
Abstract
Obesity: Molecular and Pathophysiological Features
Weight gain is influenced by several factors, such as genetics, maternal and perinatal environment, energy-dense diets, and sedentary lifestyle (3). Of great concern are the concurrent and parallel increases in the prevalence of pathological conditions associated with obesity, which include insulin resistance, type 2 diabetes, cardiovascular disease, nonalcoholic fatty liver, polycystic ovary syndrome, asthma, Alzheimer's disease, and some forms of cancer. Elucidating the causes involved in the pathophysiology of obesity-related disorders is one of the most critical endeavors in modern medical research.
Several mechanisms have emerged in the past two decades, during which obesity and especially its connection to insulin resistance have become a top-interest research topic being studied by leading groups in the field.
Ectopic-fat deposition
When the adipose tissue cannot store excess fat, lipids accumulate inappropriately in the liver, muscle, and pancreas. This lipotoxic environment, mainly mediated by diacylglycerols (DAGs), blocks correct glucose transport and insulin signaling (145). Thus, it has been postulated that any strategy that could block the entry of fatty acids (FAs) into the cell, promote fatty acid oxidation (FAO), or convert DAGs into triglycerides (TGs) could prevent insulin resistance (160).
Inflammation
The pathophysiology of obesity-induced insulin resistance has also been correlated with an increase in circulation and tissue inflammation originating in the adipocyte damage and infiltration of immune cells (107, 151). As fat accumulates in adipose tissue, adipocytes overcome their healthy size limit (157, 169) and release inflammatory cytokines and molecules known as adipokines. The excessive accumulation of lipids in the adipose tissue leads to adipocyte hypoxia (73), endoplasmic reticulum (ER) stress (132), and cell death, and causes FA spillover (145). Infiltrated immune cells also contribute to this chronic low-grade inflammatory milieu, whereby the increase in inflammatory cytokines causes insulin resistance elsewhere in the body. Thus, anti-inflammatory strategies become central as possible new treatments of insulin resistance and other complications of obesity (56).
ER stress
The fundamental role of ER is the integration of multiple metabolic signals and the maintenance of cell homeostasis (26, 58). Under stress conditions that challenge its function, ER triggers an adaptive response called uncoupled protein response (UPR) (97, 178). To resolve ER stress, UPR promotes a decrease in protein synthesis, and at the same time, an increase in protein degradation and chaperone production for protein folding. During the chronic energy surplus associated with obesity, ER cannot recover its normal function and UPR activation contributes to the development of insulin resistance through several mechanisms, such as JNK-Ap1 and NF-Kβ-IKK signaling pathways, inflammation, and oxidative stress (74).
Food intake
The central nervous system, specifically the hypothalamus, is of crucial importance in obesity-induced pathologies, since it plays a major role in the control of food intake and regulation of body weight (177). The discovery of leptin was a major breakthrough for our current knowledge of energy homeostasis (179). This adipocyte-secreted hormone acts on the hypothalamus to inhibit food intake and control body weight and has proved essential in the interaction between the brain and other organs in obesity-related disorders (51). The alteration of the circadian rhythm has also been associated with an increased risk of obesity (63, 76), proving that it is not only what you eat, but also when you eat it. Thus, advances in understanding the molecular mechanisms linking circadian rhythms and metabolism may provide new therapies for obesity and other pathologies associated with the disruption of normal sleep–wake cycles.
Lack of Efficacy of Current Anti-Obesity Drugs
Obesity develops when energy intake exceeds energy expenditure. Thus, any treatment for obesity-induced disorders must reduce energy intake, increase energy expenditure, or have an effect on both. Attempting to lose weight only by caloric restriction comes up against the problem that mammals have evolved mechanisms to store energy to survive during periods of starvation. Such homeostatic mechanisms increase caloric efficiency, thus making further weight loss even more difficult. In recent years, several anti-obesity drugs designed to limit energy intake have been withdrawn from the market due to serious adverse effects (i.e., fenfluramine, dexfenfluramine, sibutramine, and rimonabant) (36). Nowadays, only two drugs are approved specifically for weight loss by the US Food and Drug Administration (FDA): the lipase-inhibitor Orlistat that is also approved by the European Medicines Agency (EMA), but has a limited long-term effectiveness (36), and the recently approved serotonergic Lorcaserin-Belviq (84, 126). Thus, more efforts are needed to develop new anti-obesity agents. In this regard, strategies designed to increase lipid mobilization and oxidation could be very useful in the treatment of obesity and associated diseases.
Mitochondrial FAO and Bioenergetics
Hormones, such as insulin, glucagon, and noradrenaline, control the extracellular uptake and intracellular release of the main cell fuels; namely, carbohydrates and FAs. Once inside the cell, FAs are esterified, metabolized to lipid second messengers, or β-oxidized in mitochondria. The first step of the oxidative pathway is the transport of long-chain FAs (LCFAs) into the mitochondrial matrix (Fig. 1). This step is controlled by the carnitine palmitoyltransferase (CPT) system, which consists of three proteins: CPT1, acylcarnitine translocase (CACT), and CPT2 (109). Malonyl-CoA, a molecule derived from glucose metabolism and the first intermediate in lipogenesis, regulates FAO by inhibiting CPT1, thus making this enzyme the rate-limiting step in mitochondrial oxidation of FAs. Mammal tissues express three isoforms of CPT1: CPT1A (41) (liver), CPT1B (175) (muscle and heart), and CPT1C (138) (brain). Acetyl-CoA carboxylase (ACC), which controls the synthesis of malonyl-CoA; malonyl-CoA decarboxylase (MCD), which catalyzes malonyl-CoA degradation; and CPT1, which is regulated by malonyl-CoA, are components of a metabolic signaling network that perceives the level of fuel stimuli (109).
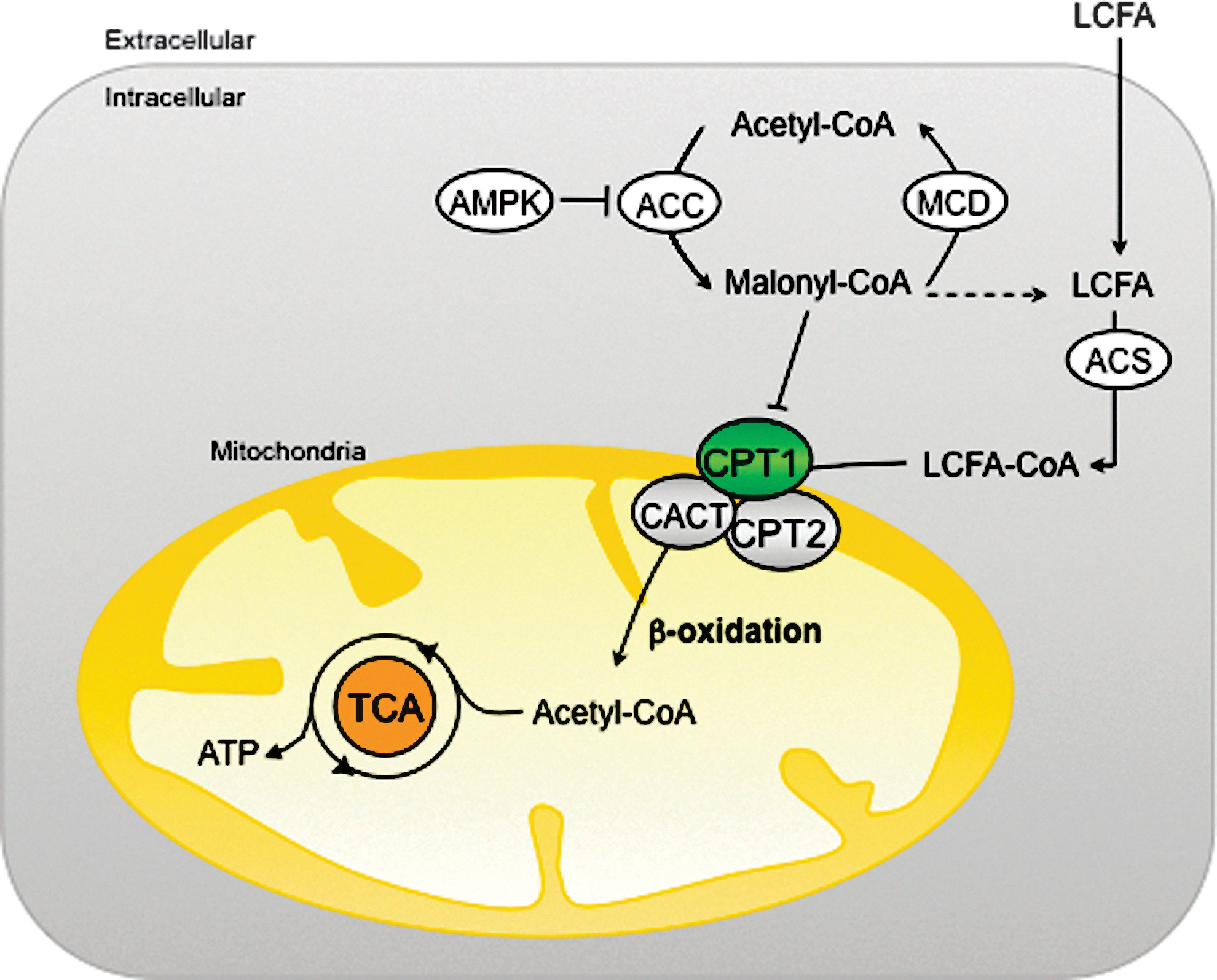
One of the main regulators of this network is the AMP-activated protein kinase (AMPK). This protein is the downstream element of a kinase cascade, activated by its phosphorylation in the Thr172 residue of the catalytic subunit (174). In general, AMPK inhibits ATP-consuming processes, while activating catabolic pathways. Active AMPK phosphorylates and inhibits ACC and reduces the expression of FA synthase, thus decreasing the flux of substrates in the FA anabolic pathway (176). In consequence, the reduction in malonyl-CoA levels leads to an increase in CPT1 activity and FAO.
The regulation of AMPK by members of the sirtuin family of NAD+-dependent protein deacetylases and ADP-ribosyltransferases (sirtuins) has been reported (94). Sirtuin 1 (SIRT1) and SIRT3 stimulate AMPK by deacetylating its upstream activator, kinase LKB1 (94, 135). In turn, the AMPK activity leads to an increase in NAD+ levels, thereby promoting deacetylation/activation of other SIRT1 targets involved in FAO, like peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α) (18). In recent years, sirtuins have emerged as critical modulators of lipid metabolism and specifically of FAO. In mammals, the seven identified members of the sirtuin family are differentially located within the cell: SIRT1, 6 and 7 are mainly located in the nucleus; SIRT3, 4 and 5 are located in the mitochondria; and SIRT2 is a cytosolic protein (111). In specific tissues, sirtuins act on different targets promoting FAO (liver and skeletal muscle [SkM]), mitochondrial respiration (brown adipose tissue [BAT]), lipolysis (white adipose tissue [WAT]), and food intake (hypothalamus) (148).
Energy flow in living cells (bioenergetics) takes place mainly in mitochondria. Energy is obtained from FAs and other nutrients in the form of ATP—the chemical currency of life—through the tricarboxylic acid (TCA) cycle, and the electron transport chain (ETC) in a process known as oxidative phosphorylation (162) (Fig. 2). This process is the main source of reactive oxygen species (ROS) in the cell. In the ETC, the energy of electrons from NADH and FADH2 is used to pump protons (H+) from the mitochondrial matrix to the intermembrane space and generate the electrochemical gradient necessary for ATP synthesis. However, when these electrons escape the ETC, ROS are produced in the mitochondria. Even under physiological conditions, the incomplete electron transfer to O2, resulting in ROS production, occurs with 0.2%–2% of oxygen molecules (21). Mitochondrial ROS production is highly regulated and important for various cell functions, as ROS can act as signaling molecules. However, high ROS levels are associated with significant cell damage and mitochondrial dysfunction in a process known as oxidative stress (122), usually associated with the etiology of obesity, insulin resistance, and type 2 diabetes (67).
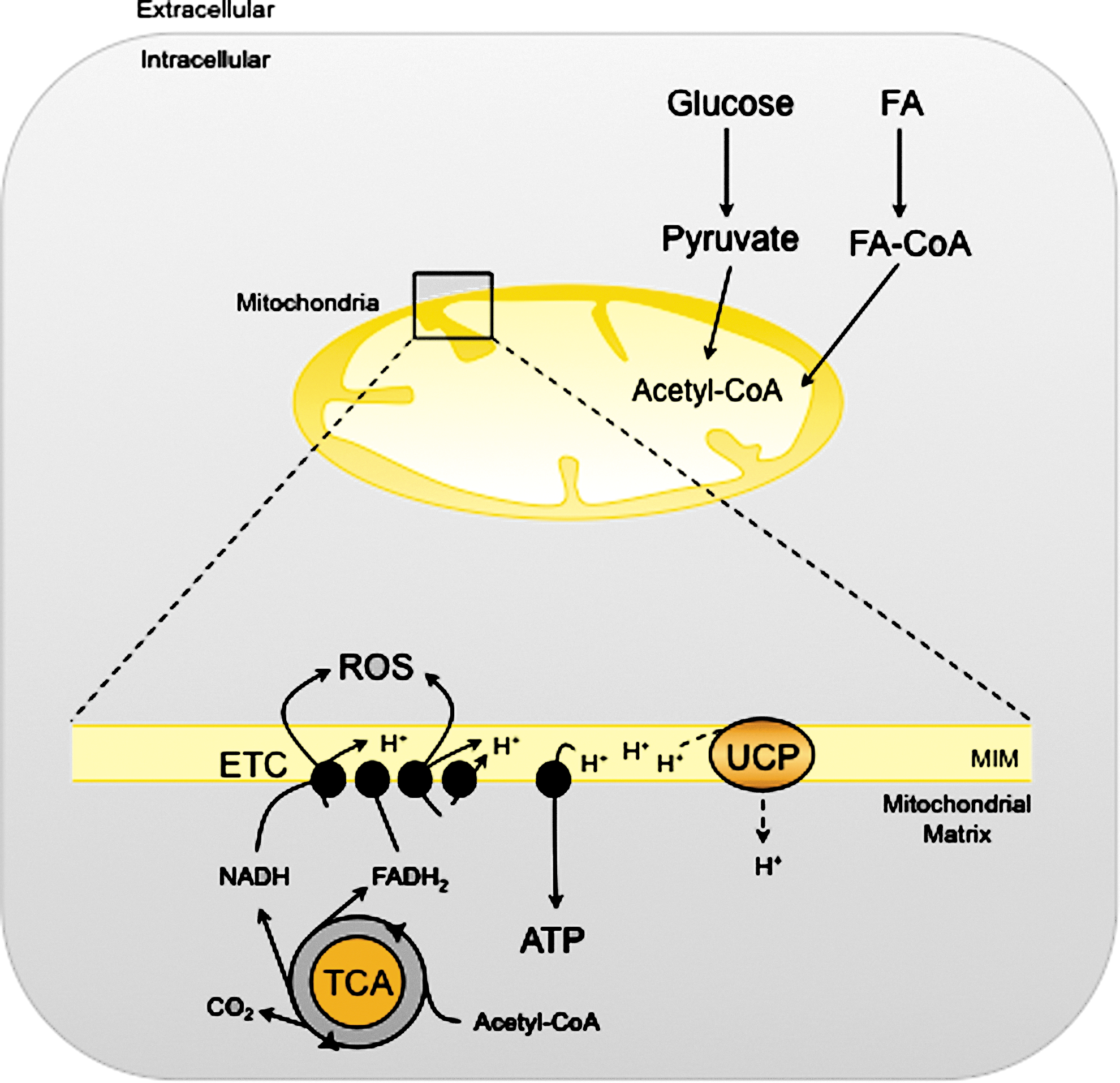
In the mitochondria, the ETC complexes remain electron-bounded when the proton gradient between the mitochondrial matrix and the intermembrane space is high, preventing the outward pumping of H+ and increasing ROS production. Mitochondrial uncoupling proteins (UCPs) uncouple ATP production from mitochondrial respiration, thereby reducing the H+ gradient across the inner mitochondrial membrane and relieving the formation of ROS (106). In BAT, UCP1 dissipates energy as heat and plays a key role in adaptive thermogenesis. In other tissues, the UCP homologues (UCP2, 3, and 4) affect ROS production and have crucial roles in energy homeostasis (106).
The Central Role of Liver in Obesity
Fatty liver and nonalcoholic steatohepatitis
The liver plays a central role in both energy expenditure and lipid/glucose homeostasis. In conditions associated with prolonged excess of energy or impaired FA metabolism, the liver stores considerable amounts of lipids in a process that leads to nonalcoholic fatty liver disease (NAFLD). A hallmark of NAFLD is the accumulation of hepatic TGs, which originate in the increased availability of free FAs (FFAs; circulating and from de novo lipogenesis), altered FAO and inadequate synthesis and export of VLDL (37, 55). The imbalance between these inputs and outputs results in lipid accumulation in hepatocytes, which causes hepatosteatosis and insulin resistance. The progression of a more severe liver disease triggers nonalcoholic steatohepatitis (NASH), a serious condition of inflamed fatty liver that can further progress to liver fibrosis and cirrhosis (32, 155). The pathogenesis of NAFLD in human and animal models has been reviewed in seminal articles (27, 161). The mechanisms underlying NAFLD to NASH progression are not completely understood. However, the alteration of FA metabolism and ROS production, which lead to mitochondrial dysfunction, and the induction of proinflammatory cytokines and fibrosis have emerged as key components that ultimately cause this liver disease (47).
Alterations of mitochondrial FAO and ROS production
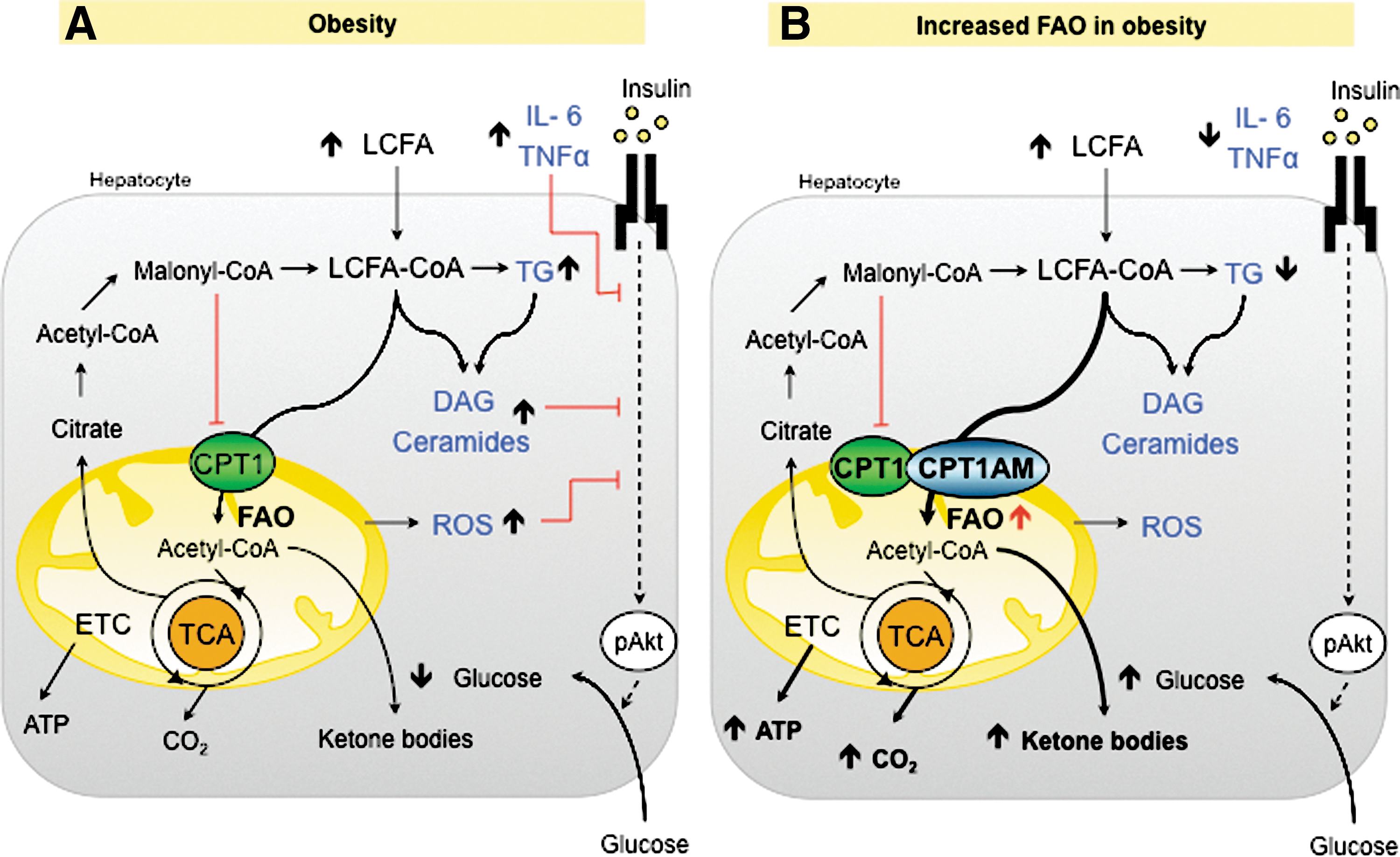
Mitochondria play a vital role in the oxidation of FAs and ROS production. In the liver, mitochondrial FAO results either in complete oxidation to carbon dioxide or in partial oxidation to ketone bodies, which are exported to provide fuel for other tissues. The key step is catalyzed by CPT1A (109). Data on the rates of mitochondrial FAO and CPT1A activity in NAFLD/NASH are not conclusive, possibly because of the use of different models and parameters. In vitro and in vivo studies of liver or hepatocytes exposed to high FFA concentrations show both increased (25, 44, 112, 116) and decreased (43) mitochondrial FAO. Similarly controversial results were obtained with CPT1A expression and activity. An increase in CPT1A expression has been reported in several rodent models (14, 134). However, a considerable decrease in the expression of this enzyme was observed in NAFLD patients (123). Several mechanisms may explain these controversial data: (i) the variation of malonyl-CoA levels that depend on the expression and activity of AAC/MCD enzymes (24, 39); (ii) the loss of CPT1 sensitivity to malonyl-CoA, which alters CPT1 activity (29, 133); and (iii) the increased pool of FFAs or lipid derivatives in hepatocytes may activate transcription factors, such as PPARs (PPARα, PPARβ/δ), which in turn may enhance CPT1 expression and FAO (19, 20, 22, 83). However, the transcriptional effect of PPARα on liver CPT1 remains controversial, since some studies have suggested that long-chain FFA regulates CPT1 expression through a PPARα-independent pathway (96, 101). Interestingly, another mechanism that modulates the CPT1 activity has recently been proposed (150). In this study, a NASH rat model fed a methionine-choline-deficient diet had a notable reduction in CPT1A activity and mitochondrial FAO despite increased CPT1A mRNA expression. The formation of a 4-hydroxynonenal-CPT1 adduct caused by lipid peroxidation as a consequence of ROS overproduction is the main cause of impaired FAO and lipid removal from hepatocytes. ROS can attack polyunsaturated FAs, initiating lipid peroxidation and the formation of aldehyde by-products (ahydroxy-2-nonenal [HNE] and malondialdehyde [MDA]), which have longer half-lives than ROS and are able to spread from their site of origin to reach distant intracellular and extracellular targets, thereby amplifiying the effects of oxidative stress (44, 170). The observations listed above suggest that CPT1 overexpression and increased CPT1 activity occur in liver during the onset of steatosis as a mechanism to compensate for increased FA levels. Accelerated mitochondrial FAO might cause excessive electron flux in the ETC and ROS overproduction. As lipids, proteins, and mitochondrial DNA are the main ROS targets, increased ROS might initiate lipid peroxidation, damage mitochondrial DNA and proteins, and alter mitochondrial morphology and function. The post-translational modifications of CPT1 caused by lipid peroxidation could, subsequently, decrease CPT1 activity and reverse initially activated FAO. This notion raises the question of whether interventions aimed at promoting mitochondrial FAO in liver would be beneficial to the treatment of NAFLD/NASH.
Several strategies have been used to promote FAO, including the use of PPAR agonists (7, 114), AMPK agonists [metformin (180), AICAR (166)], and ACC antagonists (62, 108). Genetic aproaches specifically promoting liver FAO are of considerable interest. An improvement in high-fat diet (HFD)-induced liver insulin resistance has been described in rodents, in which, FAO was indirectly enhanced by the reduction of malonyl-CoA levels through the modulation of ACC (146) and MCD (4). More interesting are the results obtained with a direct enhancement of mitochondrial FAO by increasing CPT1A expression in liver. Recent studies (117, 130) overexpressing an active and malonyl-CoA-insensitive mutant form of CPT1A (CPT1AM) (118) in obese rodents show that permanently enhanced liver FAO not only rescues impaired hepatic, muscle, and WAT insulin signaling in these animals, but also reduces steatosis, inflammation, and adiposity (Fig. 3). Furthermore, in these studies, enhanced mitochondrial FAO did not increase ROS derivatives or liver injury. Taken together, these results highlight an increase of CPT1A as a new strategy for the treatment of NAFLD/NASH pathologies. Alternatively, it has been reported that the admistration of CPT1 inhibitors reduces gluconeogenesis and improves glucose homeostasis, although chronic treatments on HFD-treated mice caused hepatic steatosis (28, 53). This side effect has interrupted the development of other systemic inhibitors, such as etomoxir and 2-tetradecylglycidic acid, as a therapeutic tool. Taken together, these data support the idea that any strategy able to switch liver FA's fate from esterification toward oxidation produces a beneficial effect on the liver and on the whole body. It seems that the liver can deal with an increased flux of FA into the mitochondria, thus escaping from liver injury. This is due to the ability of liver to flip the balance from complete oxidation to ketone body production (88). The ketone bodies produced by enhanced FAO are easily consumed by other tissues, increasing the flux of carbons from liver to other organs. Recently, a new hepatic factor, fibroblast growth factor 21 (FGF21), has emerged as a key regulator in FAO and ketogenic activation. FGF21 is induced in the liver during fasting (136) and has been increasingly pointed to as a potential therapeutic agent in obesity-induced insulin-resistant states (78).
WAT Meets Inflammation and Metabolic Disorders in Obesity
WAT has long been recognized as the main storage site for lipids derived from food intake (142). This long-term energy reservoir stores lipids mainly in the form of TGs, which can be mobilized and used to generate ATP through the mitochondrial β-oxidation pathway in peripheral organs during periods of caloric need.
WAT is composed mainly by adipocytes, but also by immune cells, such as macrophages, T cells, and mast cells (68, 104). Thus, WAT is an active and endocrine organ that secretes a large number of adipokines, cytokines, and chemokines (i.e., leptin, adiponectin, resistin, TNFα, IL-6, MCP-1, and IL-10), and plays a key role in regulating whole-body glucose and lipid metabolism (46, 151).
WAT, obesity, and inflammation
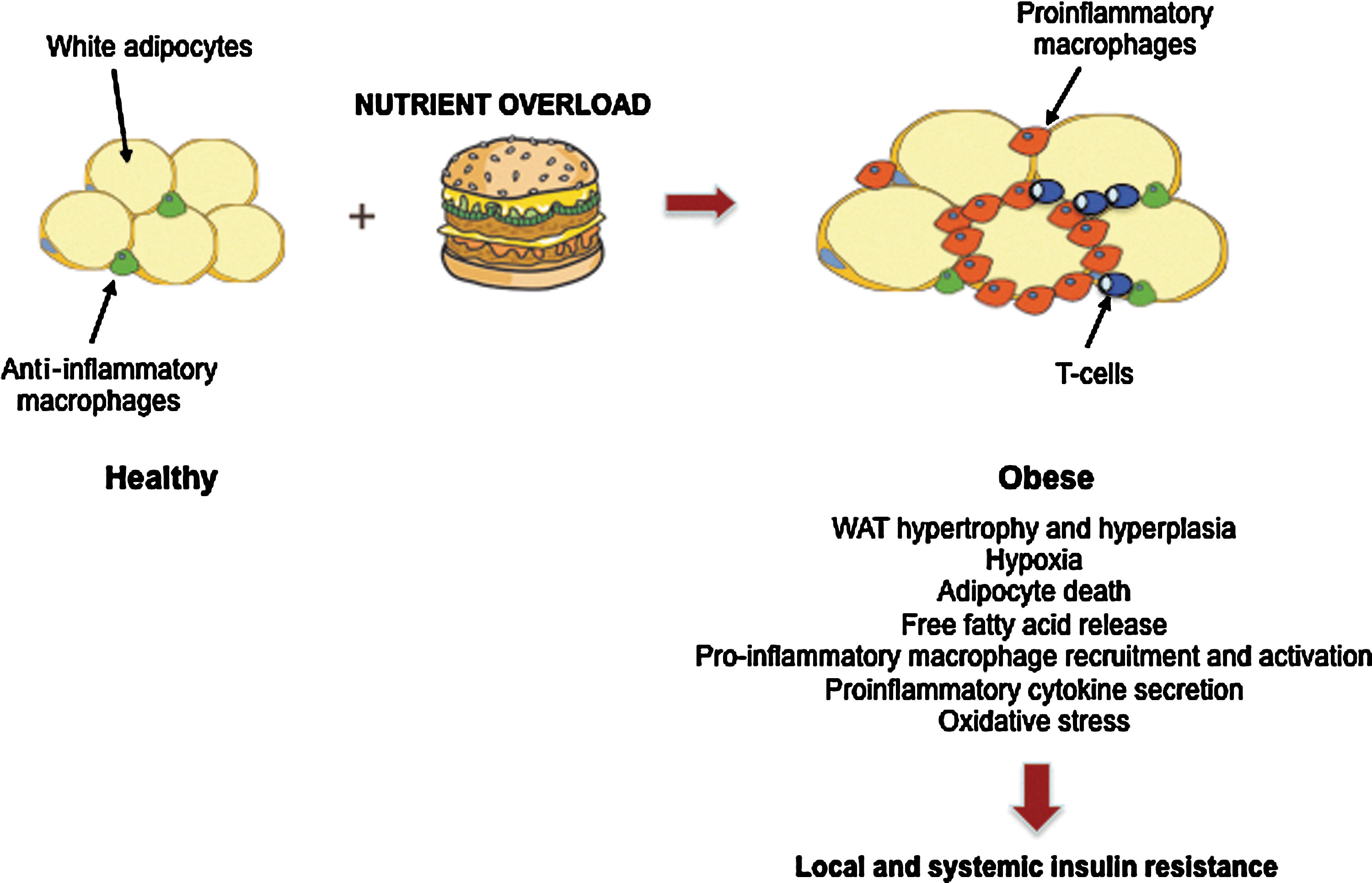
Obesity is characterized by the expansion of WAT mass due to an increase in both adipocyte number (hyperplasia) and size (hypertrophy), and it is closely associated with insulin resistance in peripheral tissues, such as SkM and liver. In fact, under excess caloric intake, WAT reaches its upper limit for further lipid storage (157, 169). Consequently, adipocytes exceed their oxygen diffusion limit, thereby promoting hypoxia (73), ER stress (132), and cell death, and increased circulating FFA and TG accumulation in ectopic sites is produced (145). The combination of microhypoxia and lipid overload triggers the recruitment of immune cells, such as macrophages in the adipose tissue and their activation (103, 129, 171). Obese adipocytes and infiltrated immune cells secrete a large amount of inflammatory mediators that promote a proinflammatory state through the activation of IKKβ-NF-Kβ and the JNK-Ap1 signaling pathways (151). The induction of JNK leads to serine phosphorylation of IRS-1 and 2, crucial molecules in insulin signaling, and consequently inhibits insulin action. These events cause insulin resistance in adipocytes, exacerbation of the inflammatory state, and systemic insulin resistance (59, 147) (Fig. 4).
FFAs, inflammation, and ROS
Physiological ROS production in adipocytes is a relevant cellular signaling mechanism in the insulin response and it depends mainly on the NADPH oxidase (NOX) family activity (9). In obesity, the excess of FFAs increases NOX-mediated ROS generation. Recently, it has been demonstrated that increased ROS in adipocytes exposed to an excess of FFAs does not depend on enhanced mitochondrial flux, but on high levels of TNFα and ER stress as well as the upregulation of aforementioned NOX enzymes (8, 60). ROS have the capacity to interfere with insulin signaling, since they activate several downstream pathways involving MAPK, JNK/IKKβ, and JAK/STAT, which are key contributors to the development of insulin resistance in obesity and type 2 diabetes (8, 75).
Enhanced FAO and improvement in insulin sensitivity
The imbalance between lipid storage and lipid utilization predisposes to adipocyte dysfunction and FFAs promote the proinflammatory response and ROS production involved in severe metabolic disorders. Although the exact physiological role of FAO in WAT remains to be determined, recent studies have shown beneficial effects of increased FAO and lipolysis in adipocytes, through direct CPT1A overexpression. In fact, this rise in lipid utilization improves insulin sensitivity in these cells and suppresses inflammatory signaling (50). However, there is still no evidence that increasing adipose tissue FAO would decrease FFA-induced ROS production. Thus, further research is required to elucidate these mechanisms and to evaluate the potential benefit of this strategy to prevent or reverse obesity and related metabolic diseases.
BAT, Turning Up the Heat
BAT morphology and function
In addition to energy-storing WAT, human fat consists of thermogenic controlling BAT. The latter has traditionally received less attention than WAT since it is less abundant and was considered exclusive to rodents and children. However, more recently, BAT have gained relevance in the mechanisms involved in obesity-related disorders.
BAT thermogenesis takes place in its numerous, densely packed mitochondria, which contain the BAT-specific UCP1. Activation of this protein uncouples aerobic respiration by producing heat instead of ATP (162). Brown adipocytes are also differentiated from white adipocytes because of their high expression of type 2 iodothyronnine deiodinase (DIO2), the transcription coregulators, PRDM16 and PGC-1α, and the lipolytic regulator, Cidea (52, 131). In rodents, BAT generates heat mainly for two reasons; namely, to protect against cold exposure via nonshivering thermogenesis and to burn the excess calories and reduce fat accumulation (54, 72). Therefore, BAT plays a crucial role in protecting mice from diet-induced obesity.
Rediscovery of human BAT
The fusion of positron emission tomography (PET) and computed tomography (CT) images allowed radiologists to see both functional and structural information in a single image. In the course of using PET-CT to detect and stage tumors in humans, active BAT was observed to increase after cold exposure (124).
However, the real breakthrough arrived in 2009 when five independent groups used PET-CT to identify the presence and study the relevance of BAT in adult humans (30, 143, 165, 168, 181). All the groups showed major depots of metabolically active fat in the cervical–supraclavicular region, a slightly different site from that in rodents and children, where BAT is found mainly situated in the interscapular area. The expression of UCP1, DIO2, and the β3-adrenergic receptor was also reported, thereby indicating the potential responsiveness of human BAT to both hormonal and pharmacological stimuli.
Interestingly, human studies showed that BAT is reduced in obese and diabetic patients, thus indicating that this tissue participates in both cold-induced and diet-induced thermogenesis (30). These observations made BAT a major breakthrough, since any strategy able to increase the mass or activity of this tissue could potentially provide hope for obese and diabetic patients.
BAT bioenergetics and mitochondrial metabolism
BAT is the only tissue to express UCP1, a protein found in the inner mitochondrial membrane that orchestrates the uncoupled reaction of allowing protons to re-enter the mitochondrial matrix without generating ATP. The dissipation of energy as heat confers BAT with the capacity to control thermogenesis. In fact, altered UCP1 expression (UCP1-deficient or transgenic mice) leads to dysregulated sensitivity to cold exposure and body weight control (40, 42, 86, 87, 98).
Body temperature changes stimulate norepinephrine release by sympathetic nervous endings that activate β-adrenergic receptors and trigger a signal transduction cascade that converts nutrients into acetyl-CoA. The TCA cycle uses this mitochondrial fuel to produce protons and electrons, which generate ATP through the ETC. However, in BAT, UCP1 allows protons to enter the mitochondrial matrix without generating ATP, that is, uncoupled, and heat is produced in this process. Thus, BAT burning power intensely clears and oxidizes circulating lipids and glucose to generate heat (17). This observation thus highlights BAT thermogenesis as an attractive therapeutic anti-obesity target.
Enhancing BAT burning power and browning of WAT as an anti-obesity strategy
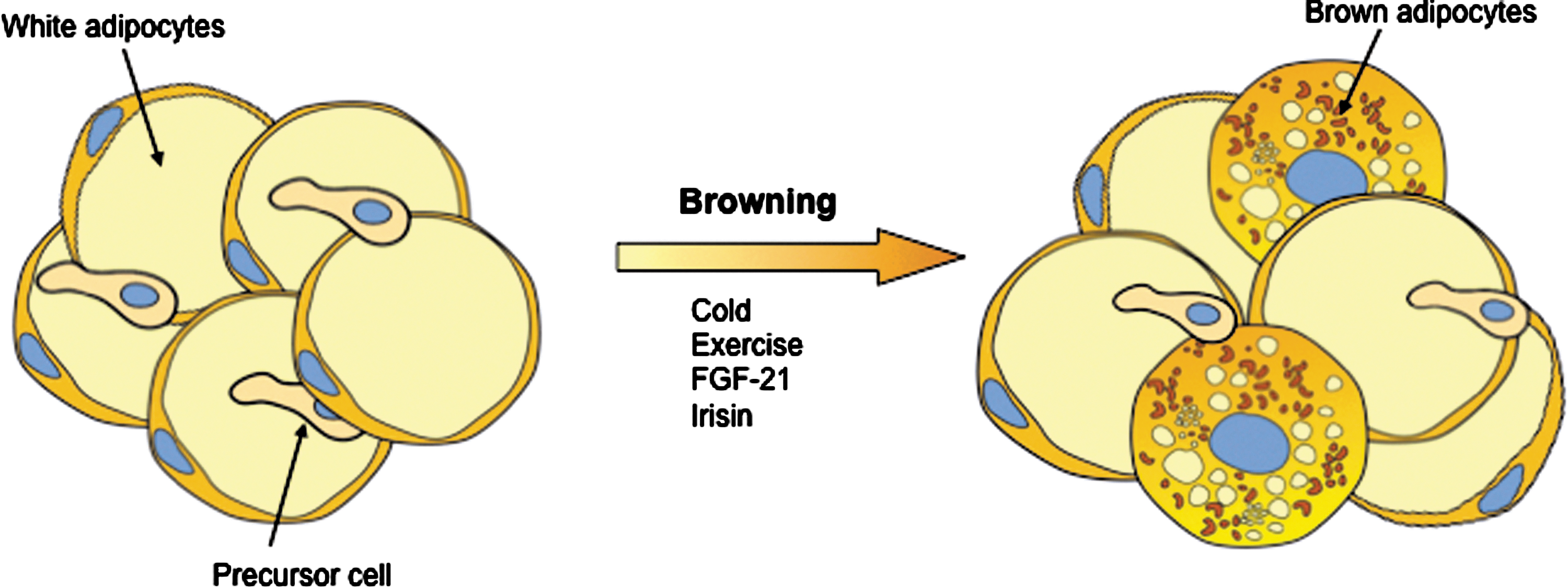
Recent landmark studies have identified novel secreted proteins, such as liver FGF21 (72), cardiac natriuretic peptides (11), and irisin (12), that stimulate brown adipocyte thermogenesis. Interestingly, a moderate increase (threefold) in irisin blood levels in mice enhanced energy expenditure and improved obesity and glucose homeostasis (12). In addition, recent articles reported that macrophages (125) and the bone morphogenetic protein BMP8B (172) have also the capacity to regulate BAT thermogenesis. Therefore, promoting BAT activation and/or brown adipocyte recruitment in white fat (browning) are approaches of considerable interest by which to develop pharmacological strategies to improve systemic metabolism by increasing energy expenditure (Fig. 5).
Role of FAO in Muscle Insulin Sensitivity During Obesity
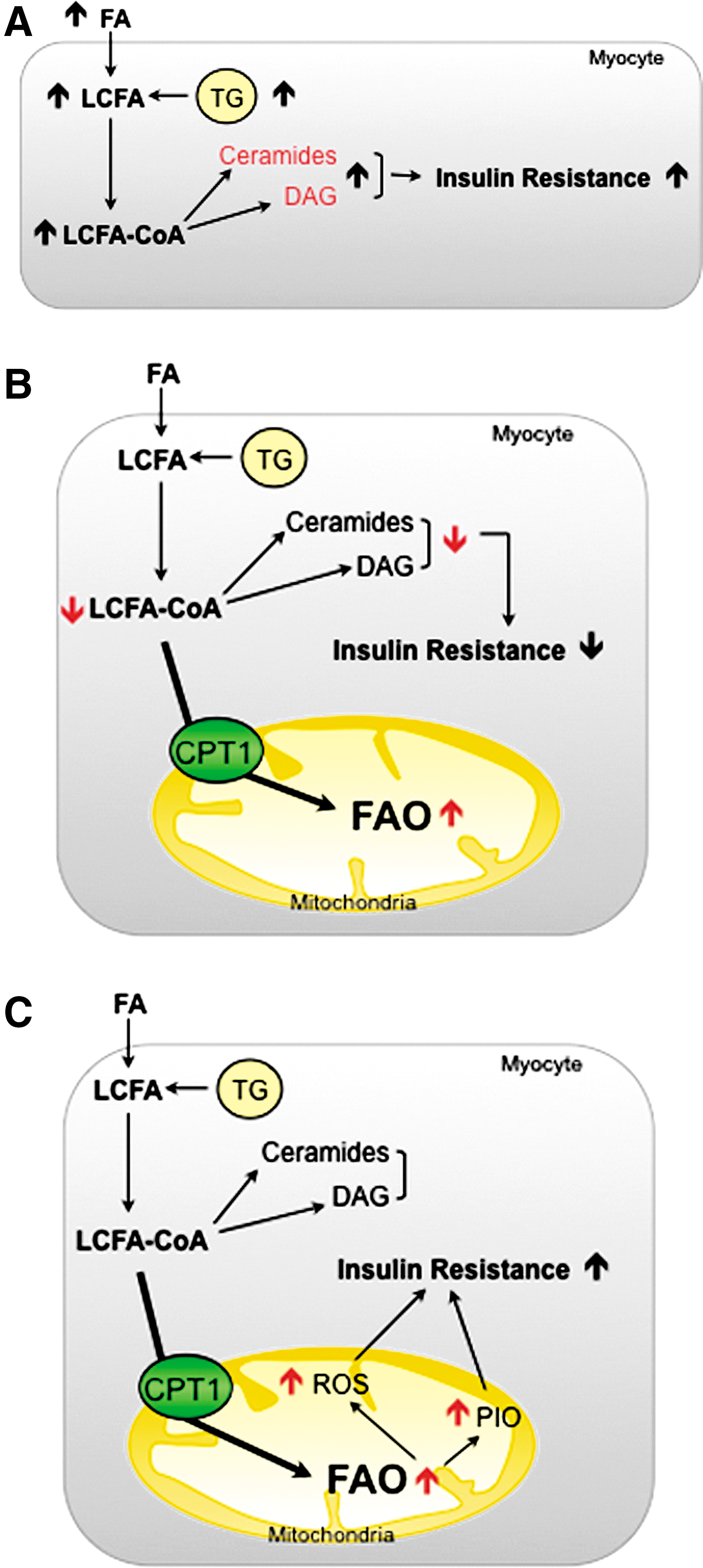
SkM is vital for the maintenance of glucose homeostasis. It accounts for ∼80% of total glucose uptake after insulin stimulation (34) and its transition to an insulin-resistant state is central for the pathogenesis of type 2 diabetes (152). Various pathways trigger obesity-induced insulin resistance (147). Both the action of fat-derived cytokines (adipocytokines) and ectopic accumulation of lipid deposition impair SkM function and play a critical role in insulin sensitivity. Obesity is characterized by a greater breakdown and uptake of FAs. Parallel with the increase in circulating lipids, the intramyocellular (IMC) lipid content appears to increase proportionally in obese humans (105) and rodents (23). The lipotoxic hypothesis proposes that FAs and their metabolites (such as DAG and ceramides) are important contributors to lipid-induced insulin resistance in SkM. These molecules activate proinflammatory and nutrient-sensing pathways that lead to the impairment of insulin action (Fig. 6A) [for further information, readers are referred to additional reviews (144, 159)].
Muscle FAO in obesity: two sides of the same coin
Several strategies have been designed to prevent IMC lipid accumulation and its deleterious effects. In SkM, these are mainly focused on the inhibition of FA uptake and esterification and/or the increase in FAO. However, whether the metabolic shift toward fat oxidation ameliorates lipid-induced SkM dysfunction is still open to debate.
Different laboratories have demonstrated that a decrease in the size and number of mitochondria, the activity of proteins in the ETC and, in general, impaired oxidative capacity in SkM are associated with obesity and insulin resistance in humans and animals (102, 139, 153). Given these observations, it is postulated that increased mitochondrial FAO could prevent lipid accumulation and, thus, improve insulin sensitivity in SkM (Fig. 6B). Several lines of evidence support this idea: (i) exercise in obese humans increases muscle mitochondrial FAO and improves glucose tolerance and insulin sensitivity (16). This effect is probably due to increased CPT1 expression and activity during exercise (163); (ii) direct CPT1 overexpression in animals and SkM cells protects muscle from FA-induced insulin resistance and apoptosis (15, 66, 149); and (iii) indirect SkM CPT1 activation, by the use of AMPK activators (77) or the ACC2 knockout (1), ameliorates insulin-stimulated glucose uptake in rodents.
Despite all the above points, the idea of mitochondrial deficiency as the main cause of diet-induced insulin resistance has been challenged in recent years. First, it is known that SkM's capacity to oxidize substrates is far in excess of what is needed to supply the energy demands of resting muscle (5). With this in mind, it seems clear that the mild (≈30%) reduction in mitochondrial content observed in obese patients does not affect the ability of resting muscle to oxidize fat. Further, despite the reduction in the mitochondrial content, insulin-resistant SkM has normal mitochondrial function (13). Secondly, since FA and glucose compete as metabolic substrates, the decrease in FAO would produce enhanced glucose utilization instead of insulin resistance. In fact, patients with severe mitochondrial deficiency have an increase in glucose uptake despite the large accumulation of IMC lipids (61). And finally, a HFD causes insulin resistance in rodents, while inducing an increase in SkM mitochondrial biogenesis and β-oxidation (164). This suggests that FAO might already be enhanced during obesity, contributing to the development of insulin resistance (121).
The main problems associated with an increase in FAO are: (i) the high rates of incomplete fat oxidation and (ii) the increase in oxidative stress associated with a mitochondrial overload (Fig. 6C) (89). Koves et al. (89) postulated that HFD induces the expression of FAO-related genes, but not those associated with the ETC and TCA cycle. This causes a mismatch between FAO and TCA cycle activity, thus leading to the accumulation of incomplete FAs and other intermediates (i.e., acylcarnitines) that correlate negatively with glucose tolerance (2). How these intermediates mediate the onset of insulin resistance is still unknown. However, studies linking overnutrition with the acylation of mitochondrial proteins are relevant to this topic and suggest a role for acylcarnitines (70). Mitochondrial members of the sirtuin family are known to deacetylate/activate several proteins involved in fat oxidation. This along with the observation that SIRT3 knockout mice have high acylcarnitines and increased acylated mitochondrial proteins and develop metabolic syndrome, implies that mitochondrial accumulation of FAO intermediates triggers metabolic complications via protein modification (71, 80).
Role of ROS in muscle insulin signaling
FAO intermediates may also create an unfavorable environment in the mitochondria, which contributes to the formation of ROS and the development of oxidative stress (119, 120). High levels of ROS and systemic oxidative stress have been associated with obesity and insulin resistance (45). However, these molecules are produced normally during mitochondrial respiration and are essential signal transducers that regulate several cell processes in SkM (8), including the insulin pathway (64, 65). Due to its dual role in insulin sensitivity, modulation of redox balance is crucial for correct cell function. On the one hand, under physiological conditions, ROS are generated in response to insulin and are required for its action (69), since the activity of different enzymes that participate in the insulin cascade are dependent on their oxidation state. In particular, the phosphatases that negatively modulate the insulin pathway are the main targets for oxidative inhibition by ROS (35). It has also been reported that ROS enhance insulin sensitivity in HFD mice (100). These molecules are also crucial for the SkM remodeling that occurs in response to exercise (31). ROS show insulin-like effects, stimulating SkM glucose uptake during muscle contraction via the activation of AMPK (81) and the induction of PGC-1α expression in vitro and in vivo (57, 79). On the other hand, during a pathophysiological state, abundant evidence indicates that a large and sustained increase in ROS impairs insulin action, through the activation of stress signaling pathways (i.e., MAPK and JNK) (85, 154), contributing to the pathogenesis of diet-induced insulin resistance (67). Therefore, clarifying the role of mitochondrial FAO, and the ROS production derived from it, in diet-induced SkM insulin-resistance is crucial to the development of new therapeutic strategies to fight against obesity and its related metabolic complications.
Hypothalamic FAO in the Regulation of Food Intake
Excess food intake in obesity is related to behavioral processes controlled by the central nervous system. Specific hypothalamic nuclei, which sense nutrients and modify energy intake and expenditure to maintain a balance, are responsible for regulating this intake (48). The mechanisms for sensing the nutritional state have not been fully discovered and less is known about lipid than glucose sensing. Nonetheless, there are two classic hypotheses on nutritional state metabolic mediators, involving malonyl-CoA and LCFA-CoA.
CPT1A on food intake control: linking the malonyl-CoA and LCFA-CoA hypotheses
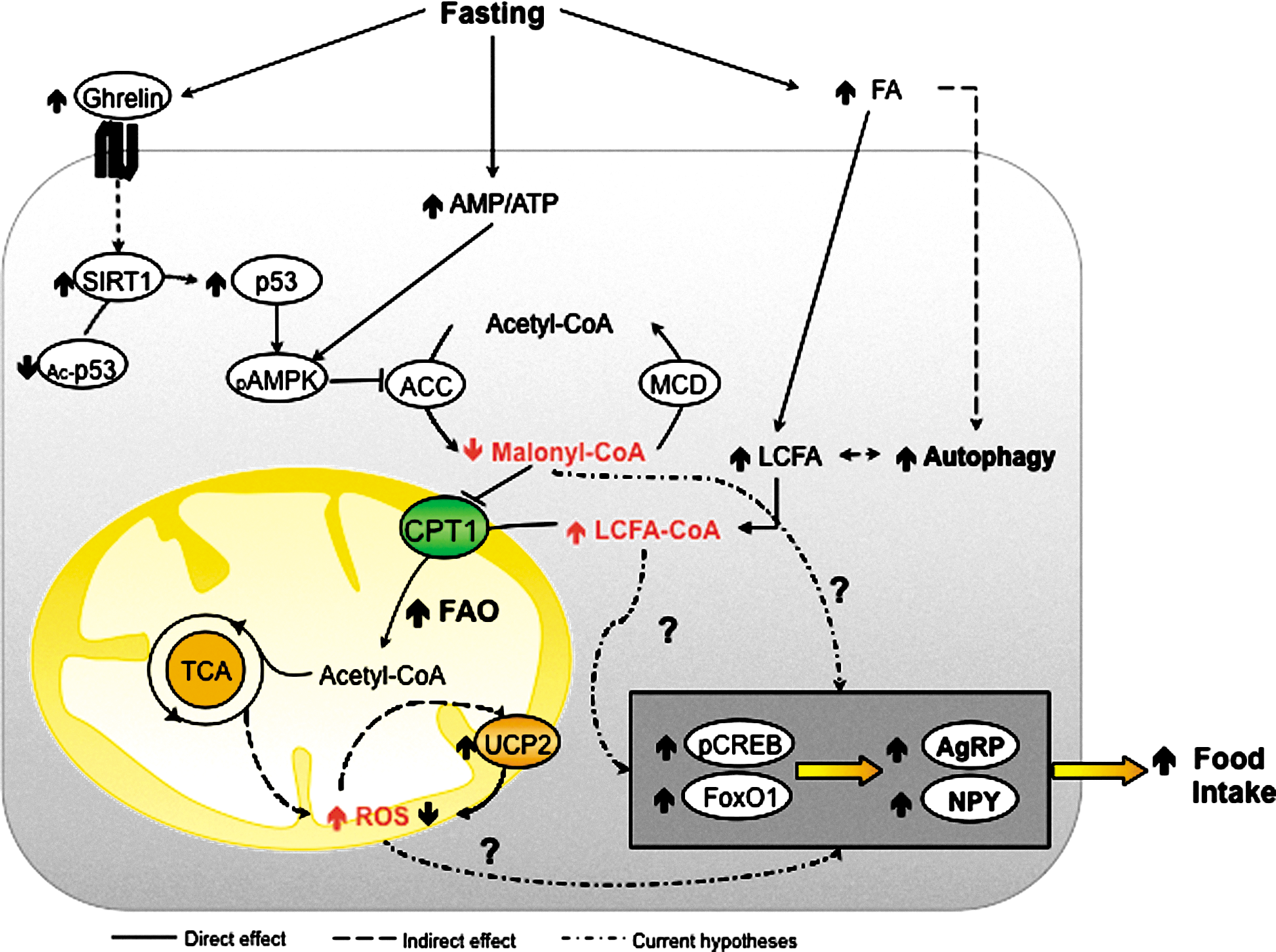
Strong evidence shows that increased malonyl-CoA levels, which are generated by activated (i.e., dephosphorylated) ACC, act as abundance indicators, thereby diminishing food intake and consequently body weight (173). During starvation, ACC phosphorylation is mainly controlled by the well-known energy sensor, AMPK (115), which acts also as an important hub, integrating in-cell energetic state sensing and its modulation due to different circulating hormones. Concretely, ghrelin-mediated activation (i.e., phosphorylation) of AMPK is done via SIRT1-mediated activation (i.e., deacetylation) of p53. It is known that the SIRT1 activity is increased in hypothalamus during starvation with high ghrelin levels (167). The mechanism of leptin action also appears to be related to increases in physiological malonyl-CoA in the arcuate nucleus, which promotes a reduction in food intake and body weight, using different mechanisms to those previously reported for ghrelin action, during energetically wealthy states (49, 141).
In addition, LCFA-CoA levels have also been put forward as mediators in nutritional state sensing. During starvation, circulating FFA increase can be sensed in hypothalamic nuclei through in-cell activation to LCFA-CoA. This increase, previous to active ghrelin rise, is an early reporter of a deficient nutritional state (156). However, unfortunately, no cytoplasmic LCFA-CoA increase in hypothalamus has been found. Even so, in the last decade, pharmacological and genetic inhibition of hypothalamic CPT1A has been reported to reduce the food intake (10, 110, 127), potentially as a result of LCFA-CoA cytoplasmic accumulation. Furthermore, the role of malonyl-CoA as a CPT1 inhibitor is worth noting, to link the two hypotheses.
Regardless of whether malonyl-CoA or LCFA-CoA is the main actor, changes in feeding behavior and peripheral metabolism are conducted via the action of neuropeptides, such as agouti-related protein (AgRP), neuropeptide Y (NPY), and melanocortins. Transcription factors affected by changes in hypothalamic FAO, such as CREB, FoxO1, and BSX, are involved in ghrelin-induced expression of NPY and AgRP (92). However, it is still unknown whether high levels of LCFA-CoA trigger NPY and AgRP via BSX and subsequent transcription factors. Probably, neither malonyl-CoA nor LCFA-CoA act as key molecular mediators per se to induce the expression of the different food intake-controller neuropeptides, but they are just two of the several pieces involved in the highly complex hypothalamic nutritional state sensing system, which is still not fully understood.
The relevance of FAO in neuronal cells is controversial because glucose is the brain's primary energy source. However, basal levels of FAO enzymes and FA transport proteins (such as FATP-1, FATP-4, and FAT/CD36) are expressed in hypothalamic neurons (93, 95). Additionally, the presence of such small, but functional FAO could be explained by its essential role in neuronal FA turnover and its action as a lipid-sensing mediator for energy homeostasis (113). There is also another controversy regarding fasting, because during fasting, high blood FFA levels presumably reach hypothalamic nuclei. This does not fit in with the satiety effect observed in early experiments with high LCFA concentration in the hypothalamus (128). Nonetheless, the direct correlation observed between fasting and FFA levels (156) and the capacity of hypothalamic neurons to sense glucose and FFA simultaneously and to integrate other nutritional and hormonal inputs (95, 99) seem to throw light on how important FAO really is in hypothalamic nuclei.
ROS, third in discord
Along with malonyl-CoA and LCFA-CoA, mitochondrial ROS have been put forward as a mediator in response to nutrient availability (140) (Fig. 7). ROS in the hypothalamus are produced chiefly in mitochondria ECT complexes. Concretely, some authors pointed to complex I as the most relevant site for single electron reduction in brain mitochondria (90). Various kinases and transcription factors, involved in neuropeptide regulation, have been reported to be modulated by redox signaling (33, 91). Further, the signaling under different levels of ROS may imply dissimilar pathophysiological changes in proteins, depending on their redox state (33). A certain level of ROS, without being excessive, is probably required to trigger neuropeptide expression. This notion may explain the importance of UCP2 as a ROS scavenger to keep a physiological ROS level that allows correct NPY and AgRP expression in the hypothalamus (6). In addition to all these considerations, the recent finding that hypothalamic autophagy is a source of endogenous FFA to regulate AgRP levels (82) could be explained by increased mitochondrial FAO and ROS signaling. However, further research is needed to establish the contribution of ROS to the hypothalamic control of food intake.
Innovation
Recent results suggest that enhancing cellular energy expenditure may be an attractive therapy to prevent or reverse the exponential growth of obesity-related disorders. We reviewed those recent discoveries regarding mitochondrial FAO and its potential as a therapy for obesity.
Conclusions
Despite considerable current efforts, the prevalence of obesity and associated diseases is rising exponentially in both industrialized and developing countries worldwide. This is especially worrying in the young. Current therapeutic strategies, focused mainly on controlling food intake, have met limited success, probably due to the inherent resistance of the human body to weight loss. However, recent approaches targeting energy expenditure and mitochondrial FAO shed light on new therapies to fight obesity.
Mitochondrial FAO is the cell source of energy from FAs. Since an excess of lipids is found in obesity and associated pathologies, a lot of research studies how to eliminate them through an increase in FAO. Beneficial effects of an increase in energy expenditure in obesity have been described in several tissues, including liver, muscle, WAT, and BAT. On the contrary, FAO therapeutic inhibition in hypothalamus seems to reduce food intake. Whether or not FAO should be modulated in the above-mentioned tissues to improve insulin resistance or to lose weight is still a subject of debate. There is no doubt regarding the involvement of ROS in pathophysiological processes related to obesity, and CPT1 seems to be a good molecular target for ROS and FAO modulation. However, several questions still need to be answered before FAO can become an obesity therapy. First, it is not known whether a long-term increase in energy expenditure would cause an enhancement of appetite as a compensatory mechanism. Second, an increase in FAO could induce pathological levels of ROS and/or other incomplete oxidation products. Third, it is not known whether FAO enhancement might reach a limit in a specific tissue, such as in BAT, in which thermogenesis is tightly adjusted to the environmental temperature. Finally, since increasing flux through β-oxidation would only make sense together with a corresponding enhancement in energy demand (121), the physiological relevance of improved mitochondrial FAO might be questioned if the individual remains sedentary (muscle, WAT, or liver) or warm (BAT). Potential mechanisms to explain the beneficial effects of targeting mitochondrial FAO could be the concomitant enhancement of hepatic ketone bodies, CO2, acid soluble products, ATP production, and endergonic processes (e.g., gluconeogenesis) seen in previous publications (38, 130, 158). Increased FAO may also decrease glucose oxidation to maintain energy homeostasis, augment mitochondrial burning capacity through an increase in the number of mitochondria and/or the increased expression of UCPs, and thus dissipate the excess of energy as heat and ATP. All of these could well alleviate the mitochondrial pressure found in lipid overload states. Thus, an increase in energy expenditure could indeed be the underlying protective mechanism against obesity-induced metabolic abnormalities.
Although more research is needed, we are encouraged that targeting of FAO and cell energy expenditure may be available in the near future as therapies to treat obesity and its associated severe diseases.
Footnotes
Acknowledgments
We thank Professor Fausto G. Hegardt for helpful, inspiring, and wise discussions and the Language Service of the University of Barcelona for valuable assistance in the preparation of the English manuscript. This study was supported by the Spanish Ministry of Science and Innovation (Grant SAF2010-20039 to L.H., Grant SAF2011-30520-C02-01 to D.S., and doctoral fellowships to M.I.M. and J.F.M.), by the CIBER Fisiopatología de la Obesidad y la Nutrición (CIBEROBN), Instituto de Salud Carlos III (Grant CB06/03/0026 to D.S. and research contract to P.M.), and by the EFSD/Lilly and EFSD/Janssen (research fellowships to L.H.).