo-antigens that bind autologous IgG triggering RBC removal (42). IgG binding has also been linked to band 3 clusters, which is triggered by the binding of denatured oxidized Hb (hemichromes) to band 3 (30, 54, 80).
The RBC membrane has a Ca-ATPase, which maintains a low intracellular concentration of free calcium (Fig. 2) (51). During aging, this calcium homeostasis is disrupted and there is a gradual increase in intracellular calcium (44, 93). Increased intra-cellular calcium activates the Gordo's potassium channel causing the leakage of potassium from the cell, resulting in cell shrinkage and impaired deformability (15, 32). Calcium also activates u-calpain, transglutaminase-2, and some caspases that can degrade/cross-link cytoskeleton proteins (79). It also inhibits phosphotyrosine phosphatase increasing band 3 phosphorylation (108).
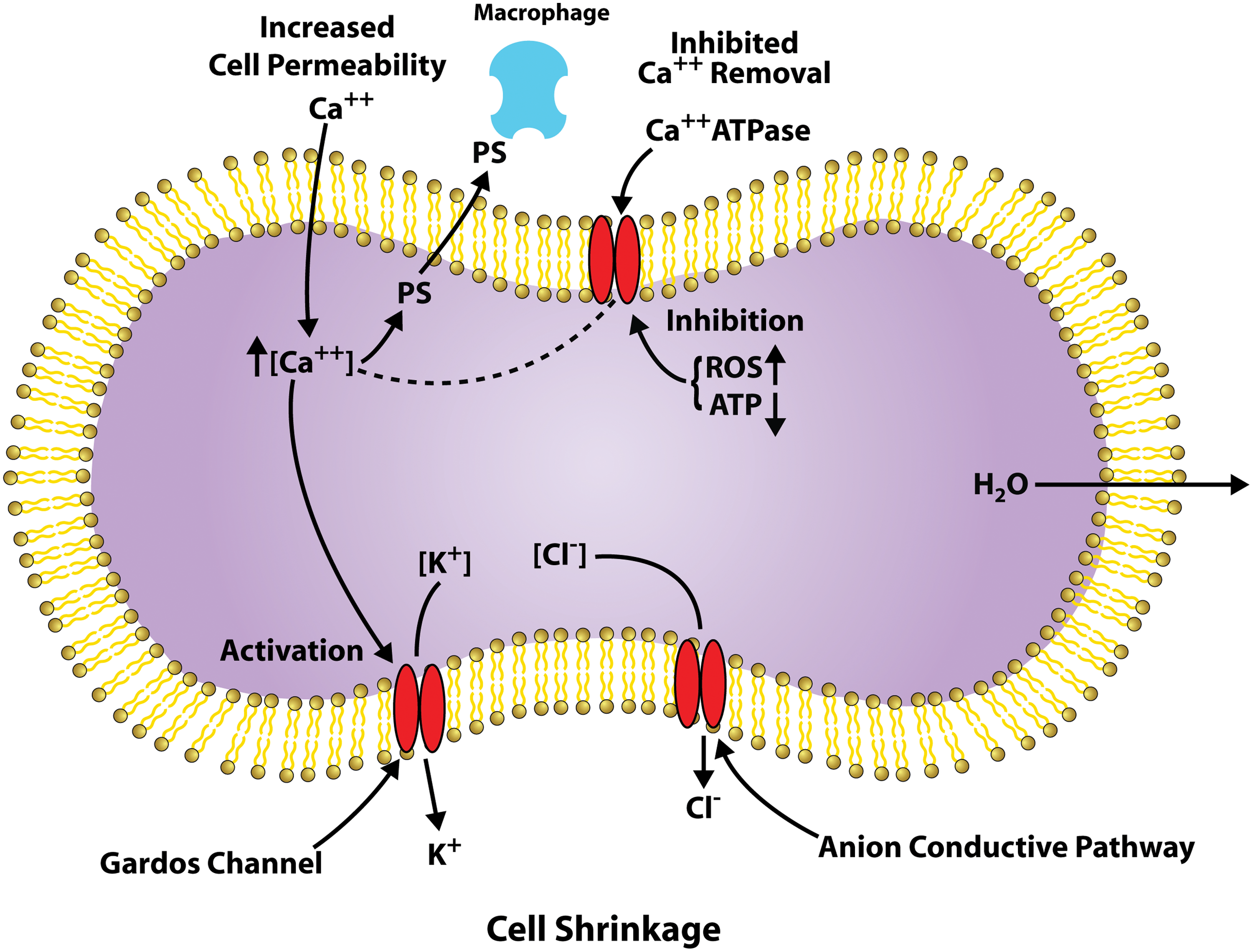
The RBC lipid bilayer contains an asymmetric distribution of phospholipids with PS being maintained on the inner surface by the competition between scramblase that randomizes the distribution and flippase that internalizes the PS. Coupled with an increase in sphingomyelinase that increases ceramide, increased intracellular calcium (Fig. 2) has been linked to the exposure of PS, which triggers interaction with macrophages and eryptosis (24, 31, 102). Caspase-3 activation, which modifies the band 3 linkage to ankyrin and the cytoskeleton, and flippase activity, has been shown to induce PS exposure.
RBC Oxidative Stress
As outlined above, there are a number of processes that damage the RBC as it ages. The relative role of each of these processes in RBC aging and the removal of the RBC from circulation is, however, not always clear. Since many of these processes are directly affected by oxidative stress, a significant role for oxidative stress in determining RBC aging must exist (Fig. 1). Examples of the contribution of oxidative stress to these processes include the following: (i) distorted calcium homeostasis (Fig. 2), which has been attributed to oxidative stress (6, 50); (ii) caspase-3 activation is clearly triggered by oxidative stress (42, 99); (iii) exposure of the neo-antigen on band 3 and band 3 clustering are triggered by oxidative stress (42, 54).
The predominant factor that determines oxidative stress in the RBC is Hb (Figs. 1 and 3). The reactive free radical species generated by Hb reactions and the interactions of Hb with membrane and cytoskeleton proteins both induce oxidative stress and are involved in RBC aging.
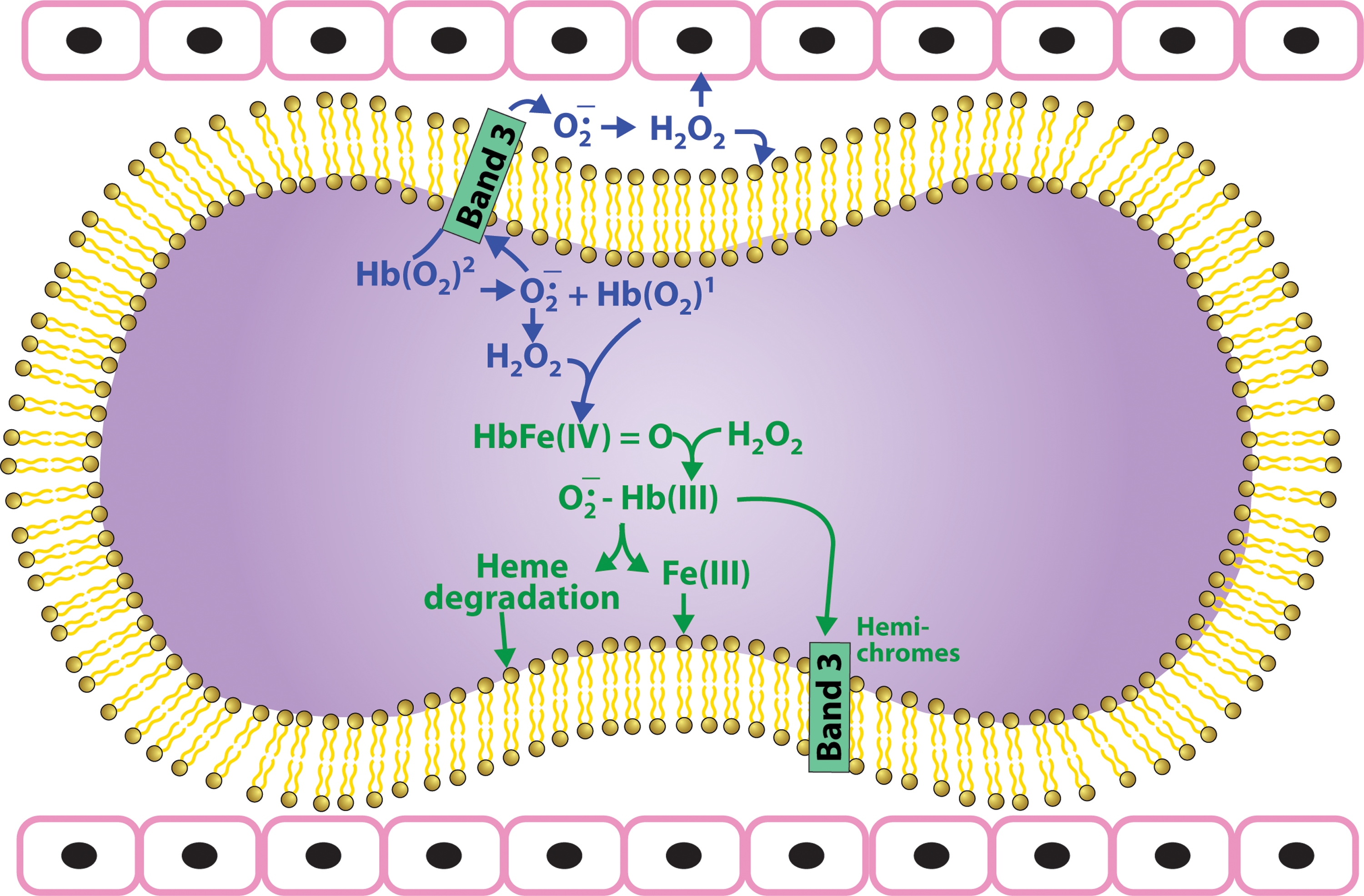
Oxygen transport by the RBC involves the reversible binding of oxygen to the 5 mM tetrameric Hb. Reversible oxygenation requires that the heme iron be in the Fe(II) reduced form. However, Fe(II) is readily oxidized to Fe(III). Although oxidation of Fe(II) is slower when the iron is incorporated into a protein, the Fe(II) heme of Hb also, although at a much slower rate, continuously undergoes a redox reaction involving the oxygen bound to Hb that oxidizes Hb producing superoxide (Fig. 3).
Although this reaction is slow under normoxic conditions (k=0.0115 h−1) (66), it is dramatically enhanced under hypoxic conditions (2). Appreciable concentrations of superoxide are, therefore, generated by high concentration of Hb, especially under hypoxic conditions found in the microcirculation (2, 4, 8, 45, 82, 86, 87). This superoxide dismutates to produce hydrogen peroxide (H2O2), a process that spontaneously occurs at a rate of 3.9×107 M −1s−1, but even more rapidly in the presence of RBC SOD. Both of these reduced oxygen species (superoxide and H2O2) are reactive and can react with both Hb and other cellular constituents.
The autoxidation reaction produces Fe(III) methemoglobin (metHb) at the same time that it produces superoxide. MetHb does not bind oxygen and cannot transport oxygen. To maintain functional Hb, most of the metHb is reduced back to Fe(II) Hb by metHb reductase. The residual non-reduced metHb also has a lower affinity for the heme prosthetic group resulting in free hemin (38). This hemin interacts with the red cell membrane disrupting skeletal protein interactions (94, 105) and reacts with membrane lipid hydroperoxides releasing lipophilic radicals (25). In the presence of hydrogen peroxide it can also induce lipid peroxidation. In the red cell glutathione, which is present in the mM concentration range scavenges hemin, inhibiting hemin induced damage (96).
In addition to the autoxidation reaction, the RBC in circulation is continuously exposed to extracellular oxidants that can be taken up by the RBC (Fig. 1) and react with various groups including Hb (7, 72).
The RBC is also exposed to nitric oxide (NO) and nitrite (Fig. 4), which can induce a new class of redox reactions involving reactive nitrogen species (RNS) (65, 83, 84, 91) that produce nitrosative stress. NO is produced by nitric oxide synthase (NOS) (47). While there is evidence of NOS in the RBC that can release NO in the RBC, most of the NO that the RBC is exposed to originates from endothelial e-NOS (49). NO in the RBC, rapidly reacts with oxyHb to form nitrate and metHb or deoxyHb to produce stable Hb(II) NO. Although these reactions are rapid (k=∼107 M −1 S−1) (21), any superoxide present due to autoxidation of oxyHb reacts with NO even faster than Hb (k=∼10 10 M −1 S−1) producing peroxynitrite (PN) (Fig. 4). PN is also generated in the interstitial spaces of the endothelium by the reaction of superoxide generated by NADPH oxidases, xanthine oxidase, and uncoupled e-NOS, which reacts with NO generated by e-NOS (33, 78). This PN generated in the vasculature spaces can diffuse into the circulation and then into RBCs. PN is a highly reactive species that can damage cellular constituents (73, 89, 90).
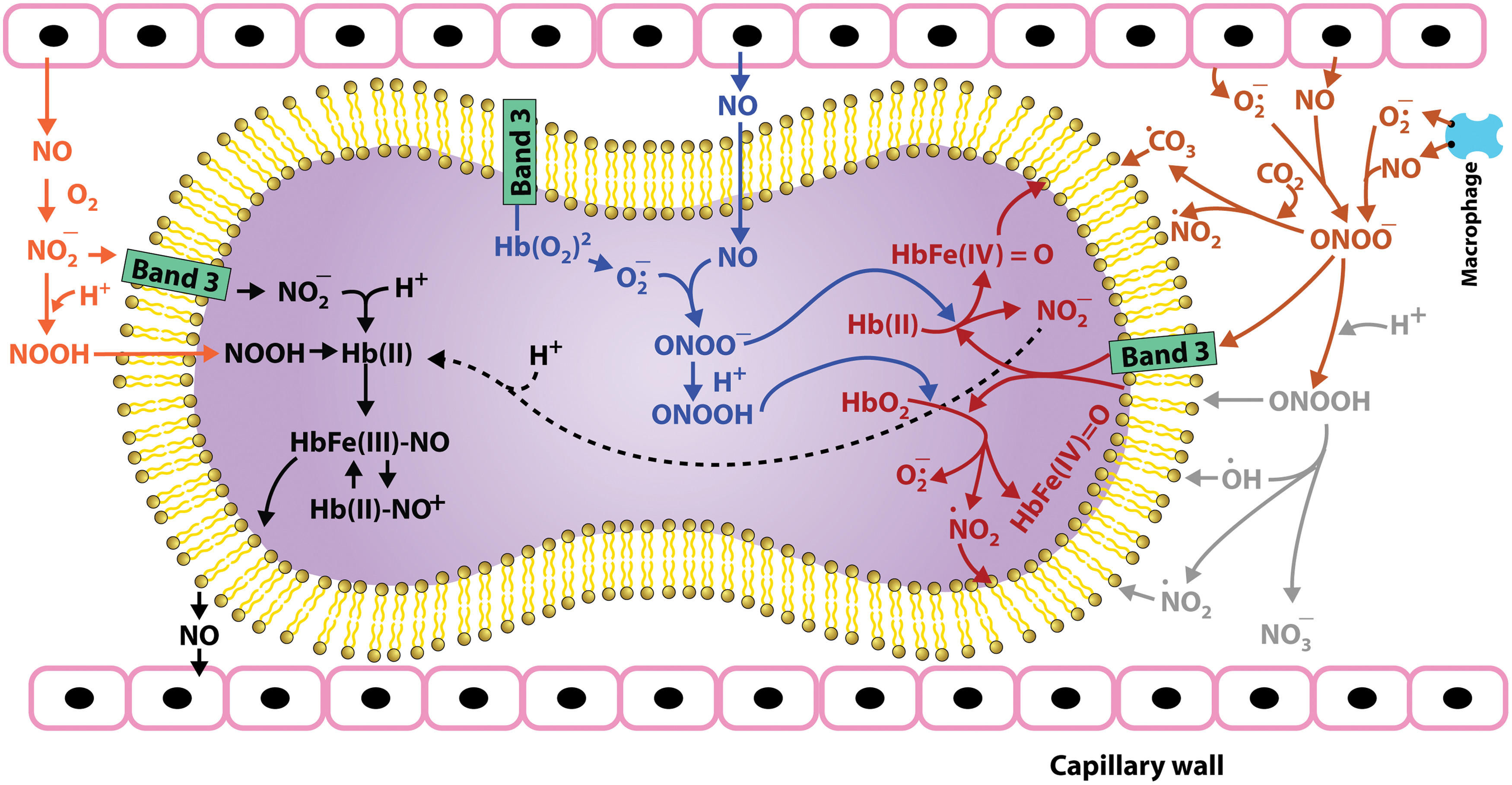
An additional source for RNS in the RBC involves reactions involving nitrite (Fig. 4), which is produced by the oxidation of NO by oxygen or metalloproteins in plasma (95). Most of plasma nitrite originates from NO oxidation, explaining the use of plasma nitrite as a measure of e-NOS activity (71). However, some of the nitrite can also originate from the nitrite and nitrate in the diet. Nitrate also contributes to the pool of nitrite, because the nitrate is reduced to nitrite by bacteria in the saliva (36). Nitrite in the RBC reacts with oxyHb to produce nitrate, which is un-reactive, and metHb (43). However, nitrite also reacts with deoxyHb (22, 28, 65). Nitrite, which has a low affinity for Fe(II) heme, helps induce a conformational change that stabilizes the bound nitrite. Once the bound nitrite is protonated, a redox reaction results in the displacement of a hydroxyl ion from the nitrite producing a nitrosonium ion bound to Fe(II), which is in equilibrium with a Fe(III) NO complex. This hybrid intermediate in the β-chain was also shown to be in equilibrium with a thiyl radical on the β-93 sulfhydryl group. These metastable intermediates retain NO bioactivity, but are not quenched by reacting with Hb or superoxide. These intermediates, thus, provide a pool of potentially bioactive NO, which can be released to the vasculature when needed (84, 91, 92).
Reactions Associated with ROS and RNS Species Formed by Hb Redox Reactions
The superoxide, H2O2, hydroxyl radicals, ferrylhb, oxoferrylHb, and PN generated by these redox reactions can damage RBC membrane proteins, lipids, and the cytoskeleton (Figs. 1, 3, and 4), which are responsible for maintaining the RBC shape and deformability. They can trigger the uptake of calcium (Fig. 2), activation of caspase, and the disruption of lipid asymmetry resulting in the exposure of PS and damage to band 3, all of which can contribute to the triggering of the removal of RBCs from circulation by macrophages (6, 31, 42, 44, 50, 57, 60).
Although superoxide is a relatively unreactive free radical species, it does have some toxic effects on the RBC. This toxicity has been established using SOD1 knockout mice, where the lifetime of superoxide formed by Hb autoxidation is extended because of the slower spontaneous dismutation of the superoxide. These mice have been reported to develop Heinz bodies and result in cells with a shorter lifespan and a shift in glucose metabolism (37).
H2O2 initiates a cascade of oxidative reactions (Fig. 3). It reacts with Fe(II) Hb taking two electrons from the heme producing the highly reactive Fe(IV) ferrylhemoglobin. It will also react with any oxidized Fe(III) Hb producing Fe(IV) oxyferrylhemoglobin where a second electron is removed from the globin forming a reactive protein radical. Ferrylhemoglobin reacts with an additional molecule of H2O2 that is reduced to form a superoxide radical that is retained in the heme pocket long enough to react with the heme, eventually resulting in the degradation of the heme (Figs. 1 and 3) releasing iron and forming several fluorescent heme degradation products (34, 67 –70). A relationship between the formation of these heme degradation products and cellular aging is indicated by the correlation between the level of heme degradation and the binding of autologous IgG to the RBC membrane (62). This effect can be due to the iron released during the formation of heme degradation products. The released iron can deposit on the membrane acting as a Fenton catalyst to oxidize lipids and proteins. An increase in redox active iron release has been observed during in vitro aging of RBCs and in cells treated with oxidizing agents. This release of iron was correlated to the binding of autologous IgG to the membrane of these cells, suggesting that iron-mediated redox reactions may be responsible for the expression of senescent antigens on the membrane (20). In addition to the effects of released iron, a possible direct effect of these degradation products cannot be ruled out.
PN (Fig. 4) has a short half-life (<10 S at neutral pH) and can directly react with various biological molecules to generate toxic species like •OH, •NO2 and •CO3 radicals (89, 90). In RBCs, PN is either scavenged by PRDX2, which is an abundant thiol protein, or reacts with Hb (58). Studies have shown that most of the PN present reacts with oxyHb to generate metHb and nitrate (90). Hence, oxyHb can act as a sink for PN. Because of the reaction of PN with oxyHb, the formation of 3-nitrotrosine, generally considered a marker for PN, is not a valid marker for PN in the RBC. However, it has been shown that PN also generates small quantities of ferrylHb and •NO2, in addition to metHb, during the reaction with oxyHb. These radical species can damage the RBC membrane and promote cellular aging (90). Recent studies have shown that the addition of PN to RBCs causes morphological alterations, decrease in cellular thiols, decrease in glycoporin A (a senescence marker), Band-3 clustering, PS exposure, and activation of caspase 2 and 3 that promotes programmed cell death in RBCs (17, 73, 76). Some of these changes are reversed by prior treatment with thiol compounds such as N-acetyl cysteine, antioxidants, and glucose (17). These results indicate that the effect of PN in RBCs involves disruption of the cellular redox system (73).
RBC Antioxidant System
To minimize the toxic effects of these redox reactions the RBC has an extensive antioxidant defense system including SOD1, which catalyzes the dismutation of superoxide to H2O2 and catalase, (GPx) and PRDX2, which scavenge peroxides and PN (53, 61). In addition, there are a number of low molecular weight antioxidants in the RBC that help to minimize oxidative stress (19).
There is extensive literature on the metabolic changes that occur during cell aging (1, 11, 39, 55, 56, 75, 77, 98, 107). Changes have been reported for the antioxidant enzymes SOD1, catalase, and GPx (35). There are, however, uncertainties regarding many of the specific changes that occur. A number of studies indicate changes in glutathione metabolism as a function of cellular age including changes in enzymes involved in the hexose monophosphate shunt (1, 35, 39, 40). This includes changes in glutathione, glutathione reductase, and glucose-6-phosphate dehydrogenase, which produces the NADPH required for glutathione reduction. However, other studies indicate that there is no change in the hexose monophosphate shunt with no change in glutathione (55). These studies do, nevertheless, indicate metabolic changes that result in a decrease in glucose metabolism. However, the change is attributed to anaerobic glycolysis (The Embden–Myerhoff pathway). This reduced glucose metabolism is primarily caused by a decrease in hexokinase activity. The decreased glucose metabolism has been shown to result in a decrease in ATP (11, 55). The reduced ATP affects many cellular processes. It can also contribute to the removal of RBCs from circulation by reducing the activity of Ca-ATPase, which limits the intracellular calcium concentration. ATP is also required for flippase activity, which retains the phospholipid asymmetry that prevents the exposure of PS (27).
The uncertainty in much of these data is attributed to several issues. (i) Many of these studies have been performed with animals and may not be directly related to humans. (ii) Most of the earlier studies made use of the separation of cells by density. While it has been shown that older cells are more dense, there does not seem to be a linear relationship between cell age and density (23). (iii) In dealing with the time-dependent changes in RBCs it is necessary to distinguish between changes attributed to reticulocytes converting into mature RBCs and the subsequent aging of mature RBCs. Since relatively large changes in many enzymes occur as the reticulocyte matures and the less dense fractions of RBCs will tend to have higher levels of reticulocytes, this can make it difficult to distinguish the two processes. It is however possible to determine the actual concentration of reticulocytes in any fraction (97). Further, these levels are usually very low in the denser fractions of RBCs. Thus, although density fractionation does not provide an accurate measure of the time-dependent changes, it can provide a measure of the changes that occur during aging.
The uncertainty involving the density fractionation was addressed (107) by transfusing biotinylated young RBCs into rabbits and following the biotinylated cells as a function of time. In this way it was possible to follow changes in activity of 19 enzymes during cellular aging. In this study, the changes occurring during reticulocyte maturing and during the aging of mature RBCs could be separated by the time dependence of the changes observed. Other recent studies have also used biotinylated cells to monitor RBC aging (100, 106).
Although there are some uncertainties about the exact changes that occur during cellular aging, it is, however, clear that as the RBC ages the cell's ability to neutralize ROS is impaired (35). This was confirmed by studies (35) where trypan blue uptake after treating cells with xanthine oxidase was determined. They found that the older cells were more readily damaged. This enhanced susceptibility to oxidative damage will produce a synergistic process where the RBC oxidative processes described above are more pronounced in older cells, further exacerbation the aging process.
The Role of Interaction of Hb with the RBC Membrane
Hb, and a number of glycolytic enzymes, binds to the amino terminus of the portion of band 3 that protrudes into the cytosol on the cytosolic side of the membrane (Figs. 3 and 4). OxyHb has a relatively low affinity for band 3, with deoxyHb having an appreciably higher affinity (18). Recent data suggest that partially oxygenated Hb, which is the dominant form of Hb in the microcirculation, may actually have a higher affinity for band 3 than deoxyHb (16). This can be attributed to the greater flexibility at the interface between Hb subunits where band 3 binding occurs. Even though band 3 accounts for a major fraction of the RBC membrane proteins, the total number of band 3 sites (1.2×106/cell) correspond to<1% of the Hb molecules and most of the redox reactions involving Hb occur in the cytosol. However, since the antioxidant enzymes are predominantly cytosolic, the reactive species that are not neutralized by the antioxidant enzymes are predominantly produced by the redox reactions of Hb that is bound to the membrane (26, 64, 85).
Partial Hb oxygenation (Fig. 3) not only increases the affinity for the RBC membrane, but also dramatically increases the rate of Hb autoxidation (2). This increase in rate of autoxidation has been attributed to increased fluctuations across the α1β1 interface that alters the distal heme pocket of the oxygenated chains increasing the probability for a nucleophilic displacement of oxygen as superoxide by the distal histidine (8). This superoxide can damage the RBC band 3 and/or leak out of the RBC through the anion channel. Once out of the cell, it will dismutate to H2O2. The diffusion of this H2O2 into capillary venules was demonstrated by perfusing lungs with hypoxic RBCs (45).
As a result of the increased affinity of partially oxygenated Hb with the membrane, hypoxia not only increases the production of ROS, but makes it more difficult to protect the cell from the ROS by antioxidant defense enzymes. This is confirmed by the elevated oxidative stress observed in RBCs of severe anemic mice, which usually experience greater hypoxia than normal RBCs (63).
The nitrite reductase activity (Fig. 4) of deoxyHb requires unliganded chains and is favored by the liganded R quaternary conformation. This reaction is, therefore, also most efficient for partially oxygenated Hb. We have also suggested that during the nitrite reaction there is a contribution from the nucleophilic interaction with the heme iron that is responsible for Hb autoxidation. In this case an analogous interaction releases NO from an Hb(II) NO+ intermediate formed during the nitrite reaction (84, 91). Redox reactions involving ROS and RNS species are, therefore, most pronounced as the cells pass through the microcirculation and begin to release oxygen.
In addition to the reversible binding to band 3 associated with the oxygenation of Hb, it has been shown that oxidized Hb and particularly hemichromes, formed by the denaturation of Hb, have a much higher affinity for the RBC membrane producing irreversible cross-linking involving both band 3 (54, 80) and spectrin (44). The formation of membrane-bound denatured Hb does not increase linearly as the cells age, but form in the oldest, dehydrated most dense cells. The in vivo band 3 association in dog RBCs was studied where the cells were completely biotinylated and the remaining biotinylated cells were separated at later times. This provided a much more reliable measure of how long the cells are in the circulation. In this study, the binding of hemichrome to band 3 was found to begin and increase exponentially after 85 days in the circulation. The formation of this denatured Hb would reflect the gradual decrease in the antioxidant systems, which impair the ability of the cells to neutralize ROS and to reduce oxidized Hb. It is perhaps only after appreciable aging that the impairment reaches the point that hemichromes and denatured Hb form (80).
The band 3 interactions with hemichromes, unlike the interaction with deoxyHb, involve a stoichiometry of 2.5:1 (Hb to band 3). This binding is also thought to disrupt the interactions of band 3 with ankyrin weakening the linkage of band 3 to the cytoskeleton triggering the clustering of band 3. Band 3 clustering has been shown to increase the binding to band 3 of IgG, which contributes to the uptake of RBCs by macrophages (30, 41).
The spectrin Hb cross-linking found in vivo is similar to that generated by H2O2 and may be related to the heme degradation products that have been found to be generated by non-neutralized H2O2 (Fig. 3). It is, thus, interesting that both spectrin-Hb binding and heme degradation formation, both of which are induced by H2O2, also induce an increase in IgG binding to the membrane (12, 102). The Hb spectrin interaction involves interactions with the spectrin head group that inhibits the dimer to tetramer association of spectrin. This disrupts the cytoskeleton and decreases RBC deformability. It has also been suggested that the spectrin globin association, which affects the spectrin cytoskeleton, disrupts band 3 interactions with the cytoskeleton and can also contribute to band 3 clustering.
Conclusion
Hb redox reactions associated, both with the endogenous autoxidation of Hb and exogenous reactive species that enter the RBC, are a source for producing ROS and RNS species that can damage RBC protein and phospholipids (59). This continuous source of oxidative stress causes accumulative damage in RBCs that have no DNA or ribosomes necessary for protein synthesis to replace damaged proteins. This accumulated damage causes metabolic impairment that exacerbates the situation by reducing the cells ability to neutralize toxic compounds and to reduce oxidized Hb. Enhanced rates for the formation of ROS and RNS occur under hypoxic conditions, where an increased fraction of Hb is bound to the RBC membrane. This facilitates damage to membrane proteins and the cytoskeleton that regulated the shape, deformability (Fig. 1), and eventual recognition for removal by macrophages.
The linkage between Hb-membrane interactions and RBC oxidative processes provides a unique way to specifically turn off RBC-induced oxidative stress. Instead of requiring large concentrations of antioxidants to neutralize the ROS species formed, blocking the interaction of Hb with the membrane will make it possible for the large supply of RBC antioxidants and antioxidant enzymes to eliminate the Hb-generated oxidants and, thereby, prevent RBC oxidative stress. Such a reduction in RBC oxidative stress will slow down RBC aging and ameliorate pathological effects associated with RBC oxidative stress.
Footnotes
Acknowledgment
This research was supported by the Intramural Research Program of National Institute on Aging, National Institutes of Health.