
Abstract
Introduction
R
Older Technologies for ROS Detection
Spin trapping
One of the key ROS in biological systems is O2 •−. This free radical has a pre-eminent role in biology and pathophysiology, because it is formed by many mammalian enzymes, has significant biological reactivity, and serves as a progenitor for formation of many other ROS, including H2O2, ONOO•−, and lipid peroxides. One of the earliest methods for O2 •− detection was spin trapping with 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) (23). It is important to distinct spin traps and spin probes. Spin traps form covalent bond with the radical by addition reaction, while spin probes are oxidized by ROS without binding (14). While DMPO and similar nitrone spin traps are very useful in studies of isolated enzymes and in chemical solutions, they react with O2 •− at very slow rate constants, between 30 and 70 M −1s−1, and have difficulty detecting O2 •− in most biological systems due to competition with superoxide dismutase (SOD) and ascorbate (55). These slow rate constants also require that DMPO and similar spin traps be used at concentrations ranging from 20 to 100 mM. These high concentrations may have off-target effects and can affect cell viability. Moreover, intracellular reductants such as ascorbate can react with the O2 •− adduct of DMPO (DMPO-OOH) and reduce it to a spin-inactive state, severely limiting the use of nitrones such as DMPO in biological systems (66). Recently, one cyclic nitrone, 5-diethoxyphosphoryl-5-methyl-1-pyrroline N-oxide (DEPMPO), has been modified by addition of a triphenylphosphonium group, promoting its selective uptake by mitochondria (27). Unfortunately, the triphenylphosphonium conjugate of DEPMPO (mito-DEPMPO) must also be used at high concentrations (50 mM), which may cause inhibition of mitochondrial ATP synthesis due to the accumulation of large amounts of the lipophilic cation in the mitochondrial matrix and alteration of the mitochondrial membrane potential (36). The use of this mito-DEPMPO spin trap is therefore likely limited by its slow rate constant for reaction with O2 •− (33), potential toxicity (34), and nonspecific effects (1), and its use in biological systems is therefore unlikely. Because of these limitations, other approaches have been developed to detect ROS in intact cells and mitochondria. These include chemiluminescence methods, fluorescent probes, and more recently, antibody-based methods that have promise. These will be covered in the following sections.
Chemiluminescent probes
A commonly used chemiluminescence technique for measurement of O2 •− is lucigenin-enhanced chemiluminescence (14). The validity of this technique has been questioned on the grounds that O2 •− production might be artificially overestimated because of a phenomenon known as redox cycling, in which the lucigenin radical reacts with oxygen to generate O2 •− (51). Other chemiluminescent probes include luminol, MCLA, and coelenterazine. Fluorescent probes are also commonly called dyes, and we have applied this term where appropriate. These likely react with a variety of ROS, and are therefore nonspecific. The lucigenin-derived redox cycling is particularly significant in artificial conditions with purified flavin proteins and NADH, but this problem may be overestimated in intact cells and tissues without NADH/NADPH supplementation. Meanwhile, questions persist about redox cycling with these agents, and they are being supplanted by other methods. None of these are specific for the mitochondria.
Fluorescent probes
Dihydroethidium and mitoSOX fluorescence
During the past decade, many research groups used dihydroethidium (DHE) and its mitochondrion-targeted form mitoSOX for cellular and mitochondrial O2 •− detection (5, 58). This approach is limited, because DHE can form two fluorescent products. One is ethidium, which is formed by nonspecific redox reactions, while the other is 2-hydroxyethidium (2-OH-E+), a specific adduct of O2 •− (58). The fluorescent spectra of ethidium and 2-OH-E+ overlap, and it is therefore difficult to use simple fluorescence detection with methods such as confocal microscopy or other fluorescence-based microscopic assays to accurately measure only 2-OH-E+. We and others have adapted high-pressure liquid chromatography (HPLC) to directly quantify 2-OH-E+ and have extensively validated this approach as a way to quantify O2 •− in biological systems (58). Careful use of specific wavelengths of excitation might allows separation of these signals, so that confocal imaging or other fluorescence-based approaches could be used (45).
Dichlorodihydrofluorescein fluorescence
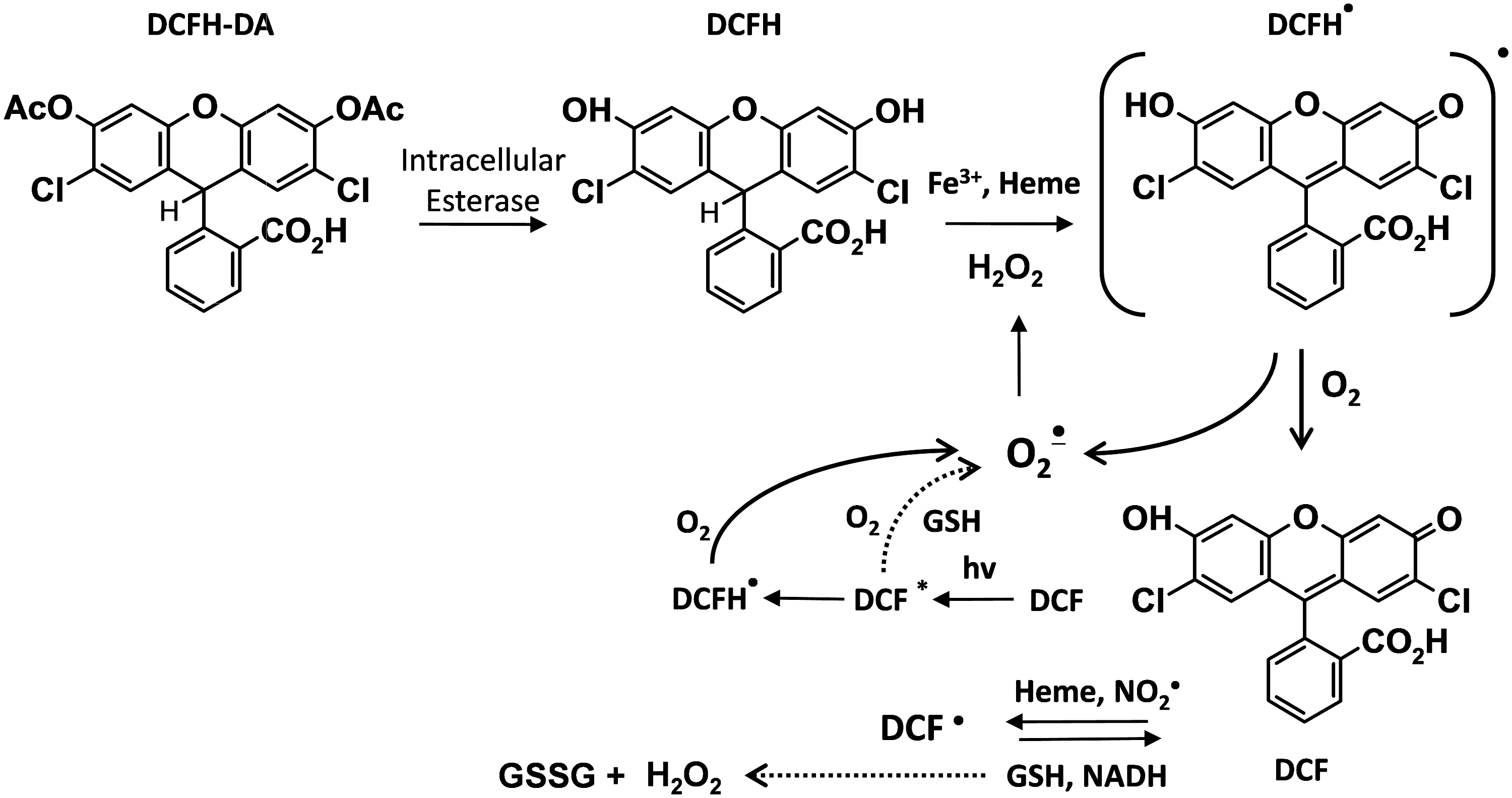
Dichlorodihydrofluorescein diacetate (DCFH-DA) is commonly used for detecting intracellular H2O2 (28). DCFH-DA is a cell-permeable ester and is hydrolyzed inside the cell to the dihydroxy-DCFH, which is retained (Fig. 1). Despite the popularity of this assay, it cannot be reliably used to measure intracellular H2O2 and other ROS for the following reasons: (i) DCFH does not directly react with H2O2; (ii) several one-electron oxidizing species will oxidize DCFH to DCF; (iii) DCF can actually produce O2 •− and H2O2 via reaction of DCF radical with the oxygen, thus artificially elevating the very ROS that it is attempting to quantify; (iv) transition metals, cytochrome c, and heme peroxidases can catalyze DCFH oxidation (32). For these reasons, the editorial board of Free Radicals in the Biology and Medicine journal stated that this agent should not be used as a reliable measure of H2O2.
Dihydrorhodamine fluorescence
Dihydrorhodamine (DHR) is commonly used for detection of ONOO•− (30). This assay is based on the oxidative conversion of DHR to its corresponding two-electron oxidized fluorescent product, rhodamine. DHR oxidation to rhodamine is not only caused by ONOO•−. The oxidative conversion of DHR to rhodamine is mediated by an intermediate DHR radical that can be reduced by thiols and ascorbic acid, leading to false-negative data. It is therefore concluded that DHR can only be used as a nonspecific indicator of intracellular ONOO•− and HOCl or other one-electron oxidant (52).
Detection of extracellular H2O2 by Amplex Red
The N-acetyl-3,7-dihydroxyphenoxazine (Amplex Red) assay, developed by Molecular Probes, is based on the horseradish peroxidase-catalyzed oxidation of the nonfluorescent molecule Amplex Red to resorufin, which when excited at 530 nm emits light at 590 nm (57). Horseradish peroxidase reacts with H2O2, generating peroxidase compound I, which produces resorufin on an equimolar basis (Fig. 2). This assay is highly specific and sensitive (63). We have used it extensively in isolated vessels and cells.
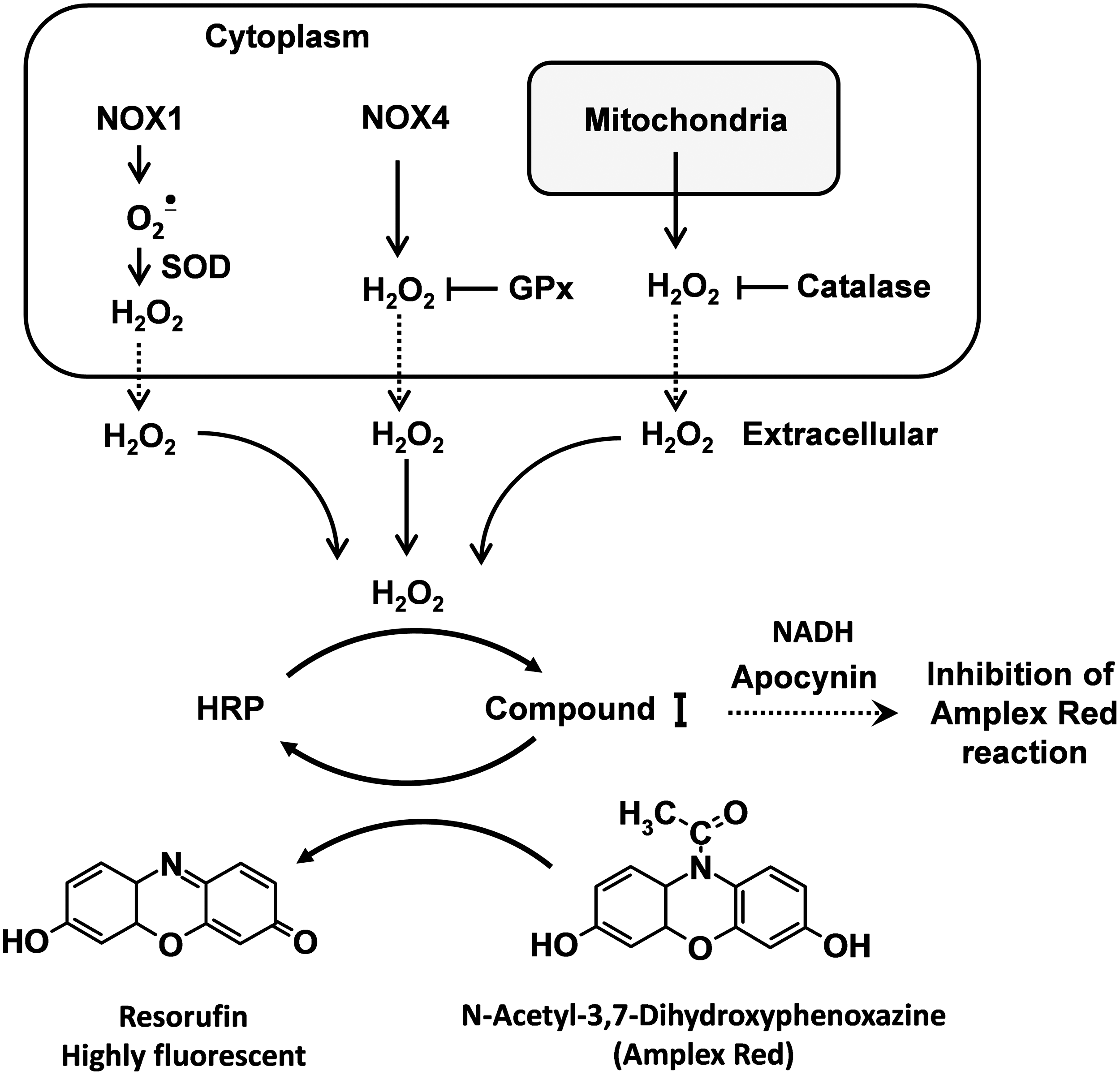
Amplex Red can be used to detect H2O2 released from isolated mitochondria (43). Superoxide produced in the mitochondrial matrix is quickly dismutated by SOD2 to H2O2, which readily diffuses through mitochondrial membranes to the surrounding medium.
A caveat for use of Amplex Red is that O2 •−can react with peroxidase either in its basal state or when the peroxidase is in the compound I or II state, and thus can lead to formation of compound III. These reactions can alter the stoichiometry of H2O2 detection. It is therefore not possible to quantitatively measure H2O2 by Amplex Red in the presence of substantial fluxes of O2 •−. This problem can be remedied by addition of Cu,Zn-SOD to the assay. This eliminates this problem and likely minimally affects the ultimate result, as O2 •− spontaneously dismutes to H2O2 even in the absence of SOD at a slightly slower rate.
The Amplex Red method may be affected by potential interference with reducing agents such as apocynin and NADH (Fig. 2), auto-oxidation of Amplex Red dye, light sensitivity (54), and inability to directly assess the intracellular H2O2 (14). The Amplex red dye is somewhat unstable. At high concentrations (50 μM), it can be auto-oxidized and produce O2 •− and H2O2. Low concentrations of Amplex Red (10 μM) minimize this problem. Amplex red does not detect intracellular H2O2. Because H2O2 is diffusible, it reaches the equilibrium with the tissue's surrounding buffer, so that values measured in the buffer should provide an index of what was originally produced by the tissue.
Superoxide detection by cytochrome c
Ferricytochrome c reduction is a time-honored and accurate method for detecting large amounts of O2 •− released by cells into the extracellular space, by isolated enzymes or by various chemical reactions. This assay however has limitations that prohibit its use in many physiological preparations. It is based on reduction of ferricytochrome c by O2 •− to ferrocytochrome c. This reaction can be followed by the spectrophotometric absorbance at 550 nm. The absorptions at the neighboring wavelengths of 540 and 560 nm are not altered, and therefore are used as isosbestic points to normalize the signal at 550 nm. Electrons donated from enzymes and other molecules can directly reduce ferricytochrome c, and the resultant change in absorbance is not specific for O2 •−. For this reason, the reduction due to O2 •− must be calculated by performing simultaneous assays with SOD added and determining the SOD-inhibitable signal. Cytochrome c reduction is suitable for quantifying O2 •− released during the respiratory burst of neutrophils or by isolated enzymes; however, it is difficult to detect the smaller quantities of O2 •− generated by nonphagocytic cells such as vascular smooth muscle cells and endothelial cells. Ferricytochrome c, being a large protein, does not gain access to the intracellular space, and therefore cannot be used to detect O2 •− released in the cytoplasm or by the mitochondria of intact cells. Finally, reactions of cytochrome c with H2O2 and various commonly used drugs such as oxypurinol, apocynin, and L-NAME can attenuate its reduction and cause artifacts in O2 •− detection.
New Methods for ROS Detection
Detection of cellular and mitochondrial O2 •− using DHE and mitochondrion-targeted probe mitoSOX
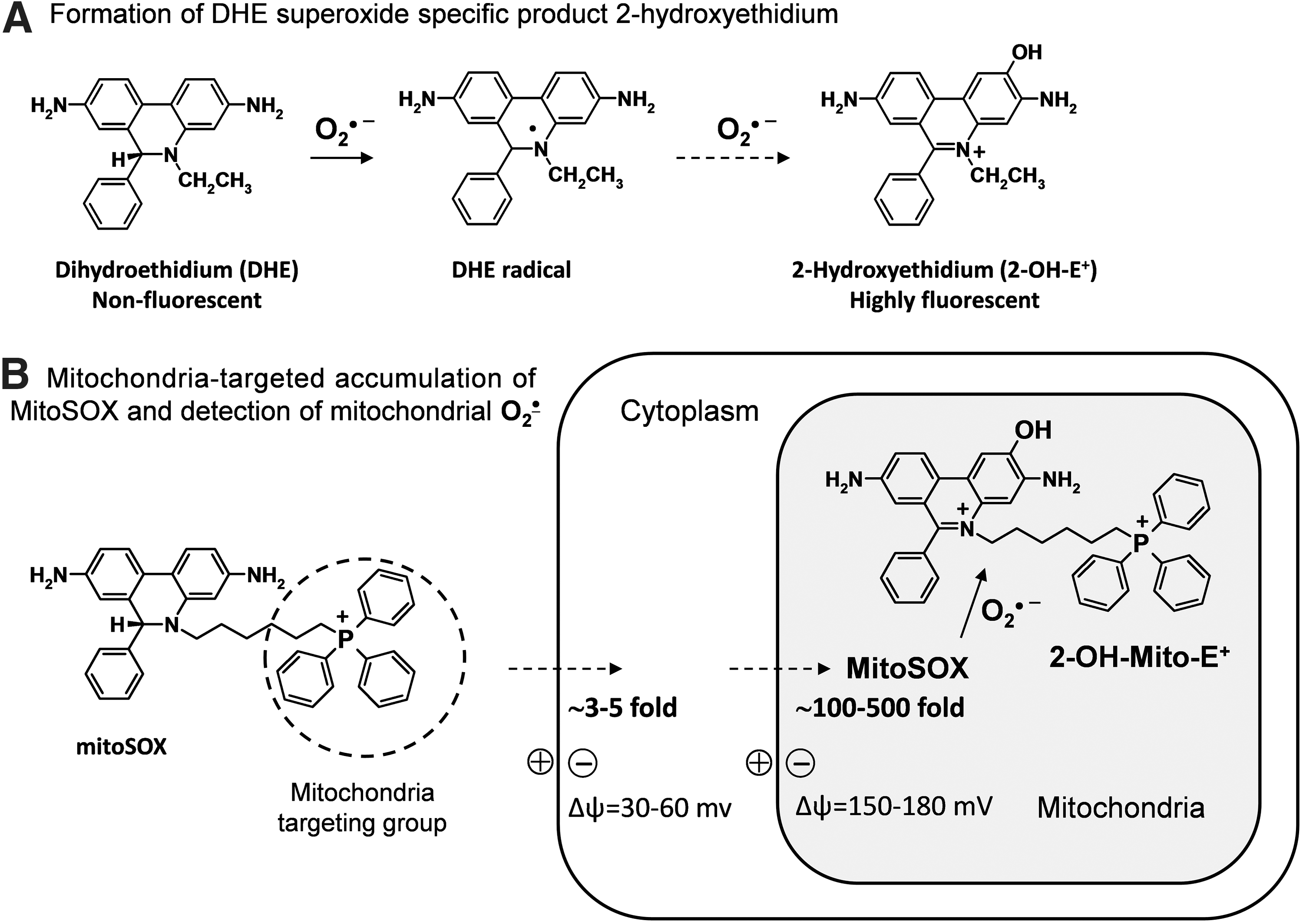
In 2003, Kalyanaraman and colleagues reported formation of a superoxide-specific product from the reaction of DHE with O2 •− (Fig. 3A) (56). Following this original report, DHE has been modified to allow detection of O2 •− in the mitochondria by addition of a triphenylphosphonium group, which promotes its accumulation in the mitochondria (14). This modified DHE analog is referred to as mitoSOX, and has become commonly used for detection of O2 •− within the mitochondria. Analogous to DHE, mitoSOX reacts with O2 •− to form 2-hydroxy-mito-ethidium (2-OH-Mito-E+) (Fig. 3B), which can be detected and quantified using HPLC (60). As an example of its specificity for mitochondrial-derived O2 •−, previous studies have shown that blockade of mitochondrial complex 3 with antimycin A increases the conversion of mitoSOX to 2-OH-Mito-E, but has no effect on conversion of DHE to 2-OH-E+. In contrast, stimulation of cytoplasmic O2 •− using phorbol-12-myristate-13-acetate (PMA) increases 2-OH-E+ formation from DHE, but does not increase formation 2-OH-Mito-E+ from mitoSOX (Fig. 4) (19). These data confirm the specificity of mitochondrial O2 •− detection by mitoSOX and illustrate that DHE detects cytoplasmic O2 •−, but not mitochondrial O2 •−. Separation of O2 •− produced in either the mitochondria or the cytoplasm is therefore possible by HPLC measurements of 2-OH-E+ or 2-OH-Mito-E+.
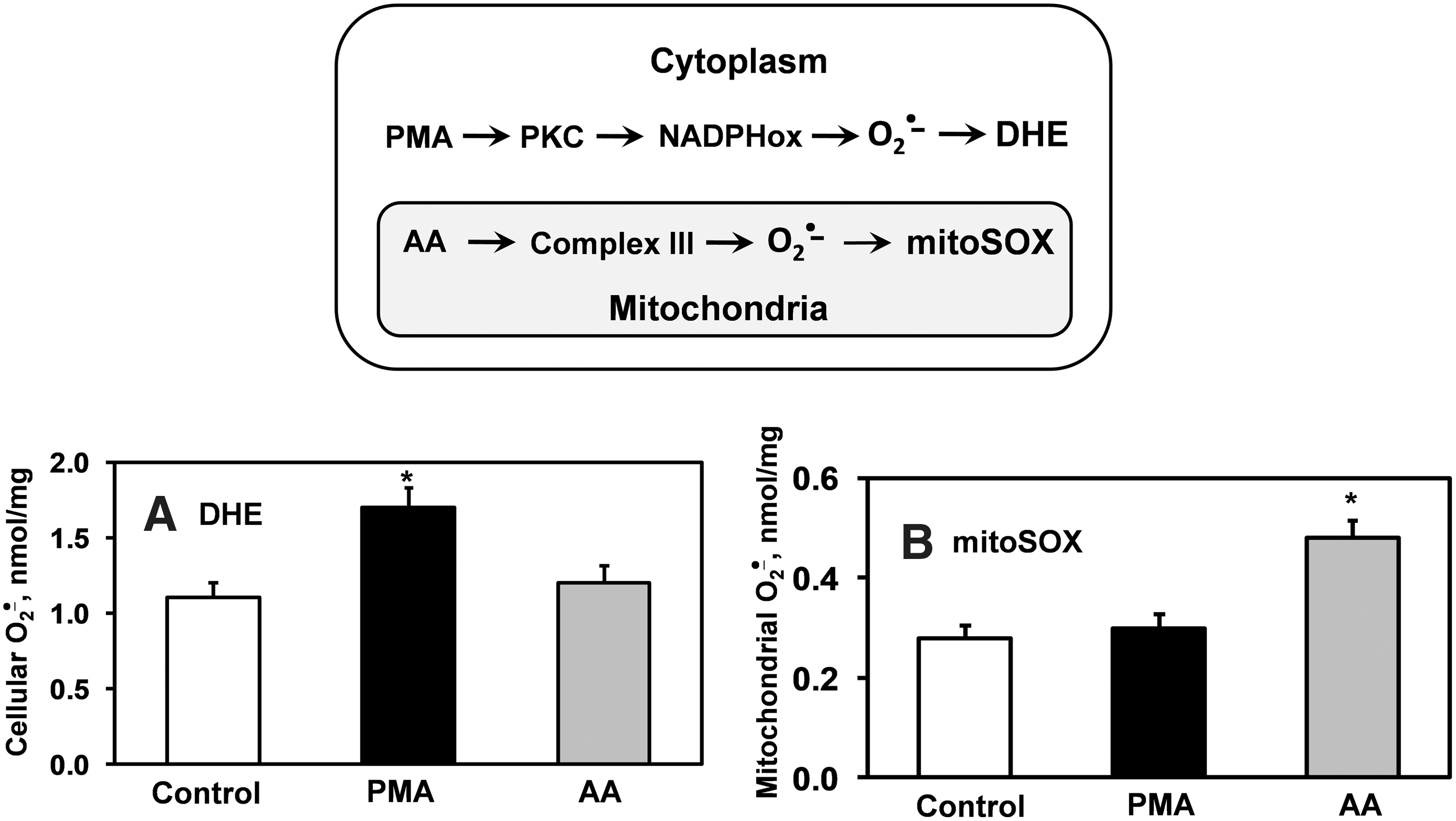
A caveat to use of mitoSOX is that high concentrations can overload the mitochondria and impair mitochondrial function (45). Furthermore, concentrations of mitoSOX exceeding 2 μM can lead to substantial cytoplasmic accumulation of mitoSOX and thus can compromise mitochondrial specificity of O2 •− detection. We therefore suggest using mitoSOX at concentrations 2 μM or less to avoid these complications.
While measurements of 2-OH-Mito-E+ are most accurately achieved by HPLC (61), Beckman and colleagues have reported that mitochondrial O2 •− can be accurately quantified in live cells using selective excitation at 385–405 nm and detection at an emission of 560 nm (45). These parameters seem to reduce signals derived from nonspecific fluorescent products. Thus, optimized fluorescence spectroscopy can be used for rapid and specific measurements of mitochondrial O2 •−; however, confirmation with HPLC analysis of mitoSOX samples is advisable.
Limitations of DHE and mitoSOX include instability of the probes and their products, complex chemistry, and potential interference with heme enzymes (60). These probes are light sensitive and are prone to auto-oxidation. They require a two-step reaction to detect O2 •− involving a free-radical intermediate (62) that can be potentially a subject for reaction with the antioxidants. Finally, formation of O2 •−-specific product can be affected by peroxidase reactions, which could compromise quantification of O2 •− measurements (21).
Detection of total cellular and mitochondrial O2 •− by cyclic hydroxylamine spin probes
We and others have found that cyclic hydroxylamines (Fig. 5A) can be used for measurement of O2 •− in cultured cells, tissues, and in vivo (13, 15, 17). These molecules are not spin traps in that they do not trap radicals, but are oxidized by O2 •− and other ROS to form EPR-detectable stable nitroxides with lifetimes of several hours in cell cultures.
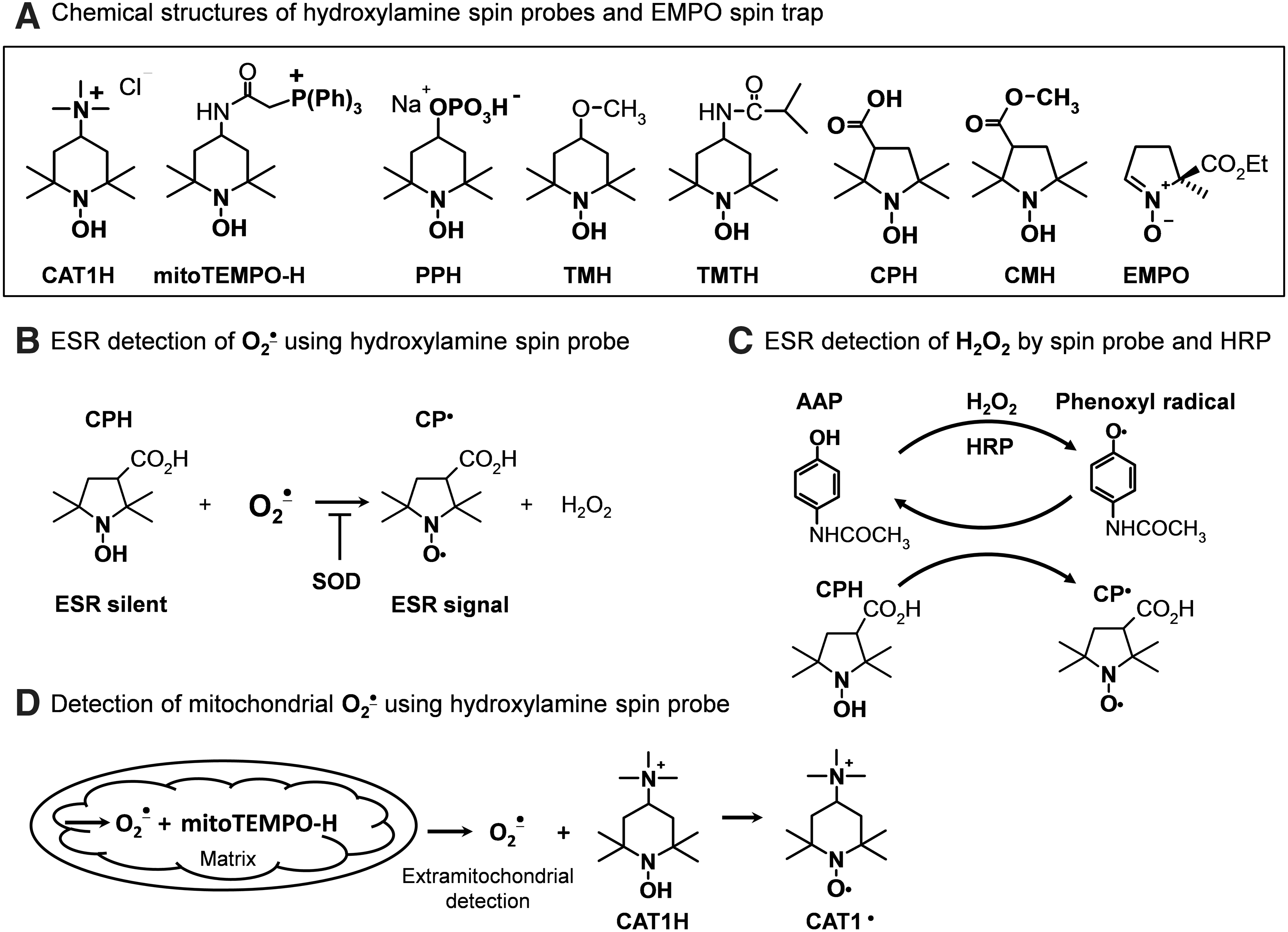
Cyclic hydroxylamines react with O2 •− with rate constants of ∼103–104 M −1s−1 (pH=7.4), which is much more rapid that the reaction of O2 •− with nitrone spin traps. This favors the competition of nitroxides with cellular antioxidants and enhances the efficiency for detection of intracellular O2 •− (Fig. 5B) (17). For this reason, hydroxylamine probes can be used at relatively low concentrations (0.05–1 mM), minimizing potential off-target effects of probes in biological systems. Another advantage of the cyclic hydroxylamines is that they react with O2 •− in a single chemical reaction. This minimizes potential artifacts that can occur with multistep redox reactions that occur with other probes (21). Unlike lucigenin or other probes, hydroxylamine is incapable of redox cycling, and therefore has no potential to artificially produce ROS (39).
There are number of cationic, anionic, and neutral hydroxylamine spin probes that have varying degrees of lipophilicity and cell permeability. By careful selection of these probes, one can separately detect intracellular ROS, extracellular ROS, or both. For example, the positively charged 1-hydroxy-2,2,6,6-tetramethylpiperidin-4-yl-trimethylammonium (CAT1H) detects only extracellular O2 •− released from intact cells and tissue or extramitochondrial O2 •− released from isolated mitochondria. In contrast, 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine (CMH) is highly cell permeable and can detect both intracellular and extracellular ROS. The phosphate-containing negatively charged 1-hydroxy-4-phosphono-oxy-2,2,6,6-tetramethylpiperidine (PPH) accumulates inside cells, presumably via active transport. The cell-permeable 1-hydroxy-3-carboxy-2,2,5,5-tetramethylpyrrolidine (CPH) and CMH accumulate in the cytoplasm. CMH also accumulates in mitochondria. Mito-TEMPO-H is a modified nitroxide that is analogous to mitoSOX, in that it contains a triphenylphosphonium group, causing it to be concentrated within the mitochondria.
There is functional evidence to support that these probes partition selectively. Upon stimulation of cytoplasmic O2 •− with PMA, both PPH and CMH provide strong signals, while the signal with mito-TEMPO-H is weak. In contrast, stimulation of mitochondrial O2 •− with rotenone only increases the CMH and mito-TEMPO-H signals (Fig. 5D). Thus, judicious use of the hydroxylamine spin probes can provide unique information about site-specific production of ROS in the extracellular, intracellular, or mitochondrial compartments.
Despite significant differences in the chemical structures, these probes have comparable rates of oxidation in response to O2 •− generated by xanthine and xanthine oxidase (18). It is important to note that conversion of cyclic hydroxylamines to their respective nitroxide radicals is not specific for O2 •−, but can be mediated by peroxynitrite and likely other oxidants. Thus, it is necessary to use specific antioxidants to probe the precise nature of the ROS involved in creating the nitroxide radical signal. As an example, overexpression of SOD2, which is targeted to the mitochondria, inhibits the CMH and mito-TEMPO-H signals caused by rotenone (18). In other cases, polyethylene glycol (PEG)-SOD has been found to reduce nitroxide radical production by 70% to 98% (18). Uric acid can be employed as a semiselective scavenger of peroxynitrite (37).
In general, there is no reaction between H2O2 and the cyclic hydroxylamines; however, H2O2 can be detected in isolated mitochondria and other subcellular fractions (Fig. 5C) using horseradish peroxidase mediated co-oxidation of CAT1H (20) or CPH (16).
A distinct advantage of the cyclic hydroxylamines is that they can be used to directly monitor cellular, subcellular, and tissue production of ROS online at room temperature by following the accumulation of nitroxide radical using an ESR spectrometer. For this, we generally run a field scan, and detect the precise g-value of the low field peak, and set the scanner to follow this g-value over time. Parallel experiments using added SOD can confirm that the signal is derived from O2 •−. Low-temperature ESR is useful for detection of ROS by intact tissues. For this purpose, samples are incubated in physiological saline containing cyclic hydroxylamines and subsequently frozen in liquid nitrogen, stored at −80°C, and analyzed later at a low temperature in quartz dewar (14).
For detection of ROS using virtually any assay, it is essential that one eliminate signals artificially generated by transition metals that often contaminate physiological buffers. This is particularly true for cyclic hydroxylamines. We have found that premixing of buffer with chelex overnight is very helpful as is the addition of deferoxamine or DTPA.
Detection of cytoplasmic and mitochondrial H2O2 by fluorescent protein-based redox probes
Recently, a revolutionary approach has been employed for detection of ROS and redox status involving the introduction of either plasmids or adenoviruses into cells. The cells then produce chimeric proteins that are capable of detecting ROS or changes in the redox status (4, 25). One such probe is HyPer, which consists of circularly permuted yellow fluorescent protein (cpYFP) inserted into the regulatory domain of the prokaryotic H2O2-sensing protein, OxyR. By molecularly recombining various signal peptides or retention sequences with HyPer, this probe has been successfully targeted to various intracellular organelles, including the nucleus, the cytoplasm, peroxisomes, the mitochondrial intermembrane space, and the mitochondrial matrix. Using these various constructs, Malinouski et al. have provided evidence supporting the presence of low levels of H2O2 in these subcellular compartments. An exception to this is the endoplasmic reticulum, where HyPer was found to be predominantly oxidized (38). This finding suggests that there is a previously unrecognized large flux of H2O2 in the endoplasmic reticulum, and might reflect the need to generate H2O2 to promote disulfide formation and protein folding in this compartment of the cell. A potential advantage of HyPer is that its fluorescence is reversible, and thus once this is loaded into cells, it can be used to follow varying fluxes of peroxide over time. This is in contrast to many commonly used fluorescent dyes, such as DCF, which are permanently activated by a pulse of H2O2. Mitochondrial-targeted redox-sensitive yellow fluorescent protein pHyPer-dMito has been used to analyze H2O2 in the mitochondria of apoptotic HeLa cells (4).
The fluorescence of HyPer can be affected by the cellular redox status and by reduction of the GFP by thioredoxins (26). To avoid these issues, Dick and colleagues have designed new probes in which a H2O2 sensor (Orp1) is conjugated to a redox sensitive reporter (roGFP2) (25). Orp1 is oxidized by H2O2, and the oxidized Orp1 then accepts an electron from cysteine residues on the roGFP2, which then emits fluorescence. This roGFP2-Orp1 construct seems to be a reliable H2O2 sensor that does not depend on the cellular redox status. This roGFP2-Orp1 redox probe may represent one of the most advanced and promising tools for specific, quantitative, dynamic, and compartment-specific H2O2 detection. roGRP2-Orp1 has also been modified to allow mitochondrial targeting.
A second chimeric construct in which roGFP2 is linked to glutathione reductase has been developed for measurement of glutathione redox potential (42). Detailed protocols for the use of these probes in both yeast and mammalian systems have been presented (42).
Although these genetically encoded redox probes can be used for quantitative in vivo mapping of the glutathione redox potential and H2O2 in the cytosol and mitochondria, overexpression of these GPx-like proteins can significantly increase H2O2 scavenging, and thus alter cellular redox signaling on their own (3). Appropriate control experiments are therefore needed to show that antioxidant side effects are not occurring as a result of the use of these probes.
Detection of H2O2 and ONOO•− by boronate-based fluorescent probes
Chang and colleagues have recently synthesized a family of boronate-based probes, including red-fluorescent peroxyresorufin-1, green-fluorescent peroxyfluor-1, and blue-fluorescent peroxyxanthone-1 (9, 41). These fluoresce in the red, green, and blue ranges, respectively, upon exposure to H2O2. These probes possess a fluorophore that is protected by boronate, and upon exposure to H2O2, the boronate undergoes a nucleophilic attack, leading to its removal from the fluorophore and allowing light emission (Fig. 6). These boronate probes are cell permeable and can detect changes in H2O2 in the micromolar range in living cells (41).

These investigators have also synthesized a boronate-based peroxy-yellow 1 probe containing a triphenylphosphonium group, again analogous to mitoSOX, which allows selective detection of H2O2 within the mitochondria (MitoPY1, Fig. 6). This new bifunctional fluorescent probe permits imaging H2O2 within the mitochondria of living cells using imaging techniques such as confocal microscopy and flow cytometry (12).
Intracellular O2 •− normally converted to H2O2 by SODs, but under inflammatory condition and oxidative stress, overproduction of O2 •− leads to very fast reaction with nitric oxide, producing peroxynitrite (ONOO−). The local ratio between NO and O2 •− has a significant impact on the formation, where ONOO•− formation is maximal at the NO/O2 •− ratio 1: 1 and is significantly declined when one of the two radicals is present in excess (59). Kalyanaraman and colleagues have recently described boronate probes that react stoichiometrically with ONOO−, yielding corresponding phenols (47, 59). One example, coumarin-7-boronic acid (CBA, rapidly reacts with ONOO•− with a rate constant of 1.1×106 M−1s−1 (Fig. 6). Using CBA and fluorescein dimethylamide boronate (FlAmBE), this group has studied ONOO•− production in endothelial cells and macrophages (63). It seems that the reaction of ONOO•− with boronate leads to a site-specific nitration of an aromatic moiety that can be followed by either HPLC or mass spectrometry (Kalyanaraman, Oxygen Radicals Gordon Conference, 2012). It is quite likely that boronate probes will provide a more reliable approach for detection of OONO− than assays using either dichlorodihydrofluorescein or DHR (31).
As suggested above, there is a potential problem related to the lack of specificity of boronate probes that can detect either ONOO•− or H2O2 (Fig. 6). The use of an ONOO•− scavenger such as uric acid or an H2O2 scavenger such as PEG-catalase might help increase specificity of boronate probes, but further studies are needed to validate and perfect such approaches.
Immuno-spin trapping
During the past decade, a new immunospin-trapping method has been developed based on the concept that DMPO will react with protein radicals, forming epitopes that can be specifically identified immunologically. Mason and colleagues have developed a panel of antibodies that reacts with DMPO–protein radical adducts and have shown that they work well for Western blot analysis, immunostaining, and immunofluorescence and even flow cytometry (44). Immunological techniques can be highly sensitive, and thus this method greatly expands the utility of the spin trapping. Moreover, this approach makes use of DMPO and potentially other spin traps possible without ESR, which, as discussed above, is of limited availability, often lacks sensitivity, and has limited applicability (40, 44). In initial studies, anti-DMPO has been used to detect DMPO–protein adducts of myoglobin and hemoglobin that occurred as a result of self-peroxidation by H2O2 (11). Immunospin trapping has been used to detect free-radical adduct formation in the mitochondria, cells, and tissue samples (24). Of note, DMPO adducts of components of mitochondrial electron transfer have been detected using immunospin trapping (10). It is likely that this approach can be widely used to examine specific targets of oxidative attack within cells and subcellular organelles. A limitation of this approach is that it does not detect free radicals such as O2 •− or OH•, but identifies modified protein adducts. It is also unclear if it will detect all radical protein adducts, and perhaps additional antibodies will need to be produced to provide broader sensitivity.
ROS Detection In Vivo Using X- and L-Band ESR Spectroscopy
While there are several approaches to detection of ROS ex vivo, a major challenge has been to detect these in vivo. It has been previously shown that O2 •− production can be detected in vivo by infusion of either nitrone spin traps (65) or cyclic hydroxylamines, followed by ex vivo analysis of the blood or tissue samples using X-band (9 GHz) ESR spectroscopy (13, 22, 35). Over the past two decades, substantial effort has been made to visualize radicals using spin probes and L-band ESR spectroscopy. X-band spectroscopy, which is most commonly used, is generally not suitable for this purpose. First, most cavities supplied for X-band will not accommodate experimental animals or even significant amounts of living tissue, but more importantly, the large amounts of water present in such samples absorb most of the microwave energy in the X-band range. L-band (1 GHz) spectroscopy overcomes some of these limitations and can provide special resolution for analysis of living tissues in online experiments (48). L-band ESR can be used for in vivo detection of short-lived free radicals in whole living animals (29). This method has been used to monitor tissue oxygen levels (2), pH, redox status, and glutathione levels (6). A downside of L-band spectroscopy is that these lower-frequency microwave energies reduce sensitivity substantially, and therefore detection of radicals such as O2 •− is limited. Efforts are being made to employ nitroxides to detect radicals in vivo with L-band (46, 50); however, these have not been successful in detecting the small amounts of radicals that are important in common pathophysiological conditions.
Antioxidant Activity of ROS Probes
The discussion above has focused principally on the use of various probes to detect ROS; however, reaction of ROS with probes may have two implications: (i) significantly reduce the steady-state levels of O2 •−, H2O2 NO, or ONOO•− during the measurements, and (ii) these agents can be of interest as antioxidants (49). Our data showed dependence of ROS detection on DHE or spin probe concentrations; therefore, one should avoid saturation of biological samples with the ROS probe to avoid side effects. Generally, it is a good idea to measure ROS by two independent methods and monitor different species in the same samples. A wide variety of spin traps and spin probes such as DMPO, PBN, TEMPOL, and mito-TEMPO protect the brain, heart, and vascular tissue under conditions of oxidative stress (8, 19, 64). Beneficial responses to these agents have been used to implicate ROS in various pathophysiological conditions.
Conclusions
Historically, studies of ROS and free radicals were strictly confined to the fields of chemistry and physics. It has however become increasingly clear that these fleeting molecules play critical roles in both normal physiology and pathophysiology. This recognition has made it essential that tools are available that allow their detection and measurement in complex biological samples, including cells, tissues, and eventually intact animals. Many initial methods such as cytochrome c reduction and spin trapping were suitable for high level of ROS production (e.g., activated neutrophils) (53); however, studies of low levels of intracellular ROS require new more sensitive methods (14). In the past decade, new methods for site-specific detection of ROS have been developed and validated, which are helping to fulfill this goal. These new methods include use of the boronates, immunospin probes, and mitochondrial-specific probes that can be adapted to other approaches such as confocal imaging, live-cell imaging, Western analysis, flow cytometry, and HPLC methods commonly employed in research laboratories. We therefore view these developments with substantial optimism with the hope that these approaches will continue to be validated and more widely used.
Footnotes
Acknowledgments
This work was supported by funding from National Institutes of Health grants PO-1 HL058000 and RO-1 HL094469.