
Abstract
Introduction
Although hemoglobin (Hb) is vital for delivery of oxygen, it needs to be strictly compartmentalized within the red blood cells (RBCs) to keep it in a reduced and nontoxic state (75). Inside the RBC, a plethora of protective mechanisms inhibit potential side reactions and further eliminate their possible products (22, 79). RBC thus contains a unique collection of enzymes and proteins that preserve Hb in a functional and safe state (66, 75, 79). However, when released from RBC, an alternative set of defense mechanisms has evolved to counteract the chemical threat of extracellular Hb to exposed cells and tissues. The balance between normal physiology and toxicity is therefore largely dependent on the physical and biochemical barriers presented by RBCs, which separate Hb from the surrounding environment. If these systems are challenged, for instance, as a consequence of intravascular hemolysis, a number of potentially toxic and dangerous side effects originating from the rich chemistry associated with the free radical reactivity of Hb, can occur.
If released from RBC, the primary Hb scavenger in plasma is haptoglobin (Hp) (46, 96). Hp, first identified and described by Polonovski and Jayle in 1938 (72), is a plasma protein that binds to and scavenges Hb. The naming of the protein is indeed appropriate and it originates from the Greek word haptein meaning to fasten, or bind. Hp is an abundant acute-phase plasma glycoprotein of the α2-globulin fraction present in the plasma in humans and other mammals, as well as in Ostrich and bony fish (94). Hp is mainly expressed in liver tissues (both mRNA and protein have been detected), but its expression has also been observed in a few other organs of the human body, including lungs (mRNA and protein detected), spleen (mRNA detected), thymus (mRNA detected), kidney (mRNA detected), and heart (mRNA detected) (60). Hp concentration in plasma varies on an individual basis, but it is also influenced by age, with a gradual increase to the age of 20 when the serum concentrations are ranging between 30–200 mg·dL−1 under normal physiological conditions in a healthy adult (97). Hp is cleared from plasma in ∼3.5–5 days, but when it forms complexes with Hb, it is rapidly removed from plasma in ∼20 min. The Hp concentration is hence often used to determine recent episodes of hemolysis, since the increased clearance of the Hp protein from plasma results in a reduced level of the protein in plasma (97). This clearance rate of the Hp-Hb complex has become controversial and large differences in the half-life have been reported (15). However, the polymorphic nature of Hp complicates the analysis, and the variations reported may be linked to differences in molecular compositions and glycosylation patterns.
Protective Mechanisms of Hp
Hp binding does not perturb heme reactivity with oxidants or gaseous ligands (18). Rates of auto-oxidation, H2O2 reactivity and NO binding and conversion all remain largely unchanged (15). However, the physiological properties of the Hb-Hp complex are drastically modified. As opposed to free Hb, the complex does not extravasate and Hp can therefore protect susceptible organs, such as the kidney, from Hb and heme exposure. In oxidatively stressed environments, the complex also protects the integrity of the Hb molecule itself by reducing globin crosslinking, heme release, and amino acid oxidation (18, 71). Lipid peroxidation by Hb–Hp is also largely reduced as compared to free Hb (47, 54, 58). Surrounding tissues and cells are thereby also protected from the native peroxidative activity of Hb when the protein is trapped within the Hp complex (37).
Several efforts have been undertaken to attempt to explain the underlying molecular mechanisms associated with the Hp shielding abilities. The obtained results give a complex picture involving protection against several naturally occurring Hb radicals. This implies that it is particularly important to examine possible electron transfer reactions in the Hb molecule. One of the key residues is αTyr42, which can both receive and donate electrons, for example, by reducing or oxidizing Met/ferryl Hb, via a radical intermediate. This appears to facilitate the detoxification of ferryl by endogenous antioxidants, such as ascorbate (77). However, this residue has also been shown to be involved in an autoreduction without the need for an external reductant (45). Deterding et al. have (25) also shown that it is possible to spin trap a radical at αTyr42 and that this site can be involved in further electron transfer pathways. The electrons may hence, subsequently be transferred to other residues. For instance, βTyr145 has been described as one of the final destinations (4), however, other Tyr residues are clearly also involved (Chakane et al., unpublished data). The mechanism behind Hp protection has therefore not been completely resolved, but it partly involves stabilization of the ferryl heme state and control of a few specific electron pathways in the Hb molecule by stabilizing the generated protein radicals (36, 54). In this context, it is also important to realize the large surface area covered in the complex between Hp and Hb, which allows shielding of several potential Hb radicals.
The Hb Scavenger Receptor CD163
The presence of free plasma Hb in the general circulation triggers the inflammatory cytokines, interleukin-1 (IL-1), interleukin-6 (IL-6), glucocorticoids, and tumor necrosis factors that enforce and induce the expression of Hp (31, 36). The Hp-Hb complex is cleared from the body with the aid of macrophages/monocytes and the iron is recycled. This complex is recognized by the macrophages/monocytes expressing the CD163 glycoprotein (or the Hp scavenger receptor), which shows high affinity to the Hb-Hp complex. After being recognized by the macrophages, the complex is engulfed by receptor-mediated endocytosis (63). The globin fraction is degraded by the lysosomes and the heme fraction is converted into less toxic compounds, which are recycled to the body for RBC production. The heme conversion is performed by heme oxygenase-1 (HO-1) inside the cytosol. Just like the Hp regulation, the expression of CD163 and HO-1 is upregulated by the action of inflammatory cytokines, notably IL-6 (62). Thus, the involvement of the triplet proteins Hb:CD163:HO-1 could control the systemic or local tissue responses to exposure of free intravascular Hb (13, 78). In contrast to the high affinity of the Hp-Hb complex to the CD163 receptor, Hp alone has no binding to the scavenger receptor, while free Hb shows a weak affinity to CD163 and it can be engulfed in the absence of Hp by the CD163-expressing macrophages (41, 62, 84).
Other Hb and Heme-Binding Proteins
In humans, there are several other Hb- and heme-detoxification systems described. For instance, a so-called Hp-related protein (Hpr) can be produced, which shows very strong homology to Hp1 (62). The HPR gene differs from the HP gene mainly in that it has a retrovirus-type sequence inserted into the first intron. The encoded protein has a serum concentration, which is ∼5%–10% of that of Hp in healthy individuals. Hpr is believed to be part of the trypanosome lytic factor (TLF), a toxic subtype of high-density lipoproteins that provides humans with an innate defense against infections by Trypanosoma brucei brucei, the parasite responsible for Nagana disease in cattle. When Hpr binds to free Hb, it kills the trypanosome via oxidative damage initiated by its peroxidase activity. Since Hp is the major serum inhibitor of the TLF, the balance between the serum concentrations of Hp and Hpr determines the degree of protection against trypanocidal infection. Hpr binds Hb with high affinity, but the Hpr-Hb complex does not bind to CD163 (61, 62, 89). In addition to Hpr, apolipoprotein L1 is bound to TLF resulting in a trypanolytic activity (61, 90).
Free heme in blood is sequestered by albumin and hemopexin (Hpx) (8, 12, 24) and the Hpx–heme complex is cleared from the circulation by CD91 (33). As indicated earlier, HO is the most essential intracellular heme catabolic protein converting heme to free iron, biliverdin, and CO. Biliverdin is then further converted to bilirubin, which is released and transported to the liver with the aid of albumin. However, other proteins are available, which normally not are discussed in this context. Particularly important is the plasma and tissue protein α1-microglobulin (A1M) (3, 66) that can also bind and degrade heme and additionally has the ability to reduce MetHb (6). Recent reports suggest that a truncated form of A1M is involved in extracellular heme catabolism (5). The heme-binding and putative heme cleavage properties of A1M, hence, provide an additional physiologic protection mechanism against extracellular heme.
Genetic Setup
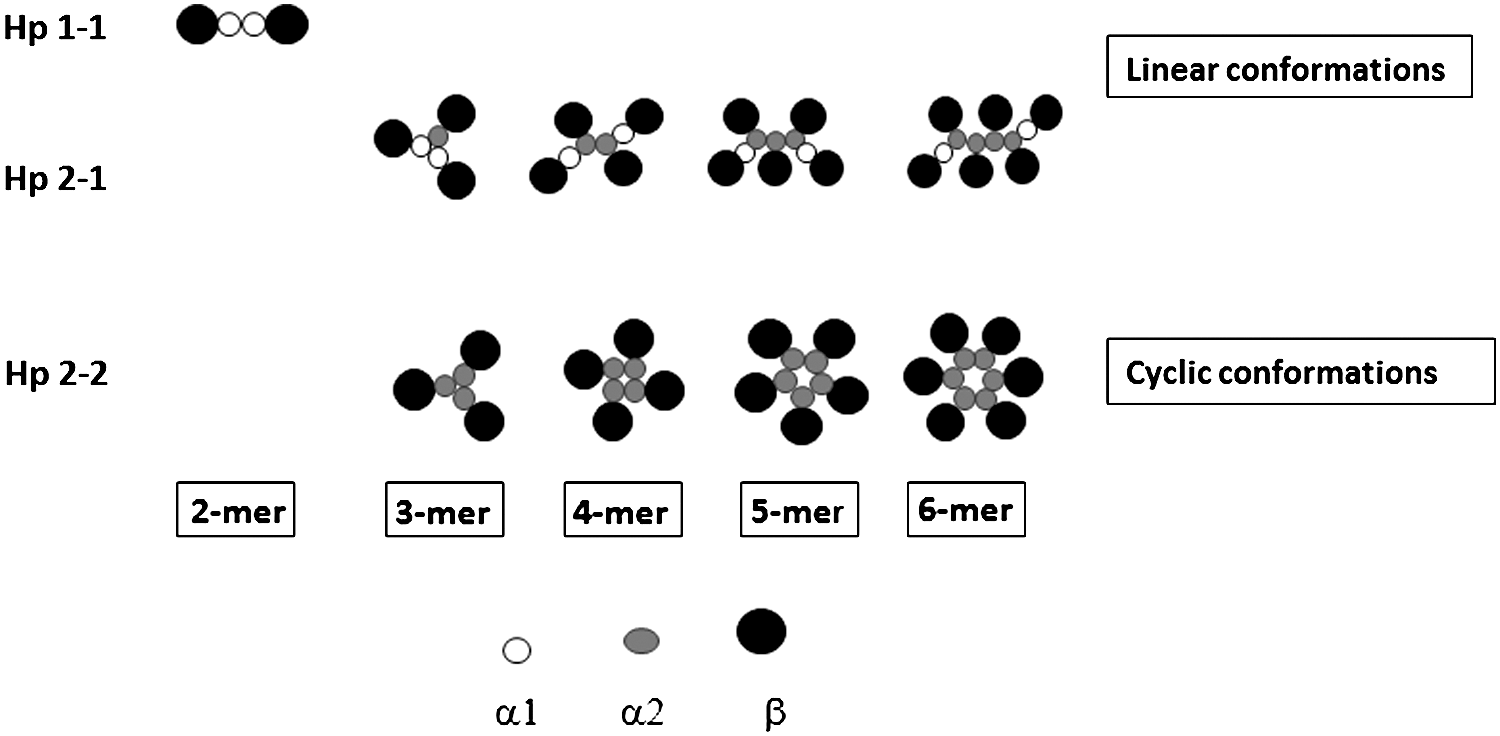
The human Hp gene locus has been mapped to the long arm of the 16q22 chromosome by in-situ hybridization analysis (96, 99). In humans, Hp occurs in three different isoforms, that is, Hp 1-1, Hp 2-1, and Hp 2-2. These polymorphic variations were first described by Jayle and Judas in 1946 and were later confirmed by Smithies in 1955 using starch gel electrophoresis (44). This polymorphism and partial gene duplication of the gene was counted as remarkable when being identified as the first example of such events among plasma proteins (39). These three Hp protein phenotypes are encoded by two major alleles, Hp1 and Hp2, where the Hp1 allele is further subdivided into Hp1F and Hp1S based on the speed of the proteins' migration on a gel; Hp1F migrates faster than the Hp1S peptide chain. There is only a single amino acid difference between Hp1S and Hp1F, the Lys54 residue in the Hp1S chain has been replaced by Glu54 in the Hp1F chain (73). The Hp2 allele has arisen from an apparent intragenic duplication of a 1.7-kb DNA fragment of the Hp1 allele (44, 53). Maeda et al. have suggested that this event occurred as a result of nonhomologous unequal crossing over of the Hp1S and Hp1F alleles resulting in an Hp2 allele encoding an extended α-chain of Hp (50, 73). It has generally been assumed that these polymorphic events only occurred in the human genome, but recently a few studies have presented evidence of similar independent events also in cow and deer (43, 52, 95). These genetic modifications have affected only the α-chain of Hp, while the β-chain remains identical in all three Hp phenotypes, that is, the α1-chain expressed by the Hp1 allele is 81 amino acids long, whereas the α2-chain expressed by the Hp2 allele is 142 amino acids. The β-chain is thus composed of 243 residues, irrespective if it is expressed from the Hp1 or Hp2 allele (44). The α1-chain is monovalent having a single SH-group, capable of forming one disulfide bond between the α and β subunit of the Hp protein resulting in a dimeric (α1β)2 Hp 1-1 protein, whereas the α2-chain (142 amino acids) having a partially duplicated amino acid sequence is divalent, thereby harboring an additional SH-group. It assembles into complexes, where the extended α2-chain either is linked to an α1-chain or another α2-chain producing a polymeric Hp 2-1 and Hp 2-2 phenotype, respectively (44), cf Fig. 1.

Hp is initially produced as a preproprotein, a single polypeptide chain containing a signal peptide, the α-chain and β-chain. The signal peptide, the first 18 amino acid residues of the protein, enables the transportation of the protein into the endoplasmic reticulum (ER), where it is proteolytically processed into its mature form. The α-chain contains the compliment control protein (CCP) domain, also denoted as the sushi domain, whereas the β-chain has a different structure resembling a chymotrypsin-like serine protease domain (94, 96). The preproprotein is further cleaved in ER into the separate α- and β-chain chains, which in the mature state of Hp are held together by disulfide bridges. The Clr-like protease complement protein is responsible for the Hp cleavage in ER (17). The S–S bond is formed between the Cys149 of α-chain and Cys266 residue of the β-chain (28, 44). There are four more disulfide bonds between the cysteine residues at 52–86, 111–145 in the α-chain and cysteine residues 309–340, 351–381 of the β-chain of Hp (42, 51, 73) (Fig. 2).
Humans possess Hp in three different isoforms, whereas most mammals carry a single Hp1-like protein, while the ruminants have a Hp2-like protein (43). However, the Hp1 and Hp2 alleles result in gene products having different physiological consequences. Hp 1-1 is a more potent antioxidant (81). The Hp 1-1/Hb complex is cleared two to three times faster than the Hp 2-1/Hb or the Hp 2-2/Hb complexes (46). On the other hand, Hp 2-2 has a higher capacity to bind to free Hb in plasma (11, 41). Several screening studies have been performed worldwide on the potential influence of the two different Hp alleles in various disease developments. There are two different properties of Hp that needs to be balanced against each other, differences in the size of the complex, which may result in a limited spatial access, and differences in the intrinsic ability of Hp to trap available free Hb. The Hp phenotypes may, therefore, be linked to different sensitivities to infectious and pathological disorders. For instance, Borsody et al. found that individuals carrying the Hp2 allele are more likely to develop cerebral artery vasospasm following hemorrhage than the patients with the Hp1 allele (16). Carriers of Hp 2-2 have also been linked to diabetic nephropathy because of larger polymeric Hp structures, which have difficulties to penetrate into the extracellular compartments (9). The Hp phenotype also appears to matter for women with preeclampsia. The prevalence of Hp1 is, hence, higher in healthy compared to preeclamptic subjects (81). Preeclampsia is a pregnancy disease shown to be associated with elevated levels of fetal hemoglobin (HbF), leaking from fetus to mother (66), and we have recently verified that Hp has a strong affinity also to HbF (Ratanasopa et al., unpublished data). Despite these apparently weaker abilities of the Hp 2-2 or Hp 2-1 proteins over Hp 1-1, it is interesting to observe the higher prevalence of the Hp2 allele in many geographical areas (Table 1). To explain this phenomenon, it has been observed that Hp 2-2 and Hp 2-1, but not Hp 1-1, are able to inhibit the growth of certain bacteria. Individuals carrying the Hp2 allele are less affected by such infections as well as show reduced incidence of malaria (10, 91). However, no doubt, more epidemiological studies are needed to make definite conclusions about the physiological consequences linked to the Hp allele.
Hp1 Allele Frequencies in Different Populations from Five Continents
The results above were calculated and further processed to create an overview of Hp1 allele occurrence across the globe from different individual studies undertaken by several independent research groups and originally compiled by Wobeto et al. (97).
Hp, haptoglobin.
Glycosylation
Hp is a secreted glycoprotein modified by post-translational events (30). The asparagine (N) residues, which are modified by N-linked glycosylation are N-184, N-207, N-211, and N-241, where complex carbohydrate residues called glycans, are incorporated into the protein enzymatically (19, 30, 48) (Fig. 2). In addition, a neutrophile-derived Hp with a high molecular weight caused by extensive glycosylation has been identified (86). Interestingly, different patterns and degrees of Hp glycosylation have been linked to several pathological conditions. Recently, the Hp glycans have been extensively characterized from several cancerous and infectious patients' sera (80). The techniques used to achieve this often represent a complex mixture of lectin arrays and mass spectrometric-based technologies. Such efforts have been undertaken to identify potentially unique glycan structures of Hp, which may be explored as biomarkers with the purpose of detecting the onset and progression of a specific disease. The Hp glycans identified so far revealed both simple and complex structures in which frequencies of the bi-antennary and tri-antennary glycans have been observed to be ∼70% and 30%, respectively, while the tetra-antennary glycans are found in less than 1%–2% of the Hp studied (14). The Hp N-linked glycans are terminally sialylated in the form of N-acetylneuraminic acid, where the glycans are either monosialylated or disialylated. Fucose residues are also detected in several instances. These residues are linked to N-acetylglucosamine (GlcNAc) moieties either in the core and/or to the antennae of the glycan (Fig. 3). However, the occurrence of the terminal sialic acid and fucose reduces is decreased in the larger multimers of particularly the Hp 2-2 isoforms; most likely this is caused by steric hindrance of the neighboring monomers in the complex preventing or reducing normal glycan incorporation (Fig. 4) (14).

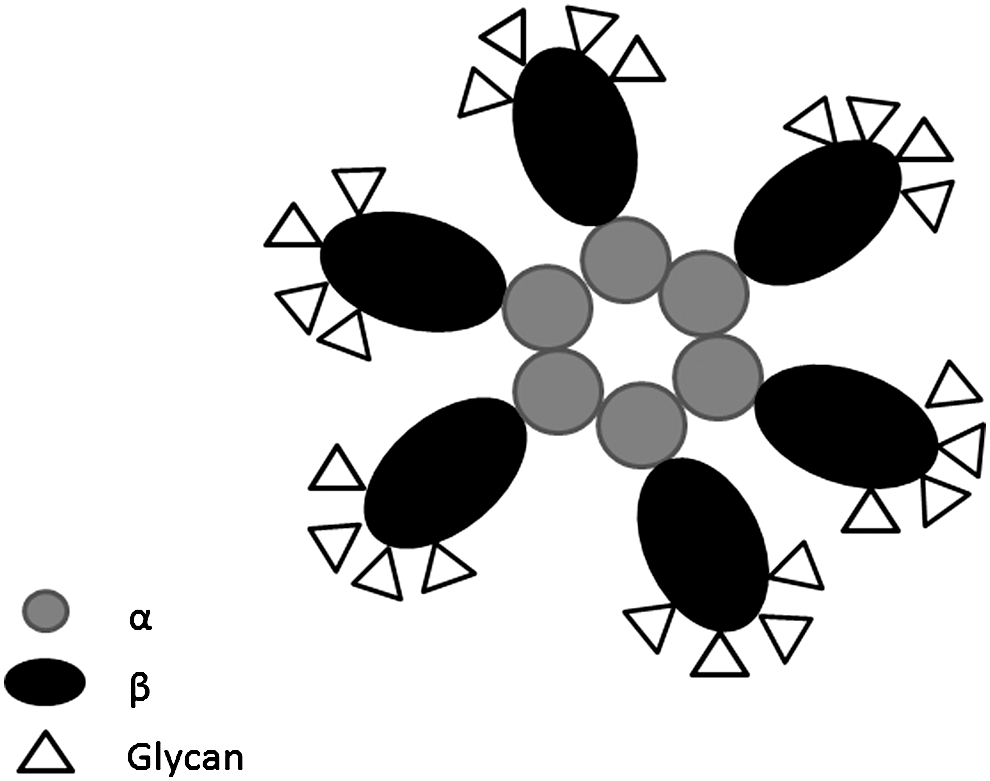
Multiple and independent studies of the glycan structures have recently been conducted on patients with different diseases (Table 2). Interesting differences have been observed, but technological difficulties in characterizing such complex carbohydrate structures still preclude extensive interpretations. Based on the observed aberrant glycan patterns, Hp will most likely become an alternative marker protein for screening of several pathological conditions. It remains to be determined if the observed variations in the glycan composition of Hp also influence the actual development of the disease by perturbing the native protective role of Hp, or it is merely a reflection of a variation of the glycosyl transferease expression systems. However, the glycans modify the intrinsic physical and redox properties of Hp. The affinities between Hp and Hb are affected as well as the stability of the complex. For instance, the glycans render the Hp-Hb complex exceptionally thermostable and it can actually be boiled without losing its activity as opposed to a deglycosylated complex or free Hp and Hb molecules. When analyzing the glycan structure of the complex, some of these chains also appear to swing out from the complex as evidenced by enzymatic deglycosylation (Ratanasopa et al., unpublished data).
List of Aberrant Haptoglobin Glycans in Relation to Different Diseases and Cancers
Elucidating the Hb-Hp Complex via Molecular Modeling
The complex glycan patterns of Hp have largely precluded the crystallization of the protein. A detailed structure of Hp, and more interestingly of the Hp-Hb complex, is therefore still lacking. Electron microscopy studies have suggested that within the complex, the Hp β-chain substitutes an αβ Hb dimer such that the complex gives the impression of being a “pseudotetramer” (92, 93). Several alternative approaches based on peptide fragmentation and protein sequencing have shed light on the amino acid residues responsible for the binding of Hp to Hb and its scavenger receptor CD163 (21). It is generally believed that the β-chain of Hp is responsible for complex binding, both with the free circulating Hb as well as with the receptor CD163 expressed on macrophages; however, some experiments suggest that the α-chain can also be involved in binding with free Hb (54). The Hp-binding site encompasses residues 121–135 (38) and 139–141 (34) of the Hb α-chain and residues 11–25, 37 and 131–146 (101) of the Hb β-chain. Likewise, the Hb-binding site comprises of 9–10, 128–131, 136–137 (49), 136, 218, 137 (22), and 234–264 (54) of the Hp β chain. Residues 1–70 on the Hp α-chain (54) have also been suggested. Despite these reports on the Hp-Hb complex, the structural details of the complex are not fully understood. To address the challenge of further uncovering the binding modalities of the interaction between Hb and Hp, we have employed various computational methodologies based on molecular dynamics (MD) to generate conformationally stable structures followed by their docking to generate Hp-Hb complexes (60).
The X-ray crystallographic structure of human dimeric Hb A (containing a single αβ dimer) obtained from the Protein Data Bank (PDB id 2DN1) and the homology model of Hp 1-1 obtained from the work of Polticelli et al. (73) and deposited in the Protein Model DataBase (accession number PM0075389) have been used as starting points in constructing Hp-Hb complex models. Appropriate protonation state of histidine and other ionisable residues have been assigned to the Hb structure neutron crystallography data (20, 40), while similar information on Hp has been calculated by PDB2PQR web server (26). Next, MD has been performed to remove possible conformational strain and relax the protein structure. Briefly, the protein structures are energetically minimized in vacuo using 5000 steps of steepest descent algorithm and the protein has been placed at the center of a rhombic dodecahedral box solvated with single point charge water molecules. The physiological ionic strength has been set to 0.15 M NaCl to neutralize the system.
Docking calculations to elucidate the Hp-Hb complex have been conducted with the HADDOCK (23, 27) server, which is an approach that incorporates experimental data to drive the docking process. Ensembles of structures from the last 10 ns of MD simulation for both proteins (representing a matrix of 10×10 possibility) have been crossdocked. Of all the putative interface residues identified, we have observed that 10, 8, and 9 residues of Hb and 7, 9, and 8 residues of Hp were classed as nonpolar, charged, and polar, respectively, as described previously (35). Taken together, these data illustrate that the percentage of nonpolar, charged, and polar residues each contributed equally to one-third of residues at the binding interface (Fig. 5, Table 3).
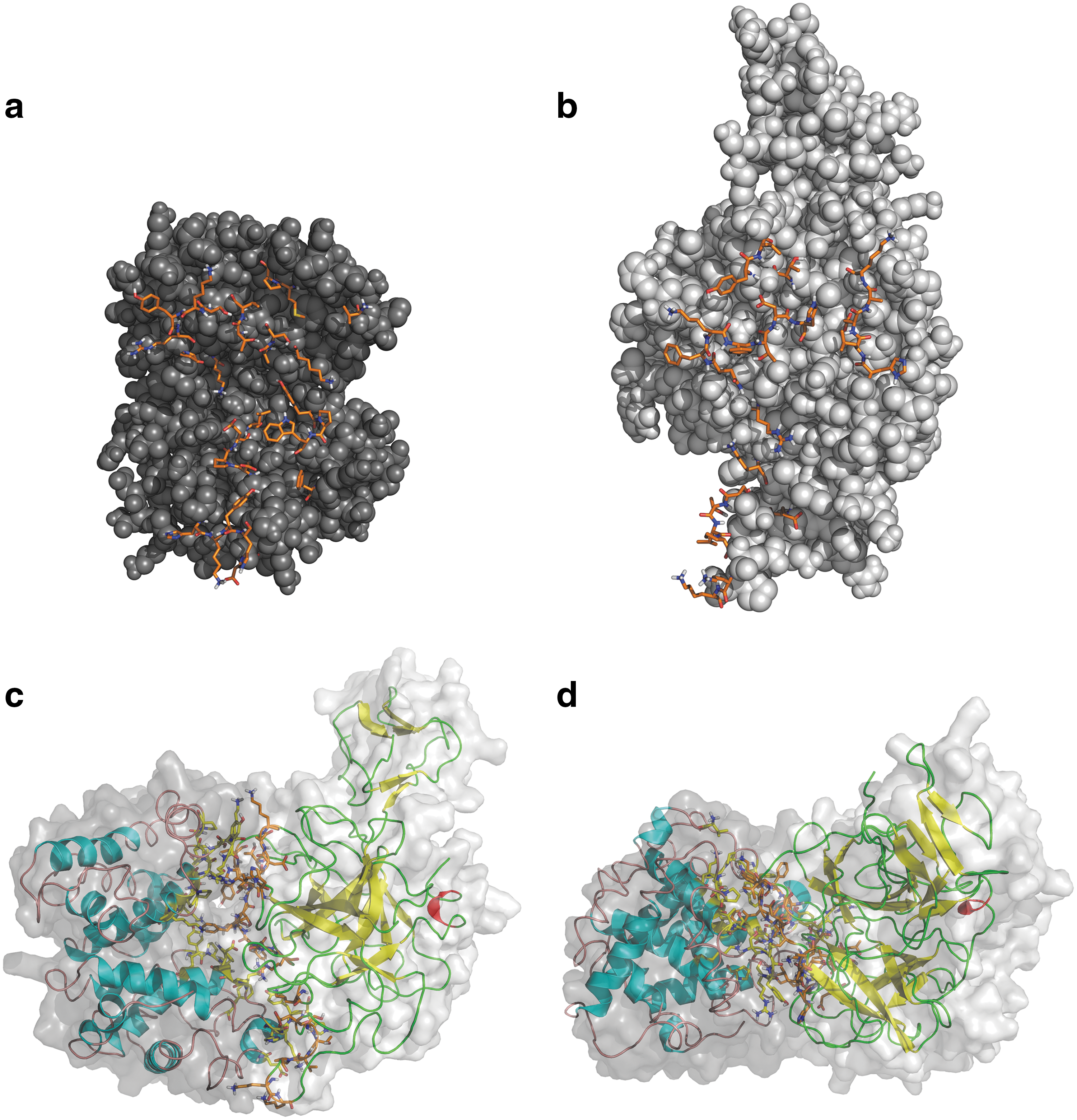
After Docking of the Hb A Dimer and Hp 1-1 Using the HADDOCK Software, the Residues at the Binding Interface of the Complex Were Determined
In this study, 10 energetically stable conformers of Hb and Hp, respectively, obtained after molecular dynamics simulations, were analyzed. Frequencies of possible residues located at the interface were determined using HADDOCK. The most prominent residues are shown.
Hb, hemoglobin.
Nooren and Thornton (64) have previously observed that the interfaces of transient complexes have small, often planar and polar interface areas than those of homodimers. Planarity as the name implies measures the shape of the binding interface or in other words the flatness of an interface. Our results show that the Hp-Hb complex has a significantly less planar surface at the interface compared to the average planarity of protein–protein interfaces (29, 88). This suggests that the interface of either protein is not flat and can therefore protrude into its respective binding partner. Our results also indicate that the secondary structure involved at the binding interface include both coils and α-helices of Hp and Hb, respectively. Tyrosine radicals have previously been shown to be involved in Hb toxicity (76, 77). When dissecting the interface in more detail, it is obvious that several tyrosine residues of Hb, particularly β145Tyr, α42Tyr, and α140Tyr, are buried in the complex and protected from further oxidative reactions (60). Such knowledge on the binding modalities of the Hp-Hb complex is anticipated to pave the way toward the design of robust Hp mimetics that may bind and mask the deleterious effects of Hb radicals.
Hp Mimics
Hb is a very abundant protein and the concentration in blood is around 150 g/L, and accordingly, an adult human carries almost 1 kg of the protein. Thus, significant amounts may escape from the RBCs even under healthy conditions and this scenario can rapidly be worsened during pathological conditions involving hemolysis. Examples are various hemolytic anemias, including inherited RBC dysfunctions and deficiencies, autoimmune transfusion reactions, and mechanical RBC disruption. Intraventricular hemorrhage and subarachnoidal bleeding result in locally high concentrations of free Hb in cerebral cavities, and these conditions are associated with severe cerebral damage and dysfunctions. Substantial amounts of cell-free Hb in the bloodstream are also found during clinical administration of artificial blood components, or hemoglobin-based oxygen carriers (HBOCs), employed for treatment of blood-loss after trauma, etc. Stimulation or induction of internal Hp production, for example, by controlled glucocorticoid treatment, or alternatively supplementation of the protein to the affected patient, may be a possible route for clinical treatment in these instances. Such conditions would require access to large amounts of clinical-grade Hp. Hp can be isolated and produced from human plasma and an Hp product is approved in Japan as a drug for the treatment of severe hemolytic conditions (98). Only small quantities are administrated to patients for such purposes; however, as indicated, many pathological situations involve abnormal levels of hemolysis and large amounts of Hp would be needed for clinical treatments. Since Hp is a large protein with multiple disulfide bonds as well as with complex and functionally essential glycosylation, recombinant Hp production in the scales needed is not realistic with present technologies. Alternatives are therefore required for allowing both a more extensive use as well as control of free Hb toxicities in the body. If the precise molecular mechanisms were understood on how Hp modulates radical chemistry within the complex and how it confines Hb-mediated oxidative responses, it will be feasible to mimic and prepare artificial Hp molecules, including small organic molecules, peptides, aptamers (85), or monoclonals that mimic the favorable activities of Hp and which could be synthesized in large enough quantities to stoichiometrically protect against free Hb.
Another fascinating option can be based on molecularly imprinted polymers (MIPs), which are artificial, template-made receptors with the ability to recognize and to specifically bind the target molecule (56, 74). The extreme stability, ease of preparation, sterilisability, and low cost of these MIPs have led to their assessment as substitutes for antibodies or enzymes in chemical sensors, catalysis, and separations. Although creating an MIP against small molecules is straightforward now, imprinting of large structures, such as extensive carbohydrate chains, proteins, and cells, is more challenging. The major problem associated with the imprinting of such large structures lies in its restricted mobility within highly cross-linked polymer networks and the poor efficiency in rebinding. Early developments included the creation of protein imprinted cavities in a thin layer of acrylic polymer or in conventional acrylamide gels, but there was only limited success. Up to now, imprinting at a polymer surface or the use of hydrogels seems to be the most promising way to overcome such difficulties. These latter materials are often significantly swollen with water and are less-heavily crosslinked than conventional MIPs that are prepared with small-molecule templates. Chayen and coworkers recently prepared hydrogel MIPs showing excellent specificity for Hb (82). These materials may be viewed as Hp mimics, which can be explored as extracorporal shunts, for example, during cardiac surgery, or intravenously in the form of biodegradable nanoparticles. In these instances, it can be beneficial to include further radical protection by adding MIPs carrying superoxide dismutase activities (70).
Conclusions
The protein complex formed between Hb and Hp involves one of the strongest molecular interactions known in nature. The aspiration to understand exactly how Hb and Hp form this complex has resulted in a number of experimental investigations. Such efforts have shed light on the Hp- and Hb-binding sites on each protein. While awaiting the structural determinations of Hp to come into place, molecular modeling of the Hp-Hb complex will no doubt be instrumental for improving our understanding of the mechanistic events hidden behind the exceptional ability of Hp to shield Hb toxicity. Such detailed molecular knowledge of both the Hp-Hb complex as well as its clearance pathways will be essential for rational development of drugs modulating Hb toxicity. In addition, it will allow for the control and modification of plasma half-life, duration of activity, and potential drug interactions. Our current knowledge is surprisingly limited and sometimes even contradictory.
Hp has several clinical applications, as a marker protein for screening and monitoring disease progression as well as for modulating Hb toxicity by direct administration. A few fields appear particularly attractive in this latter area. After a few years of pessimism in the use and exploration of HBOCs, alternative lines of development have given the area a new boost. Particularly, the use of the Hp-Hb combination appears beneficial for designing robust and safe HBOCs (4). At least when viewed on in vitro properties and on a few tests performed in animal models, the characteristics of the Hp-Hb complex could be very attractive in terms of being nonhypertensive with a long half-life in circulation. However, Hp binds dissociated Hb in its oxygenated R-state and the high oxygen affinity of dimeric Hb is locked within the complex (57). The resulting low P50 oxygen value of the complex will presumably interfere with oxygen delivery. In addition, when the structure of the Hp-Hb complex gets fully elucidated, it will be possible to remove its interaction with the CD163 scavenger receptor by site-directed mutagenesis. Protein engineering work on both the Hp and Hb moieties will therefore be needed to obtain a complex that has properties amenable for its use as HBOC. However, these findings in animals promote our understanding of the physiology behind Hb detoxification. The concept of Hb protection by natural or artificial scavenger molecules combined with the accumulated knowledge on designing Hb molecules with tailored O2 binding and radical protecting properties (32, 77) could stimulate entirely novel concepts in the HBOC field. In addition, it will be essential to scrutinize the role of the glycans, which most probably affects circulation and clearance times as well as oxidative stress properties and other systemic effects.
Footnotes
Acknowledgments
The following research funding agencies are gratefully acknowledged for supporting the research performed: Swedish Research Foundation (VR), Erasmus Mundus (to S.C.), Royal Thai Government Scholarship (to R.K.), and Swedish Research Links. This project was also supported, in part, by the Office of the Higher Education Commission and Mahidol University under the National Research Universities' Initiative (to C.N.)