
Abstract
Introduction
In recent years, energy metabolism has been rediscovered in cancer biology. The particular aerobic metabolism experienced by cancer cells has been finally recognized as an additional hallmark of the cancer cell phenotype (22, 36, 74). The abnormal high aerobic glycolysis of tumors was first observed by Otto Warburg many years ago (74). He suggested that the increased aerobic glycolysis of tumors should result from an impaired bioenergetic activity of mitochondria (95, 96). Warburg's hypothesis was heatedly debated (74) but largely neglected until its rediscovery by the implementation of tumor imaging with 18F-desoxyglucose (18FDG) by positron emission tomography (PET) in the clinical practice of oncology (53, 73).
In this review, we will emphasize the prominent role that silencing the activity of mitochondrial oxidative phosphorylation has to promote tumor development. We will show that interfering either at short- or long-term with H+-ATP synthase is enough to illustrate the actuality of the original Warburg hypothesis regarding the metabolic phenotype of cancer cells and tumors. We will highlight that the peculiar energy metabolism of the cancer cell is a “reversible trait” (i.e., in the majority of carcinomas, it is not based on permanent genetic changes). The reversibility and flexibility of energy metabolism thus widens up its value as a therapeutic approach for cancer treatment.
An Ongoing Debate
If the induction of glycolysis in the majority of carcinomas is nowadays unquestionable, the impairment of the bioenergetic function of mitochondria in cancer cells is still a matter of debate (19). Perhaps it stems from the idea that only oncogene-driven metabolic changes could explain the switch of energy metabolism in cancer and/or from the observation that some cancer cells growing in culture rely on oxidative phosphorylation as the main energy provision pathway. Indeed, the activation of oncogenes and the inhibition of tumor suppressors (myc, Akt, p53, HIF1-α, APC/C-Cdh1) certainly affect energy metabolism and could account for changes in metabolic flux (40) and in the expression of enzymes of energy metabolism (15, 22). These genetic alterations, which are present with more or less frequency in different carcinomas, could act pleiotropically to upregulate aerobic glycolysis, but are not the main underlying cause of the Warburg phenotype of carcinomas. Why? Because most prevalent carcinomas (breast, lung, colon, esophageal, …) independently of its tissue of origin, histological type, and genetic alterations all have the same protein signature of energy metabolism (1). Moreover, the Warburg phenotype is reversible and could be acquired without any genetic alteration, as we will see in the following. Since the impact of cancer genes in energy metabolism has been extensively reviewed elsewhere (15, 22), we will not refer to this point any further. In fact, we consider that emphasizing the role of cancer genes in energy metabolism provides a reductionist approach to understand the role of mitochondria in cancer biology.
On the other hand, it has been documented that increased energy production by glycolysis in response to aberrant mitochondrial respiration accompanies the acquisition of the fully transformed state (72). In apparent contrast, the transformation of human mesenchymal stem cells (hMSC) show increased dependence on oxidative phosphorylation when growing in culture (29). However, when transformed hMSC were implanted into nude mice, an increase in glycolysis and the downregulation of genes of the TCA cycle was observed (29), suggesting the relevance of the microenvironment for tumor progression. Similar findings have been obtained in H-RasV12/E1A transformed cells where the tumorigenic potential of the cells increases concurrently with an enhancement of glycolysis and a diminished activity of mitochondrial respiration (20). More recently, it has been documented that the enhanced infiltration of gliomas in the brain parenchyma parallels the suppression of mitochondrial biogenesis and an increased glycolysis (44). Consistent with these findings, in vivo tumor progression requires the previous selection of cancer cells with a cellular phenotype in which the bioenergetic activity of mitochondria is repressed (76). The acquisition of this metabolic trait is not based on a permanent genetic change, rather it represents a reversible feature that results from the metabolic adaptation of the cells to the milieu where tumor cells develop in vivo (76).
Mammalian cells show large differences in the molecular composition of their mitochondria (63). Hence, a certain degree of diversity is conceivable in the relevance of the pathways that contribute to energy provision in cancer cells. In this regard, recent findings suggest the existence of metabolic complexity, a sort of metabolic symbiosis, in energy metabolism between cancer cells of the same tumor (86) or between cancer and stromal cells (99). Moreover, it has been proposed that β-oxidation fuels energy metabolism in metastatic ovarian cancer cells by the supply of fatty acids derived from adipocytes of the omentum (65). These findings might support a different energy metabolism in primary and metastatic cancer cells. Consistent with the complex role of mitochondrial activity in tumor development, we maintain that energy metabolism represents a reversible adaptive phenotypic response of the cancer cell to hypoxia and/or other agents of the tumor microenvironment (9, 65, 80, 99) that needs to be further investigated.
Metabolic Overview of Energy Metabolism
Glucose is the preferred metabolic substrate of most mammalian cells and its breakdown in the cytoplasm and in mitochondria provides the energy, reducing power (NADPH), and building blocks required for cellular maintenance and/or proliferation (Fig. 1). The catabolism of glutamine is also relevant for cellular proliferation in some cancer cells (Fig. 1) (15, 22). Normoxic cells oxidize most of the pyruvate to CO2 in the mitochondria, and the electrons collected on NADH and FADH2 are transferred to the complexes of the respiratory chain to generate the proton electrochemical gradient used in oxidative phosphorylation for the synthesis of ATP (Fig. 1). In nonproliferating cells, the demand for glucose is low because the oxidation of glucose coupled to oxidative phosphorylation provides a high yield of ATP and NADH (15). In contrast, proliferating cancerous and noncancerous cells boost the rate of glucose consumption because glycolysis provides the metabolic intermediates and NADPH needed for biosynthetic purposes (15). In this situation, pyruvate is reduced to lactate with the purpose of regenerating the oxidized form of NAD+ for glycolysis to proceed because oxidative phosphorylation is downregulated during progression through the cell cycle (Fig. 1) (15). Similarly, when the cells have a limited supply of oxygen or have a genetic or epigenetic impairment that restrains oxidative phosphorylation, glycolysis is also enhanced. The excess of lactate produced in these situations is exported out of the cell, contributing to the acidification of the cellular environment and metastatic disease (12).
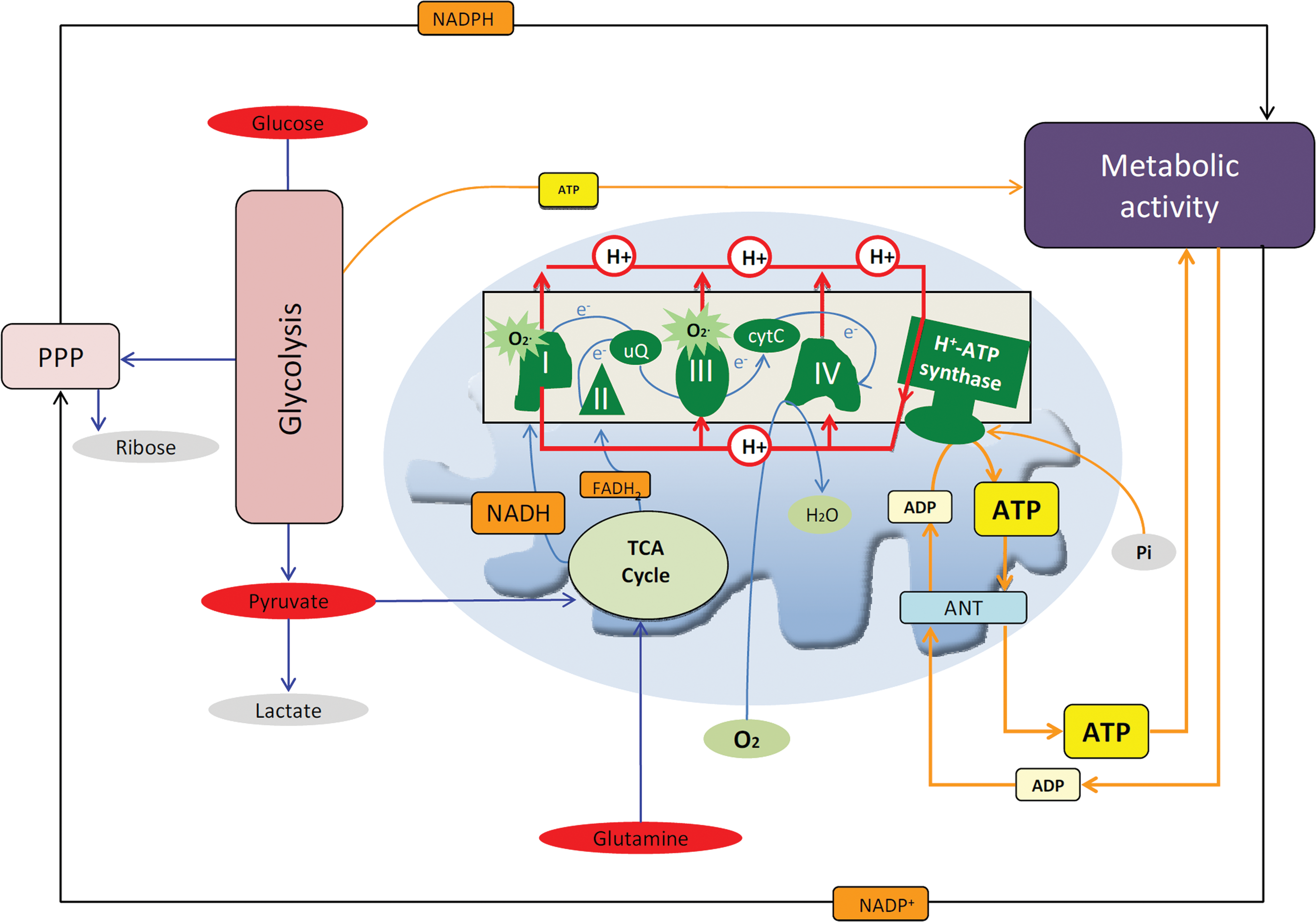
The Pivotal Role of the Mitochondrial H+-ATP Synthase in Energy Conservation
In oxidative phosphorylation, ATP is synthesized from ADP and Pi by the mitochondrial H+-ATP synthase (Fig. 1), a reversible engine of the inner mitochondrial membrane that synthesizes or hydrolyses ATP upon changes in cellular conditions (Fig. 2). An efficient oxidative phosphorylation requires an inner mitochondrial membrane with low proton conductance to prevent the waste of energy and the constant supply of ADP and Pi that are shuttled into the matrix of the organelle by the adenine nucleotide and phosphate carriers, respectively (Fig. 1).
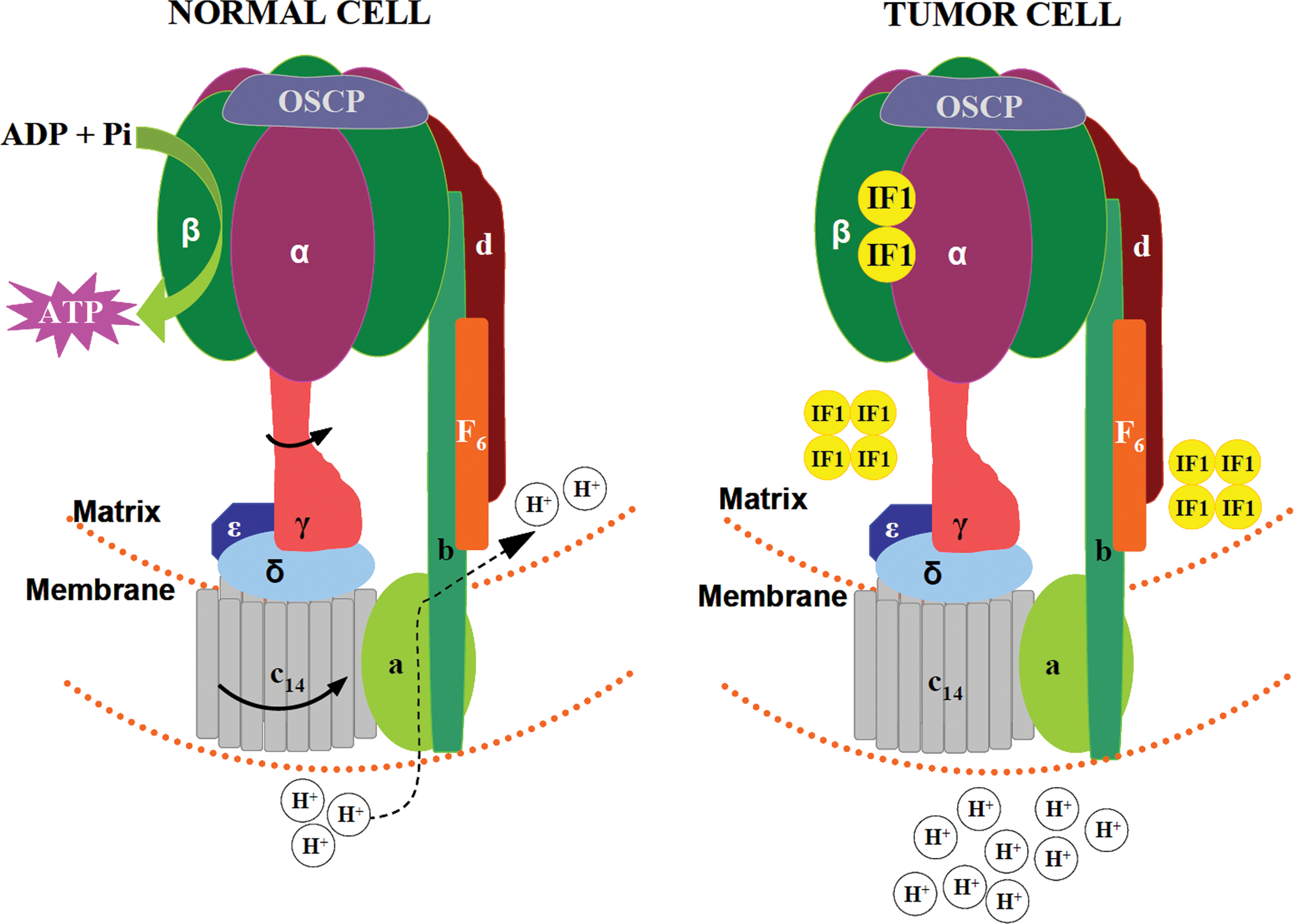
The mammalian H+-ATP synthase consists of two main domains: a membrane-bound hydrophobic F0 portion, which contains the proton channel, and a soluble F1 portion that contains the catalytic β-F1-ATPase subunit that encloses the adenine nucleotide binding sites (Fig. 2). Both regions are linked together by a central and a peripheral stalk (Fig. 2) (87). Table 1 summarizes the genes and stoichiometry of the subunits that participate in each of the F0 and F1 domains of the human H+-ATP synthase. The biogenesis of H+-ATP synthase requires the coordinated expression of nuclear and mitochondrial genes (Table 1) being the assembly of the holoenzyme assisted by ‘assembly factors' that do not take part of the final enzyme (Table 1). In normal cells, and under phosphorylating conditions, the re-entrance of protons into the mitochondrial matrix (Fig. 2) triggers the rotation of the c-ring and of the attached central stalk that induces the conformational changes in the β-F1-ATPase subunit that drive the synthesis of ATP (Fig. 2). The ATPase inhibitory factor 1 (IF1) is a physiological inhibitor of H+-ATP synthase (77). Moreover, H+-ATP synthase has stable interactions with itself to delineate a supramolecular level of organization of the membrane (88). In this superstructure, H+-ATP synthase dimers tend to align ‘in ribbons' imposing a high curvature to the inner membrane contributing to define the cristae structure of mitochondria (88).
Summary of the genes, proteins, and stoichiometry of the subunits and assembly factors of the human H+-ATP synthase. MT, indicates encoded in the mitochondrial genome.
Short-Term Regulation of Energy Metabolism
The cellular availability of ATP, NADH, and some metabolic intermediates (citrate) coordinate at short-term the activity of key enzymes of energy metabolism such as phosphofructokinase in the glycolytic pathway and various of the mitochondrial dehydrogenases (26), to limit the flux of matter and the production of biological energy as it is being demanded by the cell (Fig. 1). It has long been recognized that the efficient production of biological energy by oxidative phosphorylation determines the rates of glucose consumption in eukaryotic cells (49). In fact, this was demonstrated years ago in aerobic cells using 2,4-dinitrophenol (2,4-DNP), which uncouples the proton gradient generated by respiration from oxidative phosphorylation (49). Cells that are actively respiring when treated with 2,4-DNP re-establish the high rates of glucose consumption and lactate production that are observed when the cells are grown in anaerobiosis (Fig. 3A). Consistently, both normal and breast cancer cells significantly increase their rates of aerobic glycolysis when the proton gradient is collapsed by treatment with 2,4-DNP (Fig. 3B). The inhibition of the H+-ATP synthase with oligomycin also induces an increase in aerobic glycolysis in many cancer cells (53, 76).
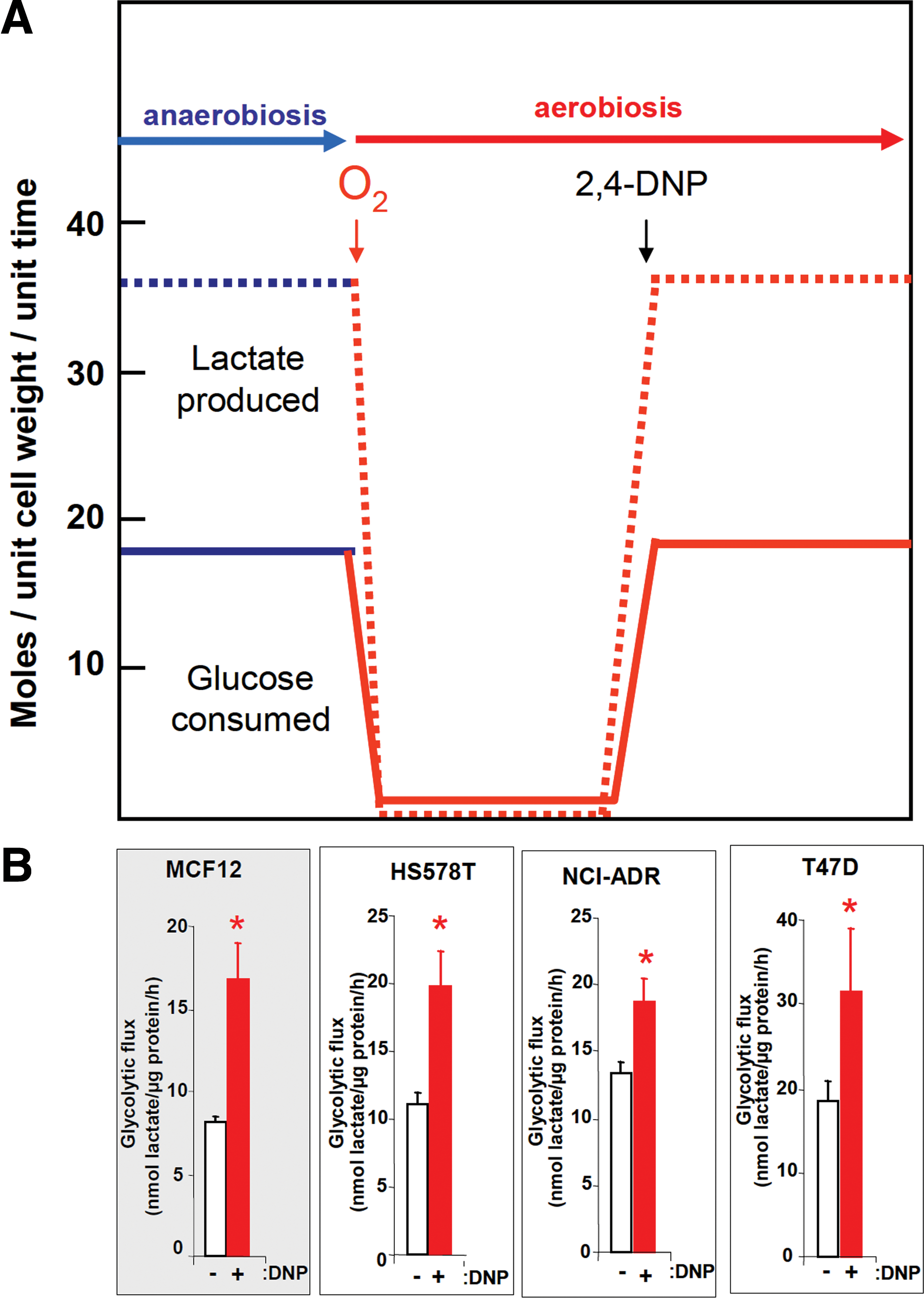
Remarkably, we have recently shown that the ATPase inhibitor IF1 whose expression is negligible in normal human breast, colon, and lung tissues is highly overexpressed in all breast, colon, and lung carcinomas (77). Moreover, the overexpression of IF1 in both normal and cancer cells triggers the inhibition of the H+-ATP synthase (Fig. 2), the metabolic switch to an enhanced aerobic glycolysis and the concurrent increase in the mitochondrial membrane potential due to the prevention of the backflow of protons into the mitochondrial matrix (Fig. 2) (77), mimicking the effects of oligomycin (77). Conversely the silencing of IF1 has the opposite metabolic effects (77). These findings strongly emphasize the relevance that the inhibition of the activity of H+-ATP synthase has for metabolic adaptation of cancer cells and tumor development. In fact, and in addition to promoting the metabolic switch observed in cancer cells IF1 simultaneously triggers a mitochondrial ROS-mediated nuclear response that switches on proliferation, invasion, and cell survival (27). The IF1-mediated adaptive response in colon cancer cells is exerted by the activation of the canonical NFκB pathway (27). Although the mechanisms that trigger the upregulation of IF1 in human carcinomas are presently unknown, its regulation in cancer provides an explanation of Warburg's original observation, further offering a promising target to regulate energy metabolism in carcinomas.
Tumor Microenvironment and the Bioenergetic Signature of Tumors
The relevance of the cellular environment in cancer progression is quite well delineated (9, 91) after the pioneer experiments from the Mintz's laboratory that showed the development of normal mice from malignant teratocarcinoma cells (61). Regarding metabolism, when short-term regulation of enzyme activities is not enough to cope with the demand imposed by proliferation or a long-lasting adaptation to a particular environment, the cells onset the gene expression programs required for adaptation. Examples that are relevant to understand the metabolic phenotype of carcinomas are the adaptations experienced by cells to hypoxia (80, 81), the metabolic and enzymatic parallelism existing between cancer cells and embryonic tissues (16), as well as the establishment of the bioenergetic function of mitochondria during adaptation of mammals to the aerobic extrauterine environment (16).
Hypoxia-inducible factor 1α (HIF-1α) is a transcription factor induced by low oxygen tension that is overexpressed in most carcinomas (80, 81). HIF-1α is involved in conferring resistance against apoptosis and in promoting angiogenesis and metastasis to cancer cells. HIF-1α promotes the transcriptional activation of many embryonic-type isoforms of the glycolytic pathway (80, 81). These “cancer/embryonic” enzymes differ in their catalytic properties and interacting partners when compared to the enzymes in normal cells. Hexokinase II (HKII), that catalyzes the first committed step of glycolysis, is highly overexpressed in most human carcinomas and is tightly bound to mitochondria by its interaction with VDAC (voltage-dependent anion channel), a protein component of the PTPC (permeability transition pore complex), linking in this way energy metabolism to the mitochondrial gate to execute cell death (28, 58, 103). The HK–VDAC interaction is stimulated by oncogenic Akt signaling (28, 62, 70), enhancing the glycolytic flux of the cancer cell by coupling the residual ATP being produced in mitochondria to the phosphorylation of glucose. This interaction and the proteins involved have a very relevant impact in metabolic reprogramming and in controlling the fate of cancer cells (28, 30, 58, 103). Moreover, HIF-1α also controls the expression of pyruvate dehydrogenase kinase 1 (PDK1) (46), a negative regulator of the utilization of carbon skeletons in mitochondria of the cancer cell.
Moreover, it is worth emphasizing the parallelism existing in the expression of pyruvate kinase M2 between cancer cells and embryos for the control of glycolytic metabolism (17, 56) and the repression of the bioenergetic function of mitochondria in the fetal hepatocyte and hepatomas (16, 21). In carcinomas and fetal hepatocytes, the complement of mitochondria is downregulated when compared to normal tissues. Moreover, mitochondria in carcinomas have less cristae than in normal cells (5, 14) consistent with the relevant role played by H+-ATP synthase in mitochondrial structure (88). In addition, transcriptomic, proteomic, functional, and structural studies in human carcinomas invariably indicate that a repressed bioenergetic activity of mitochondria parallels the activation of glycolysis required for tumor progression (for review see (15)).
Based on the observation that the expression of H+-ATP synthase inversely correlates with the activity and expression of enzymes of the glycolytic pathway during liver development, we designed a simple approach to assess the protein signature of energy metabolism in human carcinomas (14). The “bioenergetic signature” correlates the amount of the catalytic β-F1-ATPase subunit of the H+-ATP synthase to the content of the glycolytic glyceraldehyde-3-phosphate dehydrogenase (β-F1-ATPase/GAPDH ratio) in the biopsy. The bioenergetic signature was shown to be downregulated in most prevalent human carcinomas. The downregulation of the bioenergetic signature is a characteristic fulfilled by more than 97% of the carcinomas analyzed in large cohorts of tumors (3, 13, 14, 43), indicating that it represents a general phenotypic trait of cancer supporting Warburg's hypothesis. These findings have been confirmed (52) and extended to other carcinomas (see (15) for additional references). More recently, quantitative assays of the “bioenergetic signature” revealed that tumors from different tissues and/or histological types have the same signature of energy metabolism (1, 3).
A functional support to the altered mitochondrial function in human carcinomas came after the demonstration that the rates of 18FDG capture assessed by PET imaging in lung cancer patients inversely correlate with the bioenergetic signature of the tumors (53). Moreover, the cellular content of H+-ATP synthase has been shown to directly correlate with the activity of oxidative phosphorylation, whereas it inversely correlates with the rate of glucose utilization by aerobic glycolysis, strongly supporting the relevance of the Pasteur effect in cancer biology (76).
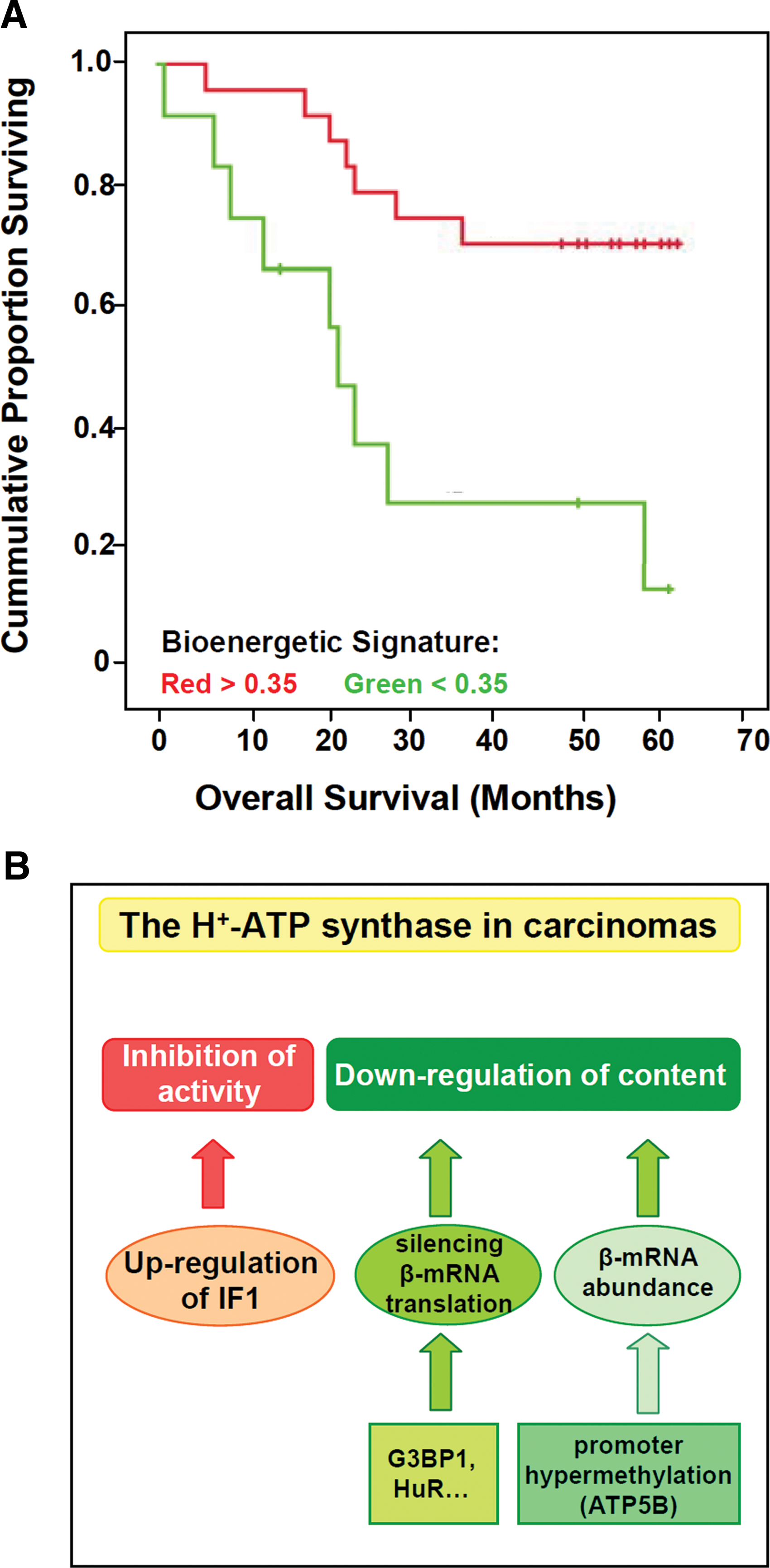
Analysis of the bioenergetic signature in large cohorts of different carcinomas have further emphasized that it affords a relevant marker for the prognosis of breast (43, 68), colon (3, 14, 52), and lung (13, 53) cancer patients (Fig. 4A). In agreement with these findings, oxidative phosphorylation has been shown to be the only metabolic pathway whose impairment correlates with a worse prognosis in colon cancer patients (83). However, we should note that only a small number of glycolytic carcinomas result from mutations in nuclear genes that are related with the metabolic and bioenergetic function of the organelle (6). Overall, the alteration of the bioenergetic signature strongly supports a relevant role for the bioenergetic arrest of mitochondria in cancer cells in order to promote progression of the disease.
Regulation of H+-ATP Synthase in Human Carcinomas
Despite the relevant role that H+-ATP synthase has in cell physiology, the regulation of its expression and activity has been scarcely studied and mostly focused on the heart enzyme. As indicated previously (Fig. 2B), the overexpression of IF1 in human carcinomas downregulates the activity of H+-ATP synthase (Fig. 4B) (27,77). The main mechanism that regulates the expression of the catalytic β-F1-ATPase subunit in fetal liver, during the cell cycle and in rat and human carcinomas is exerted at the level of translation (for recent review see (100)). The 3′UTR of β-F1-ATPase (β–mRNA) is required for efficient translation of the transcript and its translation silencing is observed both in rat and human carcinomas (Fig. 4B) (100). However, translational silencing of β-mRNA in carcinomas (101) and in development of the human liver (102) is exerted by different mechanisms.
The AU-rich element (ARE) binding protein HuR, which is a central regulator of post-transcriptional gene expression, has been identified as a 3′UTR β-mRNA interacting protein (68). Although HuR plays no relevant role in regulating β-F1-ATPase expression (68), it affords a relevant independent marker of breast cancer prognosis when studied in combination with the bioenergetic signature of the tumor, allowing the identification of patients who are at higher risk of disease recurrence (68). More recently, the Ras-GAP SH3 binding protein 1 (G3BP1) has been shown to interact with the 3′UTR of β-mRNA (Fig. 4B) (69). G3BP1 is a multifunctional ATP-dependent helicase with endoribonuclease activity that is highly overexpressed in breast carcinomas. The interaction of G3BP1 with β-mRNA specifically represses its translation by preventing recruitment of the β-mRNA into active polysomes (69). This finding suggests that G3BP1 participates, at least in breast cancer, in the downregulation of β-F1-ATPase expression and hence in promoting the metabolic switch in carcinomas. In this context, the identification of the β-mRNA binding proteins (69) and miRNAs (102) that interact and presumably hamper translation of the transcript represents a crucial step in order to understand the bioenergetic phenotype of different cancer cells (Fig. 4B).
Another mechanism used by cancer cells to silence β-F1-ATPase expression has been described in chronic myeloid leukemia (51). In this situation, downregulation of β-F1-ATPase is exerted by limiting the amount of the mRNA by hypermethylation of the promoter of the ATP5B gene (Fig. 4B) (51). In addition, it is likely that mutations that result in the inactivation of the genes involved in the biogenesis of H+-ATP synthase (Table 1), also contribute to the downregulation of this protein complex in a limited number of carcinomas. Overall, these findings illustrate the need of further basic studies aimed at characterizing the complexity of the cellular response required for mitochondrial biogenesis. These studies will allow understanding the alterations of the mitochondrial proteome that specifically affect the content/activity of the H+-ATP synthase in different carcinomas (Fig. 4B).
Oxidative Phosphorylation and Cell Death Are Molecularly and Functionally Integrated
In addition to the role of mitochondria in energy metabolism, regulation of cell death has emerged as another major function of the organelle. These two master tasks of mitochondria are molecularly and functionally integrated despite the many forms and shapes of cell death (Fig. 5). In particular, molecular components that participate in oxidative phosphorylation, such as cyt c, AIF, and subunits of H+-ATP synthase, are required for the efficient execution of cell death (Fig. 5) (55, 71, 92). Conversely, Bcl-xL, a protein that participates in preventing apoptosis (Fig. 5), has been recently shown to interact with β-F1-ATPase to regulate the efficiency of energy metabolism (2). Likewise, the tumor-suppressor p53 has been suggested to mediate the regulation of mitochondrial energy metabolism via the downregulation of parkin (104) (Fig. 5). Moreover, Bax needs H+-ATP synthase to exercise its pro-apoptotic function (55). More recently, a fundamental relationship between mitochondrial bioenergetics and the tumor response to cytotoxic chemotherapy has been elegantly documented (64) and deserves further mechanistic characterization in future studies.
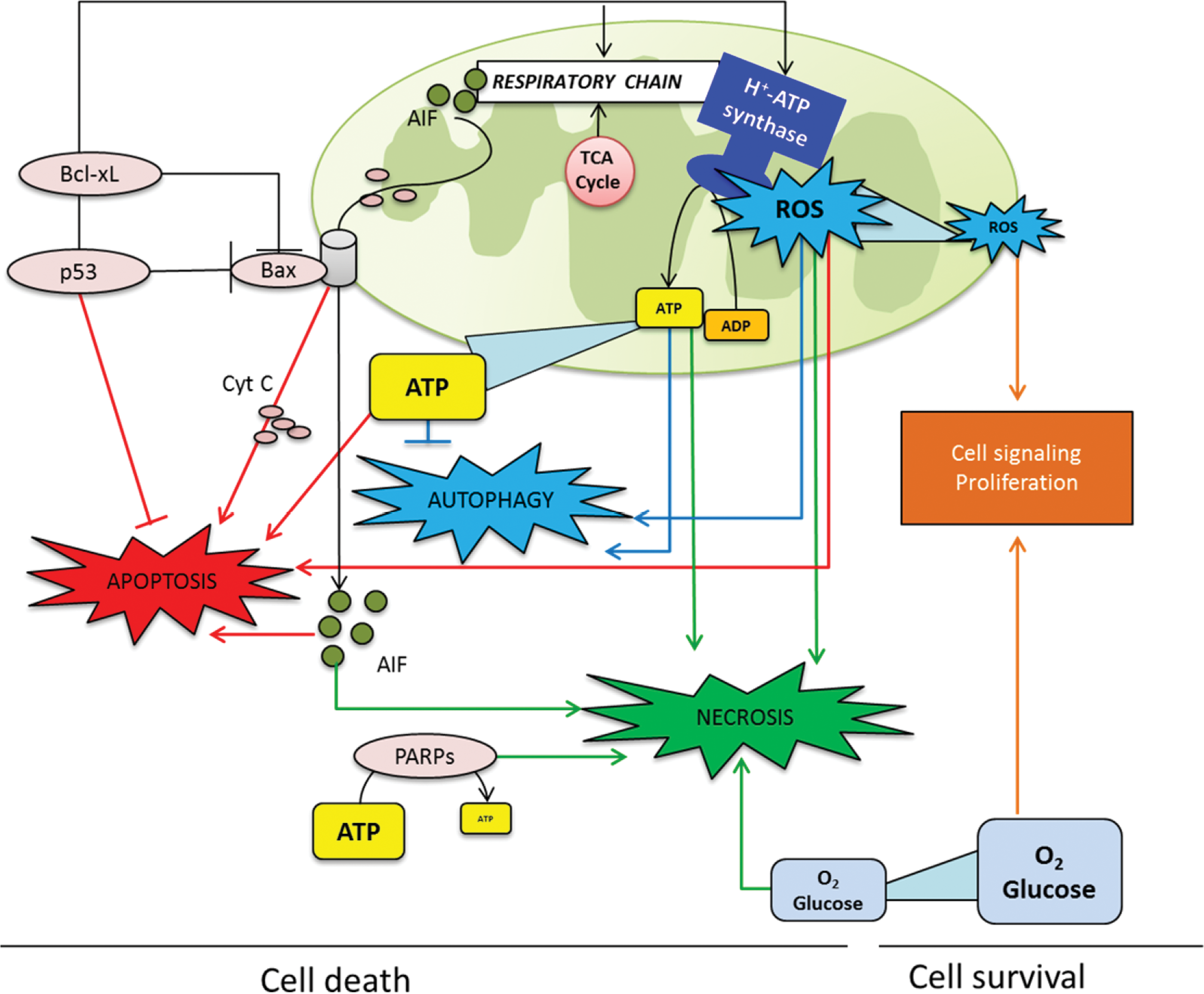
ATP and reactive oxygen species (ROS) are products of mitochondrial function (Fig. 1) that can hold the balance between apoptosis, necrosis, or other unconventional types of cell death such as autophagy (32, 34, 50, 66). In apoptosis, a sufficient supply of ATP is necessary to favor apoptosome formation to trigger the activation of caspases, chromatin condensation, and the correct execution of cell death (34) (Fig. 5). On the contrary, when ATP levels are limited, a typical apoptotic stimulus causes cell necrosis (Fig. 5). Necrosis has been considered an uncontrolled mode of cell death for years. However, rising evidence suggests that necrosis is regulated by a signaling pathway tightly connected with energy metabolism (32). Indeed, deprivation of oxygen or glucose are well known stimuli to induce necrosis in different experimental models (Fig. 5) (32). Moreover, interfering with glycolysis in cancer cells triggers massive ATP depletion and necrotic cell death (75). Upon massive DNA damage, the hyperactivation of PARP-1 leads to depletion of intracellular ATP, resulting in necrosis (Fig. 5) (25). The cell death pathway prompted by excessive PARP-1 activation has been conserved throughout evolution of multicellular organisms, perhaps for its ability to eliminate cells that have undergone excessive genotoxic stress and are therefore at risk of neoplastic transformation (25).
Low cellular ATP levels can also induce autophagy (50). It has been described that AMP-activated protein kinase (AMPK), which is activated when the H+-ATP synthase is inhibited, has critical roles both in reprogramming metabolism and in the induction of autophagy (60). A recent report linked mitochondrial morphology to the regulation of both ATP production and response to autophagy (33). In fact, when autophagy is prompted, mitochondria elongate through unobstructed mitochondrial fusion (33). Elongated mitochondria have more cristae, increased levels of dimerization and activity of H+-ATP synthase, maintain ATP production, and are protected from autophagic degradation (33). Conversely, when mitochondrial elongation is inhibited, the ATP levels fall down and the cell die by starvation-induced death, suggesting the existence of a dynamic feedback between autophagy and cellular energy balance (85).
As a product of respiration, mitochondria produce ROS at complex I and complex III of the respiratory chain (Fig. 1). ROS are highly reactive molecules that can oxidize DNA, proteins, and lipid,s and have been implicated in the oxidative damage that accompanies normal physiological ageing, cancer, and neurodegeneration. The cellular response to ROS depends on its intensity (11, 24, 66). A high ROS signal due to cellular or metabolic stress and/or in response to chemotherapy could trigger cell death by apoptosis, necrosis, or autophagy after integration of the various signaling molecules (ATP, ROS, O2, glucose, …) involved in these processes (Fig. 5). In any case, the cell fate is tightly modulated by the cellular amount of ROS, which act to tweak the balance between survival and cell death (Fig. 5) (66). In contrast, a low level of mitochondrial ROS production signals the gene transcription programs involved in cell proliferation (Fig. 5). In fact, ROS production in human carcinomas (89) leads to the activation of the signaling networks promoting tumorigenesis and metastasis (42, 97). Consistently, the low ROS produced by mitochondria in response to the overexpression of IF1 has emerged as a powerful stimulus for the induction of cellular proliferation in cancer cells (27). This implies that cancer cells exert a tight regulation of mitochondrial ROS production and of the antioxidant machinery to increase their chances of survival (11). Overall, we support that enhancement of mitochondrial metabolism acts as a tumor suppressor by promoting cell death essentially by apoptosis in normal genetic backgrounds. In contrast, mitochondrial metabolism can become tumorigenic by stimulating proliferation and metastasis when genetic alterations or the deregulation of a protein, such as IF1, participate in signaling an oncogenic phenotype via ROS production (27, 42, 97). In other words, genetic lesions and/or IF1 can promote the conversion of mitochondrial activity from a tumor suppressor into a tumor promoter, highlighting the complex role of mitochondrial activity in tumor progression.
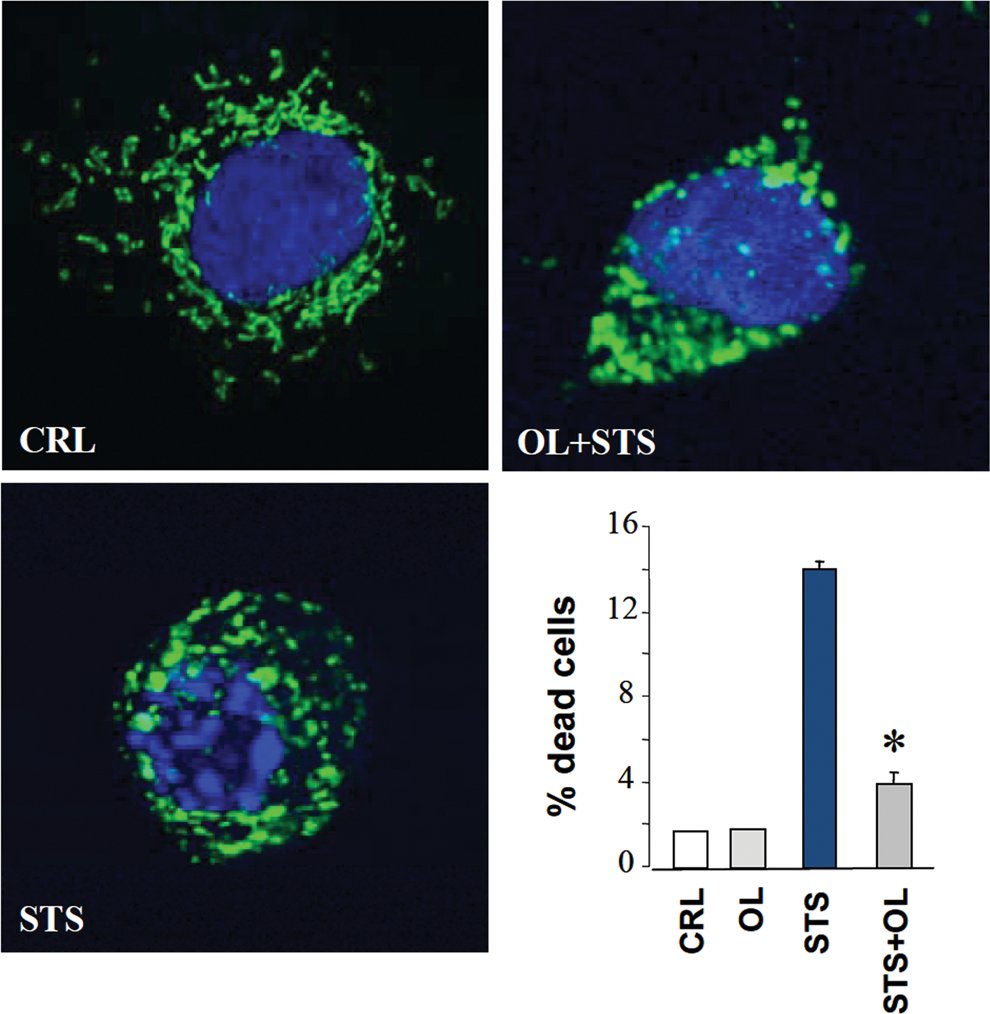
Excessive ROS production in mitochondria could result from genetic defects affecting components of the respiratory chain, by the inhibition of oxidative phosphorylation mediated by hypoxia, by different chemotherapeutic agents, or by the induction of IF1 in cancer cells (Fig. 2) (77). In these situations, the mitochondrial membrane potential (ΔΨm) is increased and the production of ROS enhanced (27, 77, 78). Therefore, ΔΨm represents an additional link between energy metabolism, ROS production, and cell fate. In fact, the induction of apoptosis by many different stressors invariably results in the collapse of ΔΨm. However, a preceding event before ΔΨm collapses is a transient mitochondrial hyperpolarization (47, 55, 78) and the subsequent production of a ROS signal (39, 78). Remarkably, oligomycin prevents mitochondrial hyperpolarization and ROS production in response to a death stimulus delaying the execution of apoptosis (Fig. 6) (55, 78). Moreover, the generated ROS oxidize cellular constituents (78), facilitating in this way the release of mitochondrial proteins that swamp the cells into death. Oligomycin prevented also these modifications (78), highlighting the deep connection that exists between energy metabolism, H+-ATP synthase, and the correct execution of cell death (Fig. 5).
The Tumor Suppressor Function of Mitochondrial Activity
Tumor suppressors such as Pten, p53, Ink4a, and Arf are known to exert a beneficial pleiotropic effect in cell physiology against aging and metabolic disease, in addition to provide protection against cancer (82). Activation of mitochondrial metabolism is a likely mechanism of their action (54, 67). Sirtuins are NAD+-dependent protein deacetylases that regulate longevity and a wide range of intracellular processes (35). Studies in mouse models with genetically modified SIRT1 levels strongly support the in vivo tumor suppressor activity of SIRT1 (see (38) and references therein). In this regard, the mitochondria-localized SIRT3 has been shown to exert its tumor suppressor function by maintaining mitochondrial integrity and metabolism (45) and, consistently, resveratrol has been shown to improve mitochondrial function and to protect against metabolic disease by activating SIRT1 and PGC-1α (48).
Consistent with the implication of mitochondrial activity and of H+-ATP synthase in cell death, there is a large body of data supporting that mitochondrial function, both under basal conditions or in response to chemotherapeutic agents, acts as a tumor suppressor. In this regard, the oxidation of mitochondrial substrates is known to halt malignant growth. The oxidation of fatty acids by the mitochondrial β-oxidation pathway is known to engage cancer cells into death (7, 98). Moreover, the inhibition of alanine aminotransferase, that slows down aerobic glycolysis and promotes an increase in cellular respiration by the oxidation of glutamine, has been shown to diminish cellular as well as tumor growth (8). Since cancer cell metabolism is characterized by a limited activity of oxidative phosphorylation to minimize oxidative stress during proliferation (10, 15), a likely mechanism that explains the preferential death of cancer cells when forced to oxidize mitochondrial substrates is by the overproduction of superoxide radical as a result of the stimulation of mitochondrial metabolism (8, 98).
Likewise, the von Hippel-Lindau (VHL) tumor suppressor activates oxidative phosphorylation and when injected into nude mice retards tumor formation and growth (41). In agreement with Warburg's postulates, a defective oxidative phosphorylation due to a deficiency in frataxin is known to favor early tumor progression in Friedreich ataxia patients, as well as in the liver of frataxin-KO mice (90). Conversely, the overexpression of frataxin in colon cancer cells triggers an increase in oxidative metabolism concurrent with a decrease in tumorigenicity (79). A dysfunctional oxidative phosphorylation has been shown to promote cellular proliferation and invasion (4, 27, 93). Conversely, the activation of pyruvate metabolism in mitochondria promotes the activity of oxidative phosphorylation and halts cellular proliferation and invasion (57) indicating the beneficial effect that restoring mitochondrial metabolism has in tumor formation.
Hence, we suggest that the tumor suppressor function of the activity of mitochondrial metabolism impinges in at least two of the hallmarks of the cancer phenotype by inhibiting cellular proliferation and activating apoptosis. Indeed, the activation of mitochondrial metabolism limits the supply of building blocks for biosynthetic purposes as a result of the burning of carbon skeletons in mitochondria, promoting energy expenditure, and preventing nutrient storage and its associated damage (67). Moreover, the activation of mitochondrial metabolism generates sufficient ROS by the functioning of mitochondrial respiration to allow the execution of cell death programs in normal genetic contexts. The molecular mechanisms involved need to be unveiled in future studies. However, both genetic (18) and pharmacological (94) studies have shown that the PGC1α-mediated improvement of mitochondrial activity and metabolism hamper cancer progression by increasing apoptosis and decreasing cellular proliferation and invasion. In particular, PGC1α protects against colon cancer by promoting ROS accumulation and the induction of mitochondria-mediated apoptosis (18), illustrating a direct connection between mitochondrial function and its tumor suppressor activity.
Taken together, the above findings support that interfering with the bioenergetic function of mitochondria predisposes to cancer development and tumor growth. Since treatment of cancer cells with dichloracetate (DCA) triggers a switch to a mitochondrial-dependent pathway of energy provision and promotes in vivo tumor regression (59, 76), the activation of mitochondrial function could represent a successful alternative for the treatment of cancer (Fig. 7). In this regard, the bioenergetic signature has been shown to predict the apoptotic response to chemotherapy in various cancer cells (37, 75, 84), as well as in colorectal carcinomas (52). Overall, these findings highlight the role of mitochondria in the epigenetic regulation of tumorigenesis and suggest that repression of oxidative phosphorylation is a prerequisite for tumor development (76). On the other hand, knocking down the glycolytic lactate dehydrogenase in mouse mammary tumor cells triggers an increase in mitochondrial function, a compromised ability to proliferate under hypoxia, and a reduction in the tumorigenicity of the cells (23). These results illustrate the tight connection that exists between glycolysis and oxidative phosphorylation and confirm that the inhibition of glycolysis could also provide a promising target to restrict proliferation of cancer cells (Fig. 7). In fact, we have shown that the bioenergetic signature of colon cancer cells predicts the potential to execute necrotic cell death in response to inhibitors of glycolysis (75).
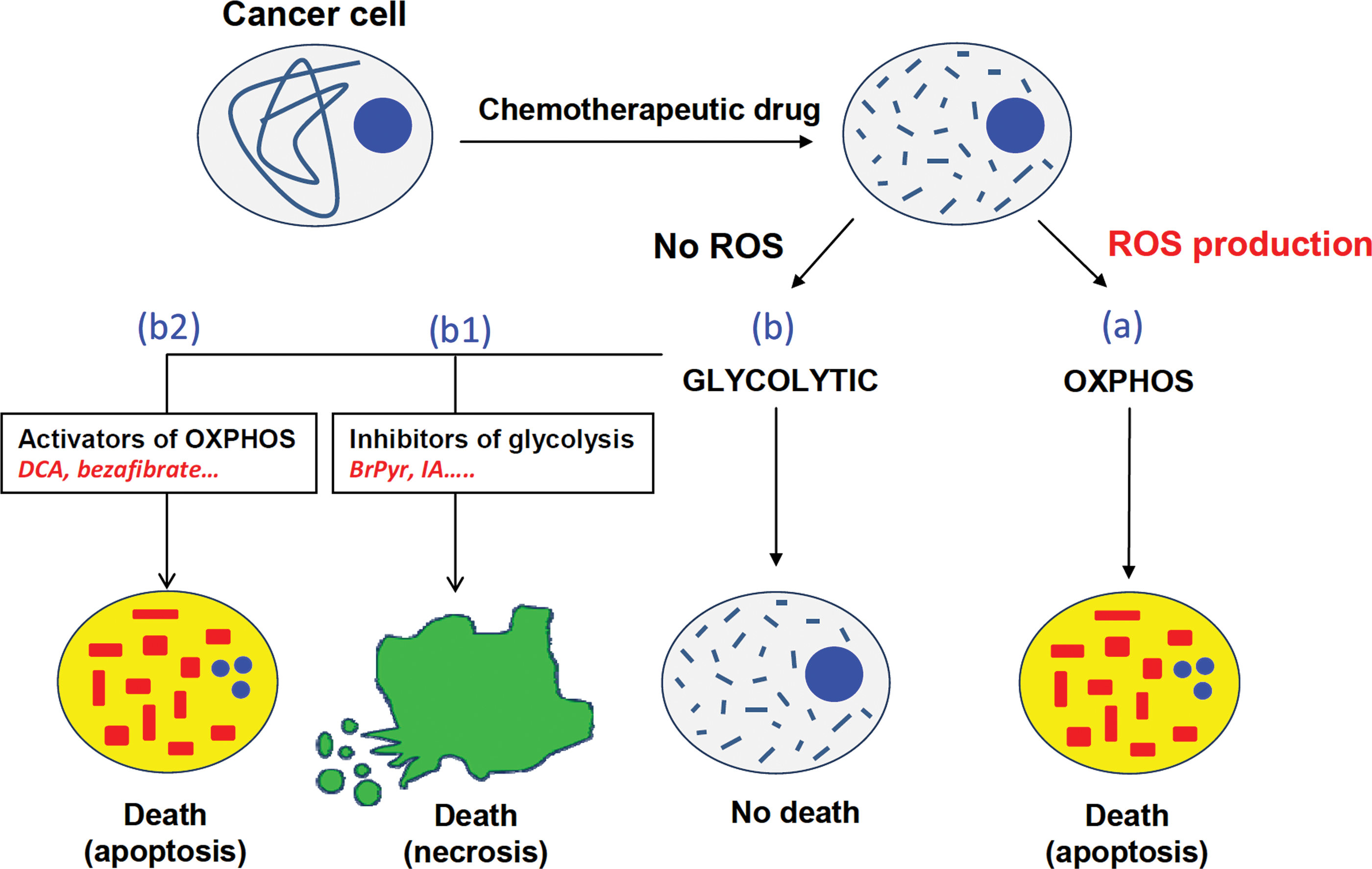
We propose that energy metabolism offers an attractive target for cancer treatment, perhaps when used in combination therapy, because it tackles a universal trait of the disease that could define the response and cell death pathway to chemotherapy (Fig. 7). Cancer cells with active oxidative phosphorylation will produce ROS in response to therapy and the resulting oxidative damage will engage cells into apoptosis (Fig. 7). In contrast, cancer cells with a predominant aerobic glycolysis are resistant to conventional therapy because of their impossibility to produce mitochondrial ROS (Fig. 7) (78). Therefore, we suggest that the strategy to follow with highly glycolytic tumors could be the activation of oxidative phosphorylation that might restore the cell sensitivity to apoptotic inducers and/or the employment of agents that by prompting a metabolic catastrophe will lead to cell necrosis (Fig. 7).
Final Remarks
Research over the last 10 years has put back into stage the relevance of energy metabolism in cancer biology. The increased aerobic glycolysis and restrained bioenergetic function of mitochondria in cancer cells is a required metabolic condition for cellular proliferation. In general, tumor energy metabolism is a reversible phenotypic trait of the cancer cell due to its adaptation to a particular environment. Limiting the bioenergetic activity of mitochondria, and specifically of H+-ATP synthase, contributes to tumor growth because oxidative phosphorylation is required for efficient execution of cell death. Two independent mechanisms specifically target the activity and the cellular content of the mitochondrial H+-ATP synthase to favor an increase in aerobic glycolysis and the restrained oxidative phosphorylation in cancer. The bioenergetic signature of the tumor has clinical utility for the prognosis of cancer patients and could aid in the establishment of new anti-tumor therapies. However, the activity and molecular composition of mitochondria is cell-type specifically expressed. Therefore, basic studies aimed at characterizing the complexities of the cellular responses needed for mitochondrial biogenesis are required to understand the alterations of the content/activity of H+-ATP synthase in cancer. Moreover, unveiling the different mechanisms and components that participate in the tumor suppressor function of mitochondrial activity is also needed. These studies could gear the establishment of new therapeutic strategies aimed at combating the progression of the disease using energy metabolism as target.
Footnotes
Acknowledgments
The authors gratefully acknowledge the technical support provided by M. Chamorro and C. Nuñez de Arenas over all these years. LF is supported by JCI2009-03918 Juan de la Cierva Grant, Ministerio de Educación y Ciencia, Spain. Work in the authors' laboratory was supported by grants from the Ministerio de Educación y Ciencia (BFU2010-18903), by the Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), ISCIII and by Comunidad de Madrid (S2011/BMD-2402), Spain. The CBMSO receives an institutional grant from Fundación Ramón Areces. We apologize to authors whose work or primary references could not be cited owing to strict space limitations.
Author Disclosure Statement
No competing financial interests exist.