
Abstract
Introduction
Topoisomerases (top) are a family of essential DNA repair enzymes that nick and religate DNA by forming a covalent enzyme-DNA intermediate between the enzyme's catalytic tyrosine residue and the end of the broken DNA (24, 200, 280). These covalent intermediates are referred to as “cleavage complexes” (200, 277, 278, 280). As a chemotherapeutic strategy, the usually transient topoisomerase cleavage complex (top cc) allows DNA to swivel during replication and repair but can be converted into a DNA lesion. Drugs such as camptothecin and etoposide trap topoisomerase by binding at the enzyme–DNA interface (23, 128, 199, 201, 202, 242). This “interfacial inhibition” effectively poisons the enzyme and converts the cleavage complex into DNA damage (201). In the continued presence of the interfacial inhibitor and unsuccessful DNA repair, cleavage complexes can be converted to DNA breaks when they are encountered by a replication fork (129). The induction of DNA breaks, stopping of DNA replication, and subsequent cell death in cancer cells are at the heart of successful antitumor activity by topoisomerase poisons.
Both iron and topoisomerase represent two distinct and mechanistically sound targets for cancer chemotherapy that have increasingly crossed paths over the past few years. Several agents identified as metal chelators have also exhibited selective topoisomerase inhibition, usually of topoisomerase 2 alpha (top2α) (Table 1). In addition, targeting both iron and topoisomerase contributed to the potent anticancer activity of these agents. Since the mechanisms of iron chelation and topoisomerase poisoning are complex, a clear reason why several structurally distinct drugs share these two targets is unclear. This review will analyze the known mechanisms of established and new iron chelators and topoisomerase inhibitors.
Dp44mT, di-2-pyridylketone-4,4,-dimethyl-3-thiosemicarbazone; top1, topoisomerase 1; top2α, topoisomerase 2 alpha; top2β, topoisomerase 2 beta; TSC, thiosemicarbazone
An agent that inhibits both DNA topoisomerase activity and iron metabolism is referred to as a dual inhibitor. This review is organized into three broad sections. The first section introduces iron and its chelation in cancer, the effects of iron-mediated cardiotoxicity, and the targeting of iron metabolism for cardioprotection and chemotherapy. The second section discusses topoisomerases, their activity in cancer, and their targeting by chemotherapeutic agents. The third section explores the overlapping mechanisms of dual inhibitors and potential biomarkers using doxorubicin, dexrazoxane, thiosemicarbazone (TSC)-24, and di-2-pyridylketone-4,4,-dimethyl-3-thiosemicarbazone (Dp44mT) as primary examples.
Iron Chelation in Cancer
Iron-mediated cardiac toxicity by anthracyclines
Cardiotoxicity induced by the anthracyclines is one of the most expected and monitored adverse events associated with an oncology agent (225). Anthracycline-induced cardiotoxicity is more prominent in female pediatric patients for reasons that are not fully understood. There is evidence that pediatric patients exposed to cardiotoxic chemotherapy early in life will, even if the cancer is completely eradicated, suffer from chronic cardiovascular disease later in life due to the cumulative adverse effects of anthracyclines (150, 261). Among the anthracyclines, doxorubicin is the most well-characterized agent due to its anticancer and cardiotoxic potential. Though preclinical studies did not detect cardiac toxicity, some early clinical studies as well as a retrospective evaluation of clinical studies of doxorubicin and daunorubicin established that cumulative doses of anthracyclines served as the main risk factor for dose-limiting cardiotoxicity (16, 66, 91, 144, 250, 271 –274).
Anthracyline cardiotoxicity is exhibited in two clinically significant forms: acute and chronic (66). Acute toxicity occurs during or immediately after anthracycline administration. It is related to rapid intravenous administration of the drug and results in vasodilation, hypotension, and transient cardiac rhythm disturbances (66). Chronic toxicity, which can be early, delayed, or late-onset, can manifest itself decades after completion of the anticancer treatment. Chronic toxicity is marked by myofibrillar loss and cytoplasmic vacuolization in cardiac myocytes, and occurs in survivors of childhood cancers who were treated with doxorubicin without immediate adverse effects (63, 66, 237, 274). According to data from in vitro studies, the damaging effects of reactive oxygen species (ROS), generated by the interaction of doxorubicin with iron, plays a critical role in the pathogenesis of the chronic cardiotoxicity (66, 237). Sub-acute and sub-chronic toxicities are uncommon (133).
The mechanism through which doxorubicin increases ROS levels and thereby induces cardiovascular changes and toxicity is reasonably well understood (56, 71, 111, 180, 237). The DNA damaging activity of doxorubicin likely plays a limited role in cardiac tissue due to the slower rate of proliferation of cardiac cells compared with tumor cells, and because of the lack of top2α in cardiac cells (21). The currently understood mechanism through which iron-mediated ROS are elevated by doxorubicin is presented in Figure 1. Being cationic, doxorubicin is preferentially taken up by mitochondria through negative membrane potential. Inside the mitochondria, doxorubicin interacts with anionic phospholipids such as cardiolipin or phosphatidylserine that are present on the inner membrane (27, 61, 74, 75, 145). Upon binding cardiolipin, doxorubicin interferes with a number of essential mitochondrial proteins such as pyruvate and cytochrome oxidase (145). Due to cardiolipin's role in the unfolding of proteins for transport across the inner membrane, its complexation with doxorubicin also indirectly inhibits the accumulation of proteins from the cytosol into the mitochondrial matrix (61). Doxorubicin is metabolized in cardiac mitochondria to a semiquinone that undergoes futile cycles of reduction and oxidation in the mitochondrial electron transport chain, leading to excess production of hydrogen peroxide (H2O2) and other oxidizing species (41, 55, 180). The quinone moiety in doxorubicin and other anthracyclines is also known for its ability to undergo iron-mediated redox cycling and produce oxygen free radicals (73, 180). Similar to other quinones, anthracyclines can be reduced enzymatically by one or two electron-transfer reactions. Two major pathways for the generation of ROS from anthracycline exposure have been proposed, one involving the Haber-Weiss and Fenton reactions and the second through the formation of anthracycline-iron complexes (77, 237). A one-electron reduction of the C-ring of doxorubicin leads to the formation of a doxorubicin semiquinone free radical. Flavoproteins, such as complex I NADH dehydrogenase, catalyze the formation of reduced semiquinone radicals by accepting electrons from NADP or NADPH and donating them to the anthracycline (labeled as mechanism I in Fig. 1). The doxorubicin thus “redox cycled” leads to the formation of superoxide free radicals (O2 •−). Superoxide dismutase catalyzes the conversion of the superoxide to H2O2. While H2O2 is stable by itself, the combination of H2O2 and O2 •− can lead to the formation of toxic hydroxyl radicals (OH•). As the first step in the Haber–Weiss reaction, ferric ion (Fe3+) is reduced to ferrous ion (Fe2+) by superoxide (47, 77). The second Fenton step between ferrous ion and H2O2 leads to the production of hydroxyl radicals. The short half-life hydroxyl radical is extremely reactive with all molecules in the cell and, hence, deleterious to the cell (47, 67, 77, 180).

The second mechanism involves the formation of an anthracycline-iron complex that could occur in the presence or absence of a reducing system. In the presence of NADH cytochrome P450 reductase or the thiols of cysteine or glutathione, anthracycline-Fe3+ is reduced to anthracycline-Fe2+ (labeled as mechanism II in Fig. 1) The ferrous ion conjugated anthracycline can react with O2 to form O2 •−, which is subsequently converted to H2O2 and could enter the Haber–Weiss reaction and produce highly reactive hydroxyl OH• radicals. Alternatively, the ferric ion-conjugated anthracyline can reduce its ferric component by either oxidation of the side chain on C9 or of the hydroquinone moiety at ring C. This results in an anthracycline free radical that is chelated with Fe2+. In the presence of O2, the anthracycline free radical can be oxidized to generate O2 •− and can react with H2O2 to yield OH•.
The iron-mediated mitochondrial generation of oxidative stress by doxorubicin damages proteins, lipids, and DNA (58, 182, 190) (Fig. 1). Cardiolipin, a major component of the inner mitochondrial membrane, is rich in polyunsaturated fatty acids, making it more susceptible to peroxidative injury by the free radicals generated by doxorubicin metabolites (94). The heart is especially sensitive to this oxidative stress because it is a mitochondria-rich organ and has lower defenses in terms of endogenous levels of superoxide dismutase and catalase than other organs such as the liver or lung (57). In addition, doxorubicin has been shown to cause a reduction in cardiac glutathione peroxidase expression (238). This enzyme functions to eliminate free radicals in the heart. The heart-specific NADH-oxidoreductase is also, at least in part, responsible for cardiac toxicity of anthracyclines (56, 186, 187). Cardiac, but not liver, mitochondria can reduce anthracyclines to semiquinones (186, 187). Thus, the heart is exquisitely sensitive to oxidative stress (111). Excess ROS produced by doxorubicin also induces protein oxidation via the carbonylation of apolipoprotein A1 in mice (5). The oxidized apolipoprotein A1 increases macrophage tumor necrosis factor alpha (TNF-α) release and is suggested to contribute to the toxicity of doxorubicin. Doxorubicin redox cycling also causes mitochondrial DNA damage that is manifested as mitochondria DNA (mtDNA) depletion, mtDNA mutations, and respiratory effects (141, 142). In addition to the known cellular consequences of excessive oxidative stress including apoptosis and necrosis, a pro-survival stress response known as autophagy also occurs as an early response to doxorubicin. This is discussed in greater detail in subsequent sections of this review (125, 267, 289).
Despite lower levels of antioxidant enzymes, cardiac mitochondria do attempt to mitigate the formation of excessive oxygen free radicals by the two-electron transfer enzyme DT diaphorase [NAD(P)H:(quinone acceptor) oxidoreductase] (26, 177). The other factors that affect the extent of anthracycline toxicity from excessive ROS are the glutathione system, low levels of labile iron, low levels of superoxide dismutase, and oxygen tension (177). Based on this information, it was proposed that a sound cardioprotective strategy might be the addition of an exogenous free radical scavenger (73).
Iron chelators as cardioprotective agents
General antioxidant strategies such as with coenzyme Q, vitamin A, carotenoids, vitamin C, vitamin E, flavonoids, polyphenols, n-acetyl cysteine, catalase, or superoxide dismutase gene therapies have been tested for their ability to ameliorate anthracycline-induced cardiotoxicity (1, 2, 11, 151, 181, 206, 218, 253, 282). Early in vitro and preclinical studies with several antioxidants showed promising cardioprotection that did not translate into clinical efficacy (31, 151, 230, 282). In some instances, the serum levels of the antioxidants required for cardioprotection were not pharmacologically achievable (253), while in others the cardioprotectants yielded a reduction in the anticancer efficacy of doxorubicin (30, 31, 230). Tissue uptake of the antioxidants, whether small molecule or gene therapy based, has also limited the clinical development of general free radical scavengers (1, 2, 151, 253).
The discovery that cardiac toxicity of anthracyclines involves iron-mediated redox cycling and cytotoxic generation of ROS spawned the investigation and development of new iron chelators, including siderophore analogs and synthetic ligands. Iron chelators have been tested for their ability as cardioprotective and/or chemotherapeutic agents. For a broader understanding of the history and chemistry of iron chelators for iron overload disorders and cancer chemotherapy, the reader is referred to other review articles (86, 132, 214, 215, 269). This section of the review will focus on the iron chelators that have shown in vivo activity as cardioprotective agents and possess some activity against topoisomerases, including dexrazoxane (ICRF-187), the TSCs, 2-hydroxy-1-naphthylaldehyde isonicotinoyl hydrazone (311), and triapine.
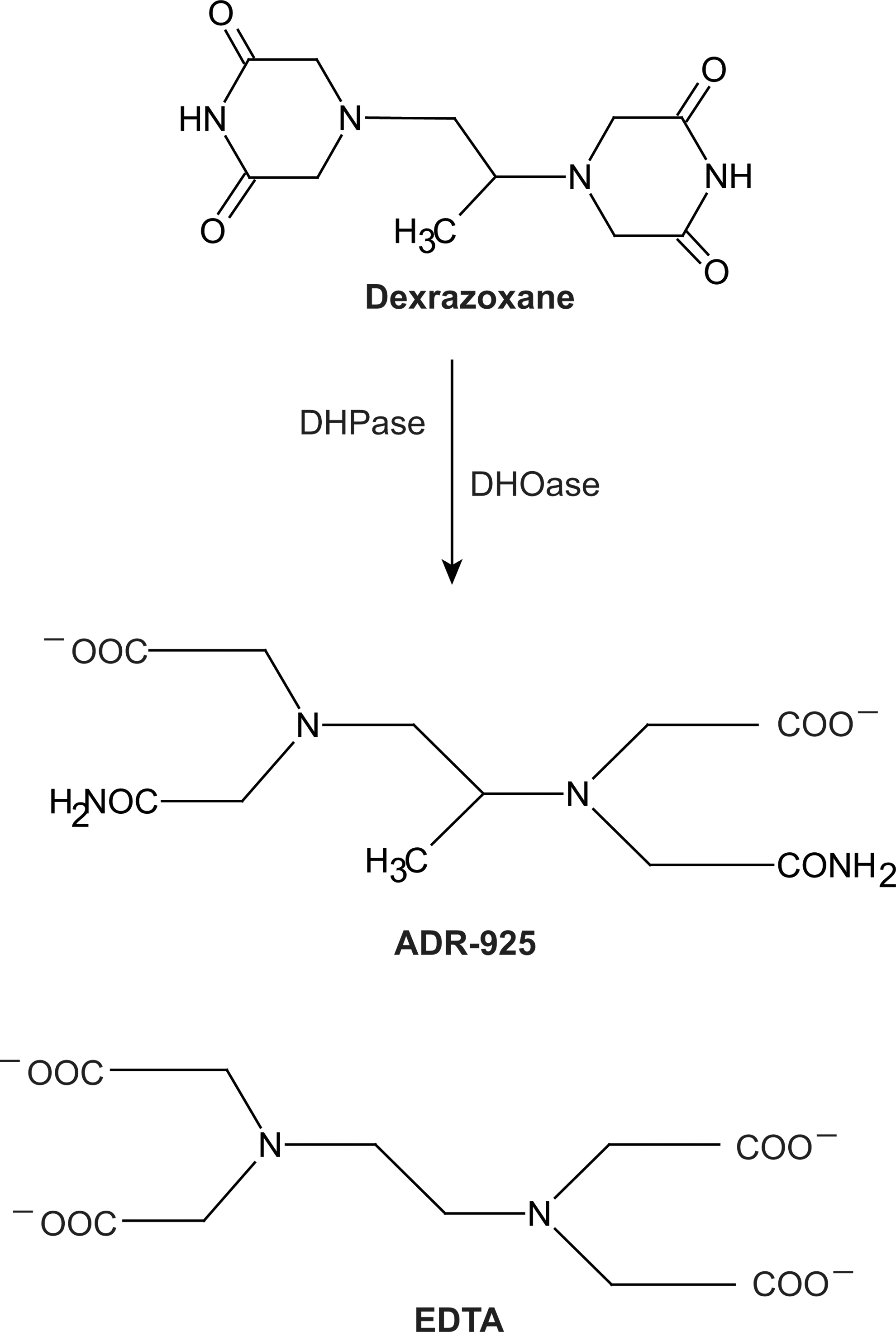
One iron chelator that has consistently shown cardioprotective ability in in vitro and in vivo test systems is dexrazoxane. Dexrazoxane is a bisdioxopiperazine that is orally active as a prodrug which is hydrolyzed to an ethylenediaminetetraacetic acid (EDTA)-like molecule, ADR-925, with iron chelating ability (Fig. 2). ADR-925 can rapidly displace iron from anthracyclines, suggesting that it has stronger affinity for iron than anthracylines. In vivo dexrazoxane has shown significant protection against doxorubicin-induced cardiotoxicity in numerous preclinical models such as mouse, rat, hamster, rabbit, and dog (81, 88 –93). In addition, the cardioprotective effects were evident in both acute and chronic models of doxorubicin-induced cardiomyopathy (90, 211).
Dexrazoxane has provided long-term cardioprotection without compromising anticancer efficacy in doxorubicin-treated children with high-risk acute lymphoblastic leukemia (62, 152, 153). The effect was greater in girls than in boys (153). Protection from anthracycline cardiotoxicity was also documented in pediatric patients with solid tumors (28, 62). Cardioprotection was accomplished in the presence of sustained anticancer activity by the combination. While there was some concern that the addition of dexrazoxane could lower the anticancer efficacy of doxorubicin (163, 248), there has so far not been any directly supportive clinical study that suggests anything but that dexrazoxane would be a pragmatic choice for sustained chemotherapy and cardioprotection (203).
The underlying mechanism by which dexrazoxane exerts its cardioprotective effect is by chelating iron, displacing iron from doxorubicin, reducing the levels of H2O2, and aiding in the up-regulation of pro-survival Akt and Erk phosphorylation pathways (84, 288). Dexrazoxane also induces protective hypoxia inducible factor (HIF)-1α and HIF-2α protein levels and activation in the cardiac cell line H9c2 (244). HIF proteins are transcription factors that are activated in response to low oxygen and regulate genes which overcome hypoxia. The anti-apoptotic effects of dexrazoxane which are observed in a rat model suggest that the overall outcome of iron chelation and prevention of oxidative damage rescues cardiac cells from the cytotoxic effects of doxorubicin (301).
Iron chelators as chemotherapeutic agents
Historically, the development of several newer generations of cardioprotective iron chelators was a pharmacological intervention in response to the cardiotoxicity by doxorubicin, which is an iron chelator with topoisomerase-inhibitory and DNA-damaging activity (291). While iron chelators were originally designed to be ROS-scavenging antioxidants and chemoprotectants in heart cells, some have been shown to induce excess ROS generation in cancer cells. As anticancer agents, iron chelators have shown marked and selective activity in several in vitro and in vivo test systems (131, 209). Doxorubicin targets top2α and topoisomerase 2 beta (top2β). top2α is absent in cardiac tissue, whereas top2β is present in most tissue, including tumors (21, 36). While the chemistry of bispiperazinedones (such as dexrazoxane) and newer TSCs (such as Dp44mT; Fig. 3) were designed and developed for iron chelation and cardioprotective ability, our understanding of these compounds in recent years has shed more light on their anticancer ability, either as single agents or in combination with anthracyclines.

Iron-chelating TSCs were first synthesized and evaluated for their anticancer activity in the late 1950s (18, 122, 123). Since then, several generations of TSCs have been tested for their chemotherapeutic potential (96, 108). In early mechanistic studies, the anticancer activity of TSCs was attributed to their ability to inhibit ribonucleotide reductase, an enzyme involved in DNA synthesis and repair (18). This inhibitory activity on ribonucleotide reductase is thought to result from the inhibition of the diferric iron core that is needed to stabilize the tyrosyl radical essential for enzyme activity (256). Recently, several TSCs have been shown to inhibit or poison topoisomerase 1 (top1), top2α, or top2β. 3-Aminopyridine-2-carboxyaldehyde thiosemicarbazone (3-AP or triapine) reduces topoisomerase I activity (25). Among those known to chelate iron and target topoisomerase IIα are dexrazoxane, (E)-N,N-dimethyl-2-(quinolin-2-ylmethylene)hydrazinecarbothioamide (TSC-24), and Dp44mT (Fig. 3) (18, 96, 108, 209). TSC-24 is considered a catalytic top2α inhibitor due to a direct interaction with the ATPase domain of top2α, which leads to a blockade of ATP hydrolysis. Interestingly, TSC-24 did not cause appreciable DNA damage as measured by neutral comet assay but rather reduced the DNA damage by the top2 inhibitor etoposide (96). Our laboratory demonstrated that Dp44mT causes DNA damage, apoptosis, and selectively poisons top2α, but not topoisomerase I or top2β, in Nalm-6 leukemic cells (209).
Topoisomerase Effects in Cancer
Types of topoisomerases and their enzymatic activity
DNA topoisomerases are a group of enzymes that resolve topological problems in the double-helical structure of DNA (24, 184, 201, 280). Topoisomerases are among some of the most well-studied enzymes for at least two reasons. First, there have been major advances in drug discovery of agents targeting topoisomerases that have successfully translated to the clinic and yielded some of the most widely used anticancer agents. Second, the crystal structures of a number of topoisomerase fragments representing nearly all known classes of topoisomerase have been solved, thereby providing molecular insights on these unique catalytic machines.
The fundamental need for this group of enzymes stems from the fact that DNA is a double-helical structure which lacks the inherent ability to rotate freely. Topoisomerases function in nearly all elements of DNA metabolism, including replication, repair, transcription, chromosome condensation and segregation, and the maintenance of genome stability (24, 201, 280). In general, these enzymes relax the positive and negative supercoiling to facilitate protein interactions with DNA and to prevent the deleterious consequences of supercoiled or unwound DNA duplexes. The structural features of all topoisomerases include hinged clamps that open and close to bind DNA, a DNA binding cavity for temporary placement of DNA fragments, and coupling of protein conformational changes to DNA rotation (24). Mechanistically, topoisomerases work by passing one strand of DNA through a break in the opposing strand (type I topoisomerases), or by passing a region of duplex DNA from the same or different DNA molecule through a double-stranded gap generated in that DNA (type II topoisomerases). All topoisomerases cleave the DNA phosphodiester backbone by nucleophilic attack from a catalytic tyrosine residue, which becomes linked to the phosphate end of the DNA break (279). These reactions do not change the DNA sequence and are reversible (201). The following section discusses therapeutically relevant classes of topoisomerases.
Type I topoisomerases work as monomers and form either 3′- or 5′-phosphotyrosyl covalent bonds. They are further divided into types IA and IB. Type IA includes the enzymes top3α and 3β and are the only enzymes that relax exclusively negative supercoiling. They form 5′-phosphotyrosyl covalent bonds with DNA. Type IB includes top1 and mitochondrial topoisomerase and form 3′-phosphotyrosyl covalent bonds. Type IIA topoisomerases work as dimers and cleave through a covalent attachment of each sub-unit of the dimer to the 5-end of the DNA through a phosphotyrosine bond (see top2 schematic in Fig. 4). In contrast to type I, the reactions of type II topoisomerases require Mg2+ and ATP hydrolysis for enzyme turnover and rapid kinetics; only one cycle of relaxation or decatenation/catenation can occur in the presence of the nonhydrolyzable analog of ATP, 5′-adenylyl-β,γ-imidodiphosphate. Type IIA topoisomerases include top2α and top2β (13). top2β has been less extensively studied compared with top2α, though they have some distinct structural features and different patterns of tissue-specific expression (13, 138, 241). The expression level of top2β is constant throughout the cell cycle, in contrast to the expression of top2α, which is low in resting cells and enhanced during proliferation (138).
Topoisomerase as a target for chemotherapy
top1, top2α, and top3α are essential in multi-cellular organisms, as their deletion is lethal in animals (4, 149, 176), which is in contrast to the other type I and IIA topoisomerases (136). top2β deletion results in developmental and neurological defects but viable embryos (164, 165). top3α is involved in resolving Holiday junctions during homologous recombination repair to prevent sister chromatid exchanges, but its application as a therapeutic target has not yet been fully explored and will not be discussed in this review (208). The essential nature of top1 and top2α is one reason that makes these enzymes attractive therapeutic targets for cancer.
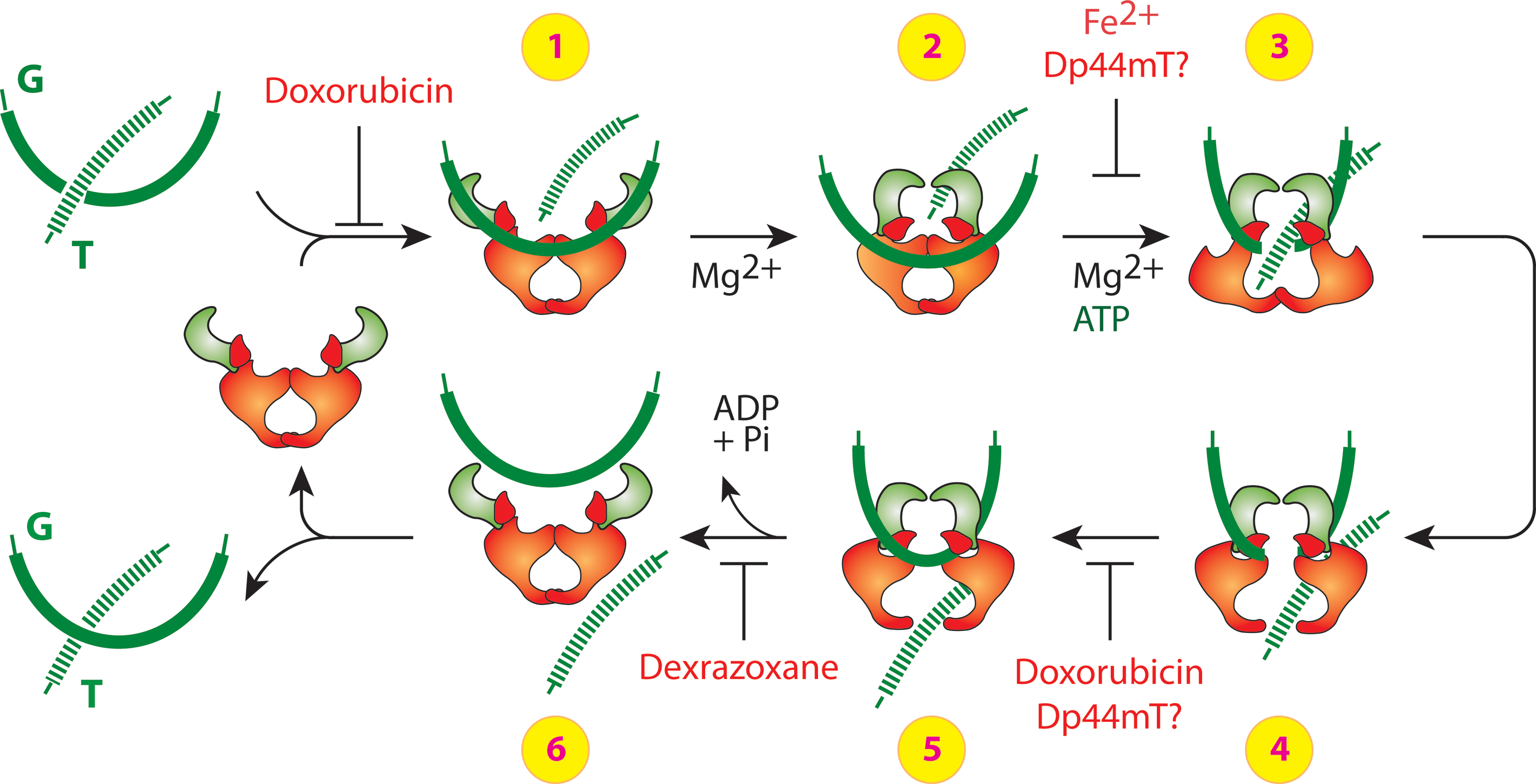
Agents that target top2 can work as catalytic inhibitors or poisons (139, 185). Catalytic inhibitors, such as dexrazoxane, can trap the top cc on DNA and inhibit its catalytic activity but do not result in a DNA strand break after a short exposure. Classical topoisomerase poisons, such as doxorubicin, increase levels of covalently bound topoisomerase-DNA complexes and generate lesions that result in a DNA double-strand break.
The mechanism through which doxorubicin and dexrazoxane target top2 is better understood than most top2 inhibitors or poisons. As illustrated in Figure 4, doxorubicin can block the catalytic cycle in two different steps. At low concentrations, doxorubicin can block DNA religation, whereas at higher concentrations, doxorubicin can interfere with top2 binding to DNA (before step 1 and between steps 4 and 5, Fig. 4) (201). The attempted DNA repair and subsequent conversion of unrepaired top2 cleavage complexes to DNA breaks by top2 poisons is discussed in the next section.
While doxorubicin can also chelate iron, its primary mechanism for anticancer cytotoxicity might not be iron mediated (163, 203, 248) and is likely due to its top2α-mediated and DNA damaging activities. Pretreatment with iron chelators dexrazoxane, desferrioximine, pyridoxal isonicotinoyl hydrazone, and salicylaldehyde isonicotinoyl hydrazone were used to show that the iron-mediated oxidative stress may not play a significant role in the cytotoxicity by doxorubicin in cancer cells.
Dexrazoxane can block ATP hydrolysis and inhibit the reopening of the ATPase domain, thereby trapping the topoisomerase complex on DNA and blocking enzyme turnover (185). The bisdioxiopiperazines dexrazoxane, ICRF-159 (rozoxane), ICRF-193, and ICRF-194 are iron chelators that enhance the formation of catalytic cleavage complexes with top2, but not top1 (7, 79, 251). Catalytic inhibitors such as dexrazoxane leave top2 trapped on DNA and interfere with DNA metabolism similar to top2 poisons but may not result in a DNA strand break in a short-term exposure (70, 100, 101, 185, 281). They act by trapping the enzyme in the form of a closed ATP-modulated protein clamp, thereby preventing the completion of the catalytic cycle (35, 82, 220). These agents are considered catalytic inhibitors and not traditional topoisomerase poisons, because not all have been shown to result in DNA double-strand breaks after the trapping of a complex with topoisomerase complex (7, 35, 79). Dexrazoxane and other bisdioxiopiperazines (ICRF-159, ICRF-193, and IRCF-194) have shown cytotoxicity in leukemic cells and cause DNA damage and apoptosis in various hematological cell lines at clinically achievable (4–5 μM) concentrations (163, 249, 293). The important role of top2α in the anticancer activity was confirmed in a transgenic mouse model with mutant TOP2A gene (76). Dexrazoxane has some clinical anticancer activity as a single agent (249, 270). Work from our laboratory and others has shown that dexrazoxane can induce DNA double-strand breaks as measured by the formation of the phosphorylated forms of the histone H2AX (termed serine 139 phosphorylated histone H2A [γ-H2AX]) in cancer cells (209, 211, 293). The DNA damage from dexrazoxane requires longer exposure times than classical DNA damaging agents such as γ-irradiation or etoposide, which is consistent with the hypothesis that dexrazoxane may function as a catalytic inhibitor of topoisomerase (185).
Expression levels of top1 and top2α are also high in cancer cells, and these expression levels correlate with the chemotherapeutic outcome from topoisomerase-targeting agents (17, 20, 33, 183, 196, 226). Levels of top1 in the NCI-60 cancer cell line panel correlate with sensitivity to the top1 poisons indenoisoquinoline and camptothecin (196, 198, 201). top2α expression is elevated in solid tumors and predicts responsiveness to anthracycline-based chemotherapy in women with primary breast cancer (33, 183, 226). Similarly, top2β levels in hematological cells correlates with sensitivity to and apoptosis by doxorubicin (117). The relationship between top2α and top2β and drug sensitivity in cancer is complex due to the role of the drug resistance proteins p-glycoprotein and multidrug resistance protein, changes in subcellular localization of top2 proteins, shared homology and catalytic activity between top2α and top2β, and phosphorylation/mutations in top2 (185).
Targeting top2β has recently been associated with increased incidences of secondary malignancies. Treatment-related acute myelocytic leukemia and myelodysplastic syndrome that progress to acute myelocytic leukemia have been reported for etoposide (158, 193). The occurrence of MLL gene transloctions has been related to the trapping of top2 cleavage complexes in the MLL gene (157, 158). The exact mechanism by which top2 cleavage complexes result in the 11q23 or 21q22 translocation in the MLL gene required for acute myelocytic leukemia is not yet clear. However, this raises the need to identify isoform-specific topoisomerase-inhibitory activity of iron chelators with potential anticancer use and the need to develop top2α-specific agents, when possible, to reduce the risk of secondary malignancies.
DNA repair responses to topoisomerase inhibition
As introduced in a previous section, the normally transient top cc can be converted to DNA lesions. Stabilization of the cleavage complex usually results from misalignment of the 5′-phosphate DNA end (200). Stabilization and misalignment can be generated by drugs bound at the interface of the enzyme and broken DNA or by alterations of the DNA substrate (such as abasic sites, mismatches, oxidized bases, and carcinogenic DNA adducts). Both top1 and top2 poisons exhibit such interfacial inhibition. For top1 poisons, the trapped enzyme generates a reversible single-strand break that is converted to a double-strand break when it is encountered by a replication fork (200). However, for top2 poisons, the two subunits of top2 can be degraded off the broken strands, yielding a double-strand break without the requirement of ongoing DNA replication (185, 254).
The repair mechanisms for top1-induced DNA damage are better understood than for top2 damage. For top1-associated DNA damage, three repair mechanisms are possible: (i) reversal of the covalent top1–DNA complex by 5′-end religation, (ii) top1 excision by tyrosyl DNA phosphodiesterase (Tdp1), and (iii) top1 excision by endonucleases. Since most iron chelators do not have an appreciable effect on top1, this review will not discuss top1 repair in greater detail. The reader is referred to other sources that discuss top1-mediated DNA damage repair in greater detail (24, 200).
The repair of top2 DNA damage requires the sensing of the top2 complex as an abnormality, the removal of the protein bound to DNA, and the repair of the DNA strand breaks. Before recognition as DNA damage, top2 covalent complexes can be reversed without any harmful cellular effects. As shown in the schematic in Figure 5, after being sensed and once repair is initiated, the top2 complex is probably irreversible, and the cells should either repair the damage or undergo cell death (185). Proteosomal degradation (26 S) or SUMOylated degradation of covalently linked top2 has been proposed to allow access of DNA repair enzymes to the broken DNA. A proteasome inhibitor, MG132, reduces the DNA double-strand breaks induced by etoposide (300). The role of the proteasome is important for the repair of both top2α and top2β damage by etoposide, as confirmed in top2β knock-out and in knock-down cells exposed to MG132 (300). Similar to the excision of top1 by Tdp1, a 5′-tyrosine phosphodiesterase Tdp2 (or TRAF and TNF receptor-associated protein, TTRAP) is capable of excising top2-DNA adducts (37). Cells depleted of Tdp2 are hypersensitive to top2 but not top1 poisons (299). Endonucleolytic cleavage can also remove top2 trapped on DNA by cleavage of DNA near the point of attachment of top2 at the 5′-DNA end. Rad27FEN1, a 5′-endonuclease, and the nucleases Rad52, Mre11, Sae2, and CtIP have been suggested to function by cleaving the top2-DNA site. Yeast strains carrying mutations in Mre11 and Rad52 orthologs are hypersensitive to top2 poisons (166).
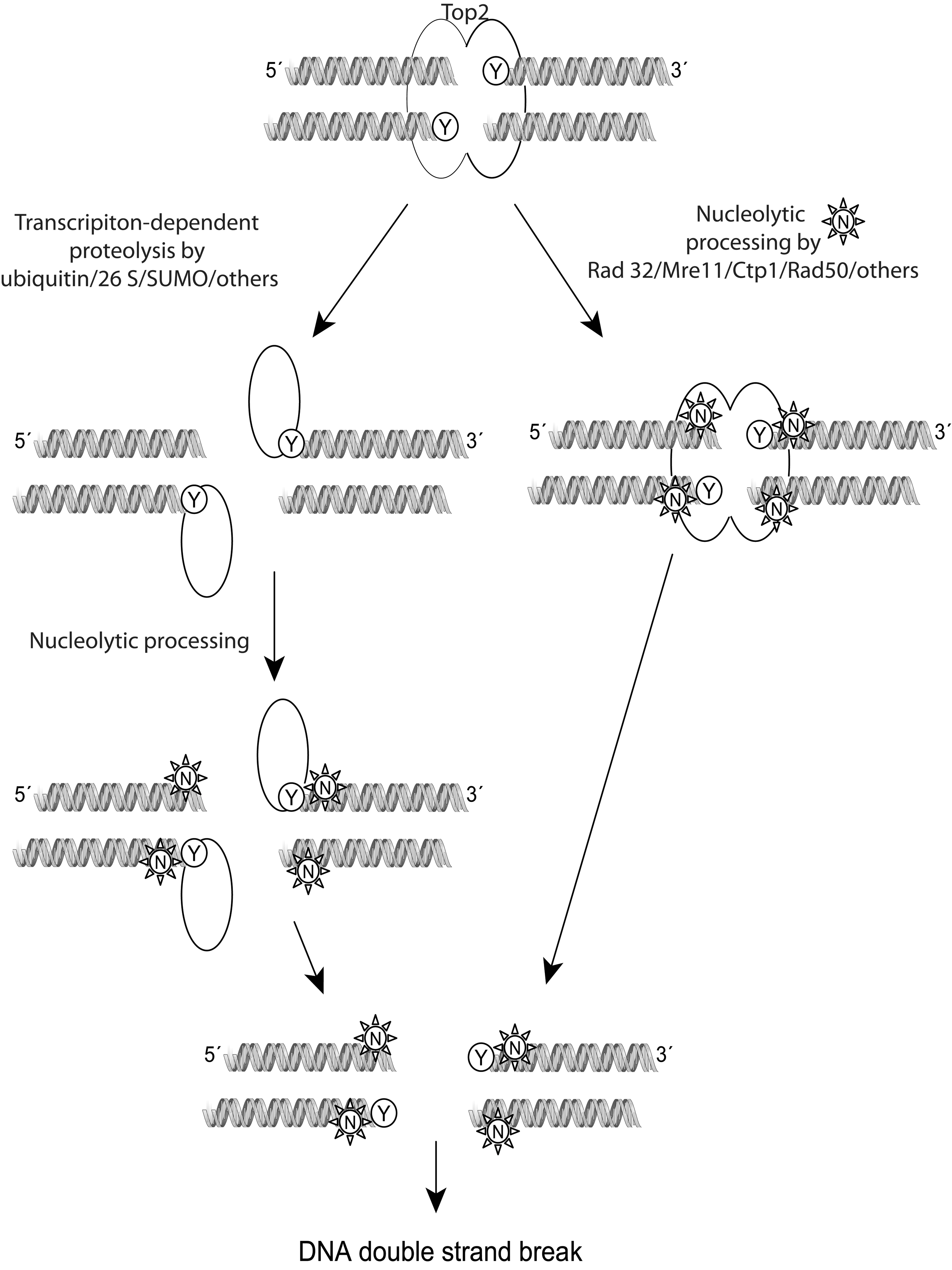
Proteolytic degradation of the top2 enzyme itself is also a potential pathway by which the cells respond to top2 DNA damage (167). This pathway is especially relevant because the top2-specific iron chelator dexrazoxane has been shown to induce the degradation of top2 (33, 167, 300). Degradation by the classical top2 poisons etoposide leads to proteolytic degradation of the enzyme by the ubiquitin/26 S proteasome pathway (167, 300). In one study, the top2β enzyme was preferentially degraded by this pathway over the top2α isozymes, while other studies using breast and lung cancer cell lines showed that both isozymes could be efficiently degraded by the proteosome in response to teniposide (167, 285). The proteolytic removal of top2 contributes to the processing of the top2 cleavage complexes into protein-free DNA breaks for subsequent activation of repair molecules, such as Chk1, Chk2, RPA, and γ-H2AX. The degradation of top2 and subsequent DNA damage signaling is attenuated, but not completely blocked, by pretreatment with proteosome and transcriptional inhibitors (64). Thus, transcription-, proteosomal-, and replication-dependent pathways may play a role in processing the top2 damage toward DNA repair.
Iron Chelators That Target Topoisomerase
Examples and significance of dual targeting anticancer agents
Several established and investigational iron chelators have been shown to act on topoisomerases (Table 1). The next section summarizes specific examples of dual targeting agents found in the literature and in our laboratory to exhibit some cytotoxic anticancer activity. It should be pointed out that in early papers the distinction between specific activity against top2α or top2β was not clear either due to experimention in Saccharomyces cerevisiae, which does not express the two isozymes, or because the top2 enzyme used was primarily top2α due to higher expression than top2β (J. Dickey [FDA], Personal Communication).
The compounds listed in Table 1 as being iron chelators that also act on topoisomerase (referred to as dual inhibitors) show varying degrees of anticancer activity in preclinical or clinical settings, with doxorubicin being the most promising as front-line therapy for breast cancer. The overall outcome from the combined activity of targeting both iron and topoisomerase appears to be a cytotoxic effect on cancer cells both in vitro and in animal models. In the case of doxorubicin, dexrazoxane, and triapine, promising results from safety testing in cancer patients warrants further studies that show efficacy in specific cancer types. Mechanistically, attacking rapidly proliferating cancer cells at two targets that are selectively up-regulated in tumors could be advantageous. Cancer cells exhibit higher levels of iron metabolism and elevated expression of topoisomerases. Hence, dual inhibitors might have novel benefits as anticancer agents. The use of dual inhibitors in combination with other anticancer agents, including other topoisomerase poisons, warrants particular consideration. In one study that compared the cardioprotective effects of different schedules of administration, dogs were exposed to dexraxozane 2 h before doxorubicin or simultaneously (90). The observations indicated that the timing of administration of dexrazoxane with regard to that of doxorubicin was an important factor, and the degree of protection was most evident when the agents were administered simultaneously. In vitro studies have suggested that the combined effect of doxorubicin and dexrazoxane is enhanced cytotoxicity in cancer cells (275, 276). A clinical study would address whether the iron chelation capabilities and metabolism of either agent interferes with the combined anticancer efficacy. Catalytic inhibitors of topoisomerase have been suggested to interfere with the DNA damaging activity of classical topoisomerase poisons, suggesting that iron chelators which are catalytic inhibitors but not poisons might benefit from being administered after the administration of a classical topoisomerase poison. This may allow the topoisomerase-induced DNA damage to occur before the iron chelation also exerts its antiproliferative effect. It is important to note that not all iron chelators interact with DNA and/or topoisomerases. Conversely, not all topoisomerase inhibitors can chelate iron to a physiologically relevant degree. Studies that have directly tested desferrioxamine and iron chelators of the pyridine-2-carboxaldehyde isonicotinoyl hydrazone class show high iron chelation potential but weak DNA binding capacity and no in vitro topoisomerase inhibitory activity (25). These agents also exhibit weaker cytotoxic effects compared with Dp44mT and doxorubicin and would not necessarily be expected to serve as anticancer agents. Some agents such as desferal are also limited in their bioavailability after oral administration and by a short half life on systemic administration. Studies designed to specially address the importance of the timing of inhibiting iron metabolism and poisoning topoisomerase are needed to fully understand the benefits of dual targeting drugs.
TSCs chelated with other divalent metal ions have also shown to inhibit topoisomerase II activity and kill cancer cells. These include Cu(II) complexed 2-acetyl-pyridyl-4N-substituted TSCs (171) and 1,2-naphthoquinone-2-thiosemicarbazone derivatives (234). Their ability to inhibit proliferation in breast cancer cell lines overexpressing top2α was greater than in cells with lower levels of top2α (171). A similar correlation between top2α levels and the ability of other copper-complexed TSCs to inhibit cancer cell proliferation has been shown in mice-bearing cancer cells with either high or low levels of top2α (284). Ruthenium(II) metal complexed TSCs have also been shown to inhibit top2α (12). Thus, TSCs and other chelators complexed with iron or other divalent metal ions have shown activity against topoisomerases that is associated with anticancer cell killing.
Possible mechanisms and caveats of dual inhibition
In this section, some possible mechanisms by which dual targeting agents may function as anti-cancer agents will be discussed. For the purpose of this review, the agents are discussed within the two broad categories: (i) iron chelators that are catalytic topoisomerase inhibitors and (ii) iron chelators that are poisons for topoisomerases and cause DNA damage. As with most pharmacological agents, this categorization is not complete and does not account for other possible mechanisms of action. Some of the agents classified as catalytic inhibitors would benefit from further investigation on their ability to directly intercalate with DNA or cause DNA-strand breaks; for instance, by using methods such as alkaline elution or induction of γ-H2AX foci (127, 197).
Iron chelators that are catalytic topoisomerase inhibitors, such as ICRF-159 or ICRF-193, could function by primarily targeting the high iron metabolism of cancer cells. These agents might have greater utility in cancer cell types where levels of iron or iron-regulatory proteins are high, in cell types that are not necessarily as high in their cell proliferation rates, or where targeting DNA synthesis has not been therapeutically beneficial. For example, an iron regulatory gene signature has been suggested to predict outcomes in breast cancer patients (170). In this study, the ferroportin-hepcidin regulatory axis was correlated to the breast cancer outcome; specifically, a decrease in ferroportin protein levels and an increase in hepcidin levels were reported in malignant breast cancer cells compared with normal mammary epithelial cells. In other studies, the levels of transferin receptor, uptake of iron, and the ratio of placental-like isoferritin to normal ferritin were higher in breast cancer cells than in healthy mammary epithelial cells (233, 295). Similarly, high concentrations of ferritin and iron metabolism have been noted in neuroblastoma cells (43). top1 activity was higher in immature neuroblastomas than in adrenal glands (266). Using desferoxamine, which does not target topoisomerase, some studies in neuroblastoma cancer models have found no benefit from chelating iron (235, 236), while the use of top1 or 2α poisons have shown antineoplastic effects in neuroblastoma patients with low tumor burden (135, 266). Future studies may benefit from identifying the contribution of DNA damage and topoisomerase poisoning in the success or failures of iron chelators in cancers with high iron metabolism. The timing of administration of iron chelating catalytic topoisomerase inhibitors with topoisomerase poisons (e.g., dexrazoxane with doxorubicin or TSC24 with doxorubicin) is also important because catalytic inhibitors could competitively interfere with the cytotoxic effects of topoisomerase poisons.
Iron chelators that are poisons for topoisomerase, such as doxorubicin and Dp44mT, could target cancer cells by working on their separate targets or by accentuating the activity of topoisomerase (Fig. 6). In the simplest scenario, dual targeting agents chelate iron and poison topoisomerase separately to stop DNA synthesis and cause DNA strand breaks and cell death. The two mechanisms may work completely independently to elicit the convergent goal of growth arrest and cell death. Such a multi-modal mechanism is likely to be beneficial in cancer cells that are resistant to one or more of these therapeutic interventions. For instance, specific populations of breast or lung cancer cells with decreased levels of top2 expression and resistance to top2 poisons might still benefit from iron chelation by dual inhibitors (155, 228).
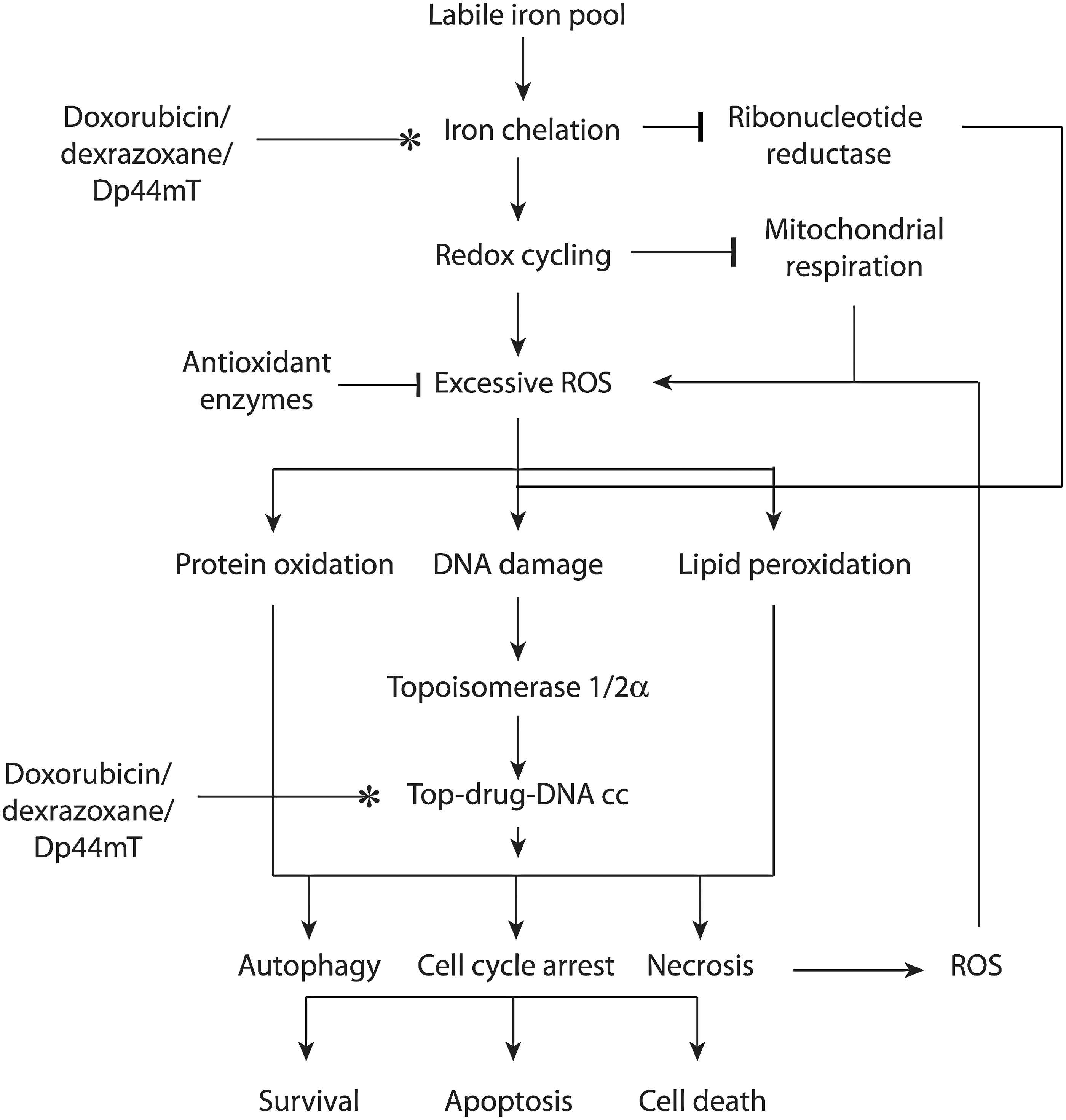
The indirect effects of chelating iron on the activity of ribonucleotide reductase could also contribute toward DNA synthesis inhibition. The indirect effects of leaching iron away from critical DNA repair enzymes also affects other proteins with iron-sulfer clusters such as the helicases XPD and FancJ, excision repair glycosylases, frataxin, and DNA methylation enzyme AlkB, each of which function to repair genotoxic DNA damage (15, 162, 212, 222).
Ironically, two critical antioxidant enzymes also contain iron or other divalent metal ions as cofactor that could, hypothetically, be leached by iron chelators and lead to decreased enzyme activity and excess ROS formation (118, 169). Both superoxide dismutase and catalase contain active site metal ions that are critical for their function (106, 264, 298). In eukaryotes, copper and zinc-dependent superoxide dismutase are most common, whereas iron-dependent superoxide dismutases are mainly found in Escherichia coli, Mycobacterium tuberculosis, and plants (34, 178, 217).
The possible contribution of other factors in drug resistance to the activity of dual inhibitors, such as the expression of multidrug resistance (MDR) genes, is still being investigated. However, it is important to note that we and others have observed sustained anticancer killing by Dp44mT in breast cancer cells that are resistant to etoposide (209, 287). Dp44mT is thought not to act as a substrate of ABC transporters such as P-glycoprotein and MDR-associated protein 1, through which cells sometimes gain resistance to the top2 poison etoposide (287).
Given the cytotoxic topoisomerase-poisoning activity of doxorubicin, the primary mechanism of action for doxorubicin in cancer cells may or may not be dependent on iron chelation. It is noteworthy that, unlike traditional iron chelators such as EDTA, doxorubicin does not mobilize iron but rather induces iron accumulation in ferritin (290). The lack of an effect of pretreatment with iron chelators such as dexrazoxane suggests that iron-mediated oxidative stress may not play a significant role in the cytotoxicity by doxorubicin in cancer cells (163). In our laboratory, the silencing of top2α significantly reduced, but did not abolish, tumor cell growth inhibition by Dp44mT (209). Dissecting the precise role of iron chelation in the cytotoxic effects of these agents needs to be addressed in vivo using side-by-side comparisons with synthetically modified drug analogs that lack the ability to bind to iron.
Another possibility is that dual targeting may accentuate topoisomerase inhibitory action. This may be possible when the iron-chelated drug can (i) act as a stronger interfacial inhibitor of topoisomerase due to conformational changes brought about by the presence of chelated iron, (ii) act as a carrier of Fe2+ and present an alternate divalent cation for mis-incorporation into the metal-binding pocket of topoisomerase, (iii) act as a carrier of Fe2+ and allow a direct interaction of iron with the DNA, which could produce DNA breaks that are sensed by topoisomerases, or (iv) undergo redox cycling and generate excess ROS and 8-oxoguanine lesions on DNA that can increase levels of top cc (see schematic in Fig. 6).
The in vitro study of agents chelated to iron and their effects on topoisomerase and DNA cleavage has been technically challenging because of the rapid degradation of plasmid DNA in the presence of iron; plasmid DNA is often used as the target substrate for these kinds of studies. Molecular docking simulations have shown that dual agents such as TSC-24 and dexrazoxane can bind to the ATP-binding site of top2α (82, 96, 163). These theoretical simulations were confirmed by competitive inhibition assays (96). Dexrazoxane itself can form a tight complex with the ATPase domain of both top2α and top2β. The amino acid side chains involved in the interactions between dexrazoxane and both top2 isozymes are identical (163). In a seminal study, Hasinoff et al. (82) compared the ability of the bisdioxopiperazines (including dexrazoxane) to inhibit topoisomerase catalytic activity, and a two-log range in the inhibitory concentration 50 values for inhibiting topoisomerase was reported. It was suggested that the bisdioxopiperazine binding site on topoisomerase II is large enough or flexible enough to accommodate the methyl group in either enantiomer of racemic ICRF-159. The cytotoxicity of the bisdioxipiperazines toward Chinese hamster ovary cells is highly correlated with their inhibition of the catalytic activity of type II topoisomerase. In our laboratory, in vivo studies with Dp44mT used an assay that extracted topoisomerase-drug-DNA complexes from whole cells, separated the DNA containing fractions, and probed the complexes with an anti-topoisomerase antibody. In this experimental setup, it can be assumed that some fraction of the drug is chelated to iron in the cell, but we could not estimate what fraction of the cellular drug-iron chelates was complexed with topoisomerase on the DNA. Cell-free studies with iron-complexed dexrazoxane, doxorubicin, or Dp44mT are needed to compare their ability to interface with topoisomerase and to iron-free drug.
Both type IA and type II topoisomerases require a divalent metal ion to function (51, 227, 239). For top2α, in vivo evidence has suggested that the DNA cleavage reaction requires Mg2+ as a cofactor for DNA scission (143). The solved crystal structure of top2α also showed a single Mg2+ atom per active site. In a series of in vitro studies, the ability of several divalent ions to support DNA cleavage by topoisomerases was examined in detail (48, 227, 239). The divalent ions Ca2+, Mg2+, Co2+, and Mn2+ were examined for their ability to affect topoisomerase function, and it was reported that top2α requires two divalent metal ions for DNA cleavage (227). The two metal ions are apparently necessary for DNA cleavage/ligation activities of the enzyme (51). One metal ion makes a critical interaction with the bridging atom of the scissile phosphate, which is most likely needed to stabilize the leaving 3′-oxygen and facilitate cleavage. The second metal ion is required for DNA scission; this ion is most likely needed to stabilize the DNA transition state and/or assist with the deprotonation of an active site tyrosine [see Fig. 7, adapted with permission from refs. (51, 227)]. A separate metal ion is required for the interaction with ATP. The exact role played by the metal ions is still being explored and the importance of their interaction with other sites in the primase domain, for instance, remains unclear (143). Divalent ions Mn2+, Ca2+, and Co2+ generated higher levels of DNA cleavage than Mg2+ (49). Fe2+ was not reported in these studies, probably because of technical challenges due to direct DNA damage by iron. However, previous studies suggest that other divalent metal ions can be substituted for Mg2+ in binding to topoisomerases.
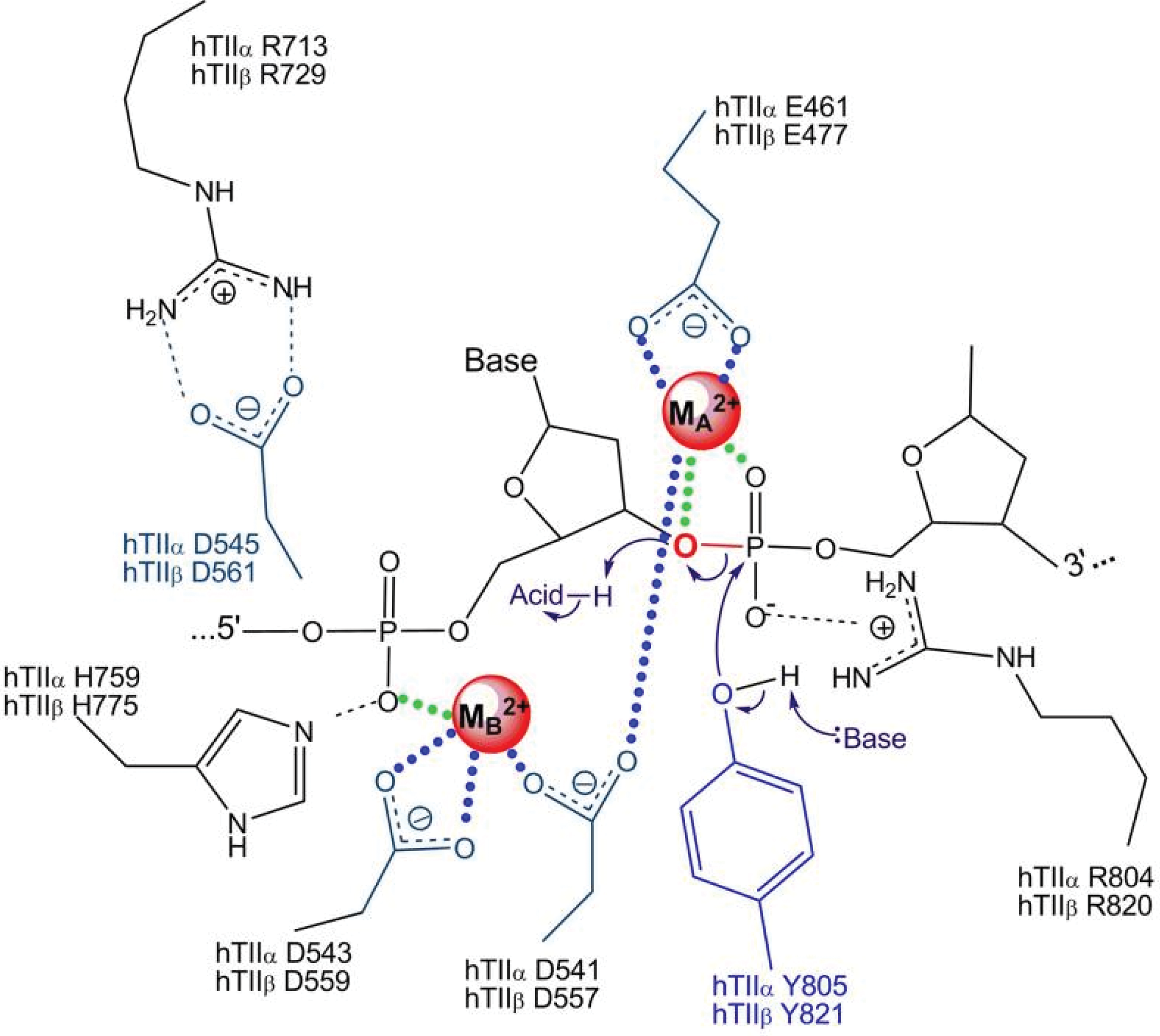
An examination of the amino acids in top2α that are in the active metal binding sites and postulated to interact with Mg2+ (D543, D541, Y805, R804, E461, R713, and H759), and the comparative stability constants of various metal ions for binding to these amino acids (aspartic acid, tyrosine, arginine, glutamic acid, and histidine) (40, 51, 60, 98, 168, 294) suggest that it is theoretically possible for iron to form a stable complex with top2α. In most of the studies cited, stability constants were determined in cell-free systems using free amino acids, under aqueous conditions, and using the formula K=[ML]/[M][L], where K is the stability constant, and M and L are the metal and ligand concentrations. This does not necessarily correlate with how metals behave when interacting with amino acids within native protein conformations, or how they would behave when chelated to other small molecules (including free amino acids). However, it highlights the need for further studies examining how other metal complexes, such as with iron, can affect topoisomerase structure and function. To add to this argument, topoisomerase I in E. coli was recently suggested to be an iron- and zinc-binding protein (161). Using inductively coupled plasma-emission spectroscopy on purified proteins and the iron indicator Ferrozine to determine metal content, the authors found that top1 isolated from E. coli is an iron-binding protein; earlier, it had been suggested to be only a zinc-binding protein. The iron-bound top1 had little or no activity compared with the zinc-bound top1, suggesting that iron could be bound to top1 but not substituted for zinc in supporting its catalytic activity. The physical ability, but catalytic inability, of iron to substitute for zinc is also seen in estrogen receptor zinc finger protein and transcription factor for polymerase III A (32). In contrast, this metal substitution is catalytically functional in the case of neural zinc finger factor 1, tristetraprolin-2D, and GATA-1 zinc finger proteins (14, 54, 191). It may be possible that the mis-incorporated iron in eukaryotic top2α could lead to catalytic inactivity of the enzyme or DNA damage. This incorporation could also trigger metal-catalyzed oxidation of topoisomerase by the formation of hydroxyl radicals. The hydroxyl radicals generated by the redox active topoisomerase could then cause damage to adjacent DNA. The iron metal-catalyzed oxidation could also inactivate the enzymatic activity of topoisomerase and trap the enzyme on the DNA, similar to the iron metal-catalyzed oxidation and inactivation of alkaline phosphatase, glutamine synthase, or coenzyme Q1 (173, 174, 246).
The paradigm of accentuating or diminishing DNA damage is important for iron- and other metal-chelating drugs that could alter the expected responses from concurrently administered topoisomerase poisons. These include certain antibiotics that have antitumor activity and nonferrous metal-binding abilities such as 4′[(9-acridinyl)-amino]methanesulphon-m-anisidide, (mAMSA) netropsin, and tetracylines (87). For instance, mAMSA exerts copper-dependent oxidative and top2-mediated DNA cleavage (87). Hence, the role of chelated iron, its effects on topoisomerases and on the resultant cleavage complex formation, and anticancer outcomes require further investigation.
The final two possibilities for the actions of the dual inhibitors discussed here involve inducing DNA breaks. Doxorubicin is the most intensively studied example, and it is one of the few with direct proof of being an iron chelator with topoisomerase poisoning activity that can directly interact with DNA and chromatin (229, 307). The doxorubicin C-9 side chain plays an important role in determining the strength and specificity of the anthracycline–DNA interaction. The propensities of other chelators to bind directly to DNA have shown varying degrees of ability, all of which are lower than doxorubicin (25). The ability of chelators to interact directly with DNA has typically been determined by the measurement of DNA-mediated hypochromicity and hyperchromicity in the UV-vis spectra of the DNA ligand and iron complex. The decrease in absorbance (hypochromicity) or increase in absorbance (hyperchromicity) on addition of DNA to the compound was considered indicative of an interaction between the two (25, 38). Whether Dp44mT, dexrazoxane, and the newer dual inhibitors directly interact with DNA remains to be investigated.
Acting as a carrier of the metal ion to DNA, iron chelators could also damage DNA through direct hydrolysis of the phosphodiester backbone catalyzed by the metal ion (192, 232, 240, 259). Iron in its ferrous form and in an aqueous environment, can induce nonenzymatic strand cleavage and alkali-labile lesions (107, 240). Iron can incorporate into DNA and bind to it, possibly more so in the ferrous than ferric form (232, 263), although other studies suggest that the ferric form exerts the major stereochemical effect (6). The direct role of iron in mediating such DNA breaks was also documented as a part of the mechanism for the anticancer drug bleomycin, which complexes with iron (126, 194, 195, 240). It has also been proposed that superoxide promotes hydroxyl radical formation and oxidative DNA damage by releasing iron from storage proteins with enzymatic iron-sulfer clusters (118, 146). Such leaching of iron, or other mechanisms, may provide a pathway for DNA damage.
Finally, iron chelated drugs can undergo redox cycling and generate excess ROS (hydroxyl radicals) and 7′,8′-dihydro-8-oxoguanine (8-oxoG) lesions on DNA that can increase the formation of top cc (154). The mechanism for generation of excess hydroxyl radicals by doxorubicin has been previously described in this review. The iron chelator Dp44mT has been shown to induce excess ROS using 2′,7′-dichlorodihydrofluorescein dye, in one study using M109 cells and in our laboratory using MDA-MB-231 cells (unpublished results) (297). While ADR-925 has been shown to mediate hydroxyl radical formation, dexrazoxane has been primarily shown to reduce oxygen radical formation (78). Within cells, if the net result of the redox cycling of metal-bound chelators was to generate excess hydroxyl radicals, they could target DNA to produce 8-oxoG, the most abundant oxidative modifications in DNA. Using oligonucleotides containing 8-oxoG modifications, a significant increase in top1-mediate cleavage was found when the 8-oxoG was at the +1 or +2 positions relative to the cleavage site (204). The oxidative lesion 5′-hydroxycytosine also enhanced top1 cleavage by two-fold. The trapping of top1 adjacent to an 8-oxoG was enhanced when asparagine adjacent to the catalytic tyrosine was mutated to histidine, suggesting a direct interaction between the asparagine and the DNA major groove. Quenching of excess ROS by N-acetyl cysteine abrogates the formation of top1 cleavage complexes by the top1 poison staurosporine (243). Interestingly, the exposure of leukemic cells to H2O2 can also induce top1 cleavage complexes in DNA (39). In other cancer cell lines, H2O2 or 8-oxoG lesions induced top2 cleavage complexes in DNA (148, 223). ROS-induced formation of top cc is likely a secondary effect of dual inhibitors but one that would have cytotoxic consequences in the presence of sustained excess ROS. A recent report suggests a lack of cell cycle arrest, apoptosis, DNA damage, or top2α inhibition by Dp44mT at early time points and proposes that the top2α inhibition observed by 24 h could be due to iron-Dp44mT complex-induced oxidative DNA damage which can stimulate top2α-DNA complexes (292). Although cell line, technical, or reagent differences could also be probable causes for the observed lack of top2 inhibition, cell cycle arrest, or apoptosis, metal-complexed Dp44mT has been shown to have redox activity that generates hydroxyl radicals (105, 156). While we cannot presently rule out that top2α-directed activity of Dp44mT might be due to its redox-active and metal- complexed form, it nevertheless contributes to the irreversible cell killing consistently demonstrated in different cancer cells by several other groups (188, 209, 258, 286, 297).
Cellular responses to dual inhibition
Dual inhibitors may cause cell cycle arrest, apoptosis, necrosis, and/or autophagy in cancer cells. The ultimate cellular outcome most likely depends on the concentration and duration of drug exposure. Transient cell cycle arrest and autophagy could aid the cell in survival, while permanent cell cycle arrest could lead to apoptosis and/or necrosis. The targets of dual inhibitors discussed in this review trigger at least two overlapping downstream pathways that initiate a cytotoxic cellular response, namely excess ROS generation and DNA damage. Excess ROS could originate from redox cycling of iron-chelated drugs, metal-catalyzed oxidation of proteins, or from the activation of immune cells as an inflammatory response to drug treatment [e.g., formation of the oxidant taurine chloramine by from activated neutophils (121)]. Generation of excess ROS has been explored as a strategy for selective anticancer activity (121, 302). The deleterious effects of excess ROS would be via the oxidative modification of essential proteins, lipids, and DNA. DNA damage could also ensue from the unrepaired topoisomerase-DNA cleavage complex formation, or from direct DNA binding. Apoptosis induction by iron chelators or by anthracylines is covered elsewhere in this issue of the journal. Necrosis induction by iron dual inhibitors has been poorly defined, although it has been documented as one of the cellular responses to iron overloading, iron chelation, and in response to doxorubicin (260, 268). The remaining part of this section will focus on cell cycle arrest and autophagy.
Dual inhibitors have shown some variability in the phase of cell cycle arrest they elicit in cancer cells. Dp44mT and TSC-24 arrest cells in G1-S phase, which is also a feature of iron chelators with no topoisomerase-inhibitory activity such as desferal (95, 96, 188, 209). On the other hand, doxorubicin and dexrazoxane arrest cancer cells in the G2-M phase of the cell cycle, which is a feature of classical topoisomerase poisons such as etoposide (175, 275). The reason for this is not clear, although it is thought that chelation of iron inhibits DNA synthesis so that iron chelators induce an S-phase arrest. Inhibition of ribonucleotide reductase, up-regulation of the GADD family members, and increases in p53 levels are, at least in part, responsible for the S-phase arrest (224). top2 plays an essential role in sister chromatid separation at anaphase of mitosis, which could explain why top2 poisons cause an M-phase arrest. Both protein levels and catalytic activity of top2 peak during the G2-M phase of the cell cycle (99, 140). The top2 checkpoint monitors the catenation status of sister chromatids after the S-phase and prevents exit from G2 until the sister chromatids are sufficiently decatenated (59). Cell cycle arrest by dual inhibitors is likely due to the primary mechanism of action of the agent or the stage of the cell cycle in which the cells encountered the dual inhibitors. An analysis of synchronized populations of cells (for instance, in G1-S using aphidicolin or M phase using nocadazole) would be beneficial to identify the most sensitive cell cycle state for cancer cells exposed to dual inhibitors. Such studies could also help deliniate the primary target of the dual inhibitors.
The induction of autophagy by dual inhibitors can occur as a survival mechanism to overcome oxidative or chromatin damage. The exact role of iron in the formation of autophagosomes is unclear; however, iron chelators do induce the formation of catabolic autophagosomes (45). Interestingly, since lysosomes contain most of the cellular supply of labile iron, iron binding proteins can be sequestered in autophagosomes as a strategy to minimize the damage from free iron (134). In some circumstances, iron chelators can prevent autophagy by sequestering the iron used in the formation of autophagosomes in response to H2O2 (22, 134). Dp44mT can also cause autophagy, which is one pathway proposed to explain selective toxicity to cancer cells versus healthy cells (156, 209). Lovejoy et al. have proposed that since autophagic pathways are abnormal in cancer cells by virtue of monoallelic deletion of the autophagic regulator, beclin1, apoptosis, and cell death would be the favored pathway in cancer cells (156). Doxorubicin also induces autophagy as one of its cellular responses (205). This has been exploited with the autophagy inhibitor, resveratol, to attenuate the cardiotoxic effects of doxorubicin (289). We have observed increased autophagy in response to both doxorubicin and dexrazoxane without an appreciable decrease in the levels of the autophagy-associated protein myosin-binding light chain protein (LC3)-II in cancer cells (unpublished observations).
Future Directions
Potential biomarkers for iron chelators and topoisomerase inhibitors
Future studies that utilize dual inhibitors as tools for research or for development of therapeutic interventions in cancer could benefit from selecting a set of biomarkers which reflect the known mechanisms of such dual targeting agents. This section summarizes characteristics that can be objectively measured and evaluated as indicators of biological response to dual inhibitors. The list of biomarkers discussed here is not suggested to be absolute, but rather a starting point for exploratory studies. The assays listed were considered physiologically relevant, technically feasible, and translatable across multiple laboratories. The endpoints are also illustrated in Figure 8 alongside the relevant mechanism.

The unknown iron chelating capacity of known topoisomerase inhibitors could be probed using the fluorescent dye calcein-acetomethoxy derivative (10). The displacement of preloaded iron from calcein, as indicated by a change in the floruorescence of trapped intracellular calcein-iron complex over time, can be used as a marker of iron chelation. The iron chelation abilities of Dp44mT and other molecules have been shown using this method and appear to correlate with electron paramagnetic resonance spin trapping experiments with ferric and ferrous ion complexed with a chelator (83).
Endpoints of excess free radical formation are most frequently studied using redox-sensitive probes. The high performance liquid chromatography (HPLC)-based detection of superoxide-generated products of hydroethidine (HE) can be used to measure excess ROS (306). The quantification of 2-OH-E(+), the product of the reaction between HE and superoxide, can be measured in cell lysates as well as cell-free systems (210, 303). Mitochondrially targeted Mito-HE is capable of undergoing similar redox chemistry and may be useful in identifying mitochondrial ROS (109, 219). 2′,7′-Dichlorfluorescein-diacetate (DCFH-DA) is another fluorescent probe that is used for measuring intracellular H2O2 and oxidative stress (109). However, this probe may not be specific for H2O2 and could give misleading results due to oxidation by lipid hydroperoxides and H2O2, as mediated by cellular iron-sensing molecules (304, 305). Oxidant signaling by redox-active iron, heme proteins, or cytosolic, enzymatically active cytochrome C had been implicated in the changes observed in DCFH-DA (305). Karlsson et al. reported that the oxidation of this dye was neither dependent on Fenton-type reactions nor on unspecific enzymatic oxidation by cytochrome c, because neither superoxide nor H2O2 directly oxidizes the dye (112). Hence, the conversion of this dye might be indicative of lysosomal membrane permeabilization and the relocation of intracellular, lysosomal iron, and of mitochondrial cytochrome c rather than H2O2/ROS levels or general ‘oxidative stress’. Since doxorubicin itself is fluorescent, the analysis of any fluorescent probe should account for the background and specific wavelength of fluorescence emission of the redox dye as well as doxorubicin itself. Hence, results with DCFH-DA, especially those using fluorescent iron chelators, should be interpreted with caution. For comprehensive reviews of other redox dyes and their chemistry, the reader is referred to other publications (110, 308).
Robust quantitative measurements of protein oxidation and lipid peroxidation are still being optimized, though several assay platforms provide a good starting point (53, 147, 231). Protein carbonylation and disulfide-bridged dimer formation are two of the major types of protein oxidation that can be initiated by ROS. Protein carbonylation refers to the formation of reactive ketones or aldehydes on side chains of lysine, arginine, proline, and threonine residues; these react with 2,3-dinitrophenylhydrazine (DNPH) to generate hydrazones (DNP) (147, 231). Antibodies against DNP allow the detection of oxidized proteins via immunological methods (53, 231). Disufilde-bridge linked dimers can be formed between highly reactive cysteine residues. The oxidized protein dimers can be detected using a gel-shift assay under reducing and nonreducing conditions (210). Lipid peroxidation can be detected via the formation of lipid adducts such as malondialdehyde and 4-hydroxynonenal using mass spectrometry or antibody-based detection (213, 245).
The appearance of top cc using the immune complex with enzyme bioassay method indicates in vivo topoisomerase-directed poisoning activity (247). This method utilizes the collection of DNA-containing fractions from sarkosyl-lysed cells separated by a cesium chloride gradient. The fractions are loaded on a slot blot and probed with top1, top2α, or top2β antibody to indicate complex formation between the DNA, drug, and topoisomerase.
Two widely used assays for measuring single- or double-strand DNA breaks are the single-cell gel electrophoresis (Comet) assay and the phosphorylated histone H2AX (γ-H2AX) nuclear foci assay. The comet assay measures DNA damage as a “tail moment” or “comet” formed by the broken DNA of cells that are electrophoresed under alkaline or neutral conditions (189). Under alkaline conditions, structures in addition to single-stranded breaks such as double-strand breaks and abasic sites are also detected. The induction of γ-H2AX can be measured using antibodies which are specifically raised against the phosphorylated form of histone H2AX that are formed after DNA double-strand breaks (197). Microscopic and high-throughput formats of the assay have been developed and used for testing the DNA damaging actions of topoisomerase poisons (9, 120, 207). To measure oxidative modifications associated with DNA breaks, the comet assay has been modified to detect oxidatively clustered DNA lesions that might be informative for dual inhibitors which cause excess ROS (69). 8-oxoG oxidative DNA lesions have also been measured with antibodies or sensitive HPLC coupled with electrochemical detectors (115).
The cellular outcomes of cell cycle arrest and apoptosis have been measured as populations of cells in specific cell cycle phases or by annexin-V-positivity (124). While the activation of a noncaspase-mediated cell killing is probably suggestive of necrosis, a definitive biochemical marker of necrosis has not emerged. Studies are ongoing to define specific markers of necrosis, such as cyclophilin A, that could be utilized in drug discovery studies (29, 116). Future studies could benefit from identifying the contribution of autophagy toward resistance or sensitivity to dual inhibitors. Autophagy is a catabolic process that utilizes members of the Atg family and other proteins to engulf cellular contents for recycling. During autophagic flux, the cyctoplasmic form of LC3-I (Atg8) is lipidated and proteolysed to LC3-II during autophagosome formation. An increase in LC3-II can be measured by Western blotting and is indicative of the number of autophagosomes. When measuring autophagic flux by LC3-II, it is important to interpret the levels of LC3-II with caution because LC3-II itself is degraded during autophagy (124, 172). Hence, the levels of LC3-II should be measured in the absence and presence of lysosomal protease inhibitors such as E64d and pepstatin A (124, 210). The LC3-II, thus, measured would be indicative of autophagic flux and lysosomal turnover of LC3-II, rather than a transient increase in LC3-II (252).
Conclusions
The rapid evolution of stronger iron chelators, topoisomerase inhibitors, and a growing list of agents that target both has prompted an examination of the commonalities, opportunities, and caveats of developing dual inhibitors as anticancer drugs. Doxorubicin is a front-line drug against breast cancer, while dexrazoxane serves as a cardioprotectant that can be used with doxorubicin. Newer agents such as TSC-24 and Dp44mT and their analogs are still being explored but appear promising in preclinical models. This review presents arguments addressing the possibility for enhanced therapeutic activity through the use of agents that target both labile iron pools and elevated topoisomerase levels in cancer cells. Whether the theoretical rationale for greater cytotoxicity by dual-targeting agents translates into the clinic will have to be tested. The known mechanisms of excess ROS generation and DNA damage by these agents are highlighted as opportunities for a cytotoxic therapeutic outcome. The arguments against their use as anticancer agents include the high concentrations required for cytotoxicity, no appreciable anticancer cytotoxicity by some cardioprotective iron chelators, secondary malignancies associated with top2β activity, and decreased cytotoxicity in combination with other topoisomerase poisons. In vitro biochemical studies are needed to dissect exactly how iron binding and topoisomerase cleavage-complex formation are related in rapidly dividing cells. Gaps in knowledge are identified in the mechanisms of these agents, and where possible, avenues for pragmatic drug development studies are presented for future studies.
Footnotes
Acknowledgments
V.A.R. is thankful to the Food and Drug Administration Critical Path Initiative and the National Cancer Institute for research support. The author wishes to thank Drs. Emily Shacter (FDA), Eugene Herman (FDA), and Yves Pommier (NCI) for insightful discussions and encouragement. Drs. John Nittis (University of Illinois), Neil Osheroff (Vanderbilt University), and Tomas Simunek (Charles University in Prague) are acknowledged for graciously agreeing to allow an adaptation of their published figures. Drs. Jennifer Dickey (FDA) and Melanie Simpson (NCI) are thanked for their critical reading of the article.
Author Disclosure Statement
No competing financial interests exist. The opinions expressed in this article are those of the author alone and do not necessarily reflect the official position or policy of the U.S. Food and Drug Administration.