
Abstract
Introduction
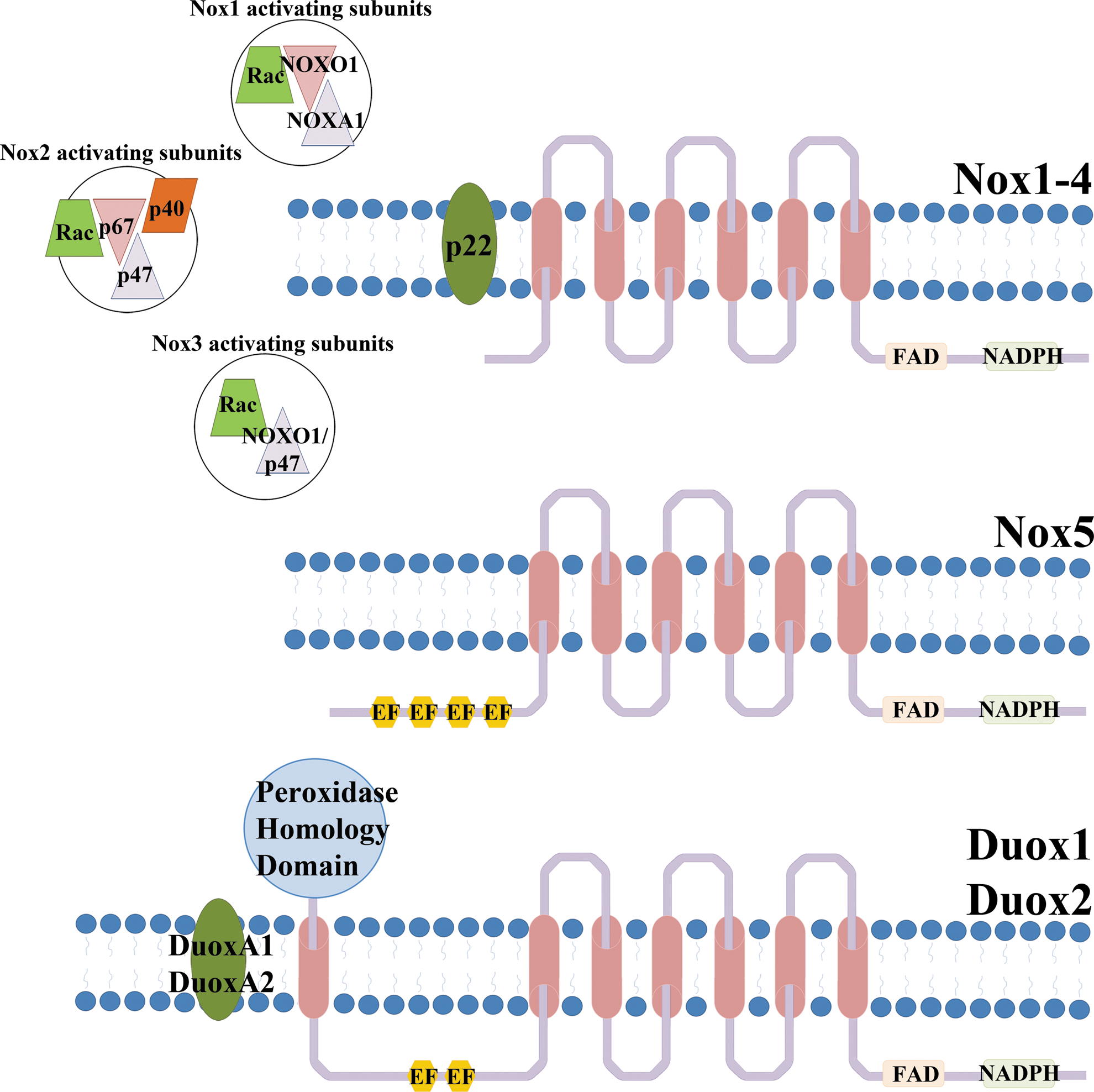
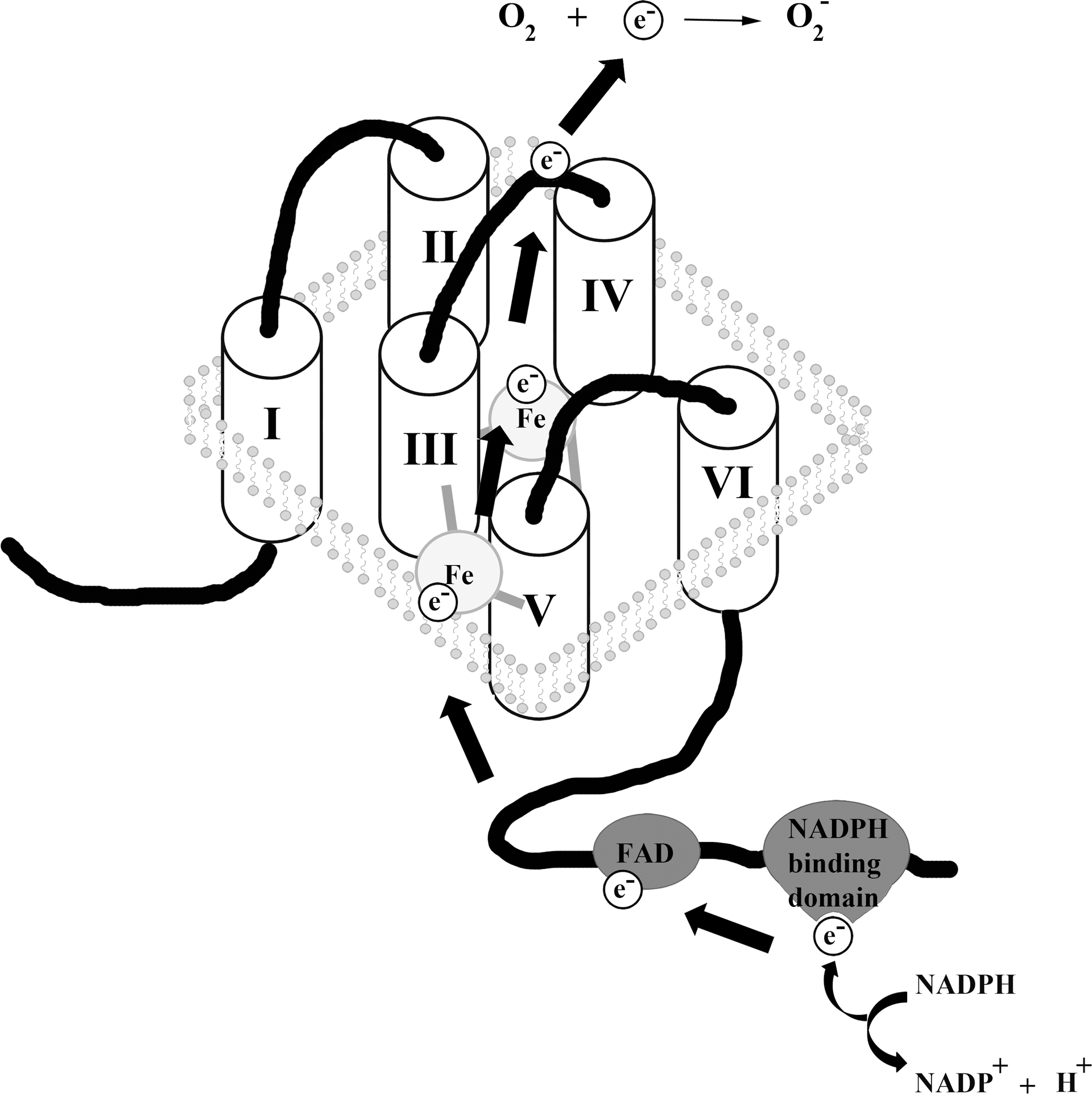
The Nox/Duox family has wide tissue distribution, ranging from the innate immune system, kidney, brain, thyroid, and skeletal muscle to testis and inner ear (32). Almost all cell types and tissues express at least one Nox, either constitutively or inducible, with many cells harboring several oxidases. The subcellular localization of these enzymes, when active, is usually the plasma membrane or membranes forming specialized vesicles (redox signalosomes). Unusual locations, such as mitochondria and nuclear compartments have been reported (33, 55). Interestingly, not all of the Nox/Duox enzymes release the same ROS. For instance, Nox4 and the Duox enzymes seem to generate only hydrogen peroxide in comparison to the superoxide that the other oxidases produce. This is unusual considering the shared catalytic domain structure of these enzymes. The core structure of Nox is composed of six transmembrane domains, two of which contain histidine residues required for heme coordination. The loops between these membrane-embedded segments contain regions that are required for assembly and activity of the oxidase. On the intracellular side, the carboxyl terminus harbors the NADPH and FAD binding domains. The electrons they accept are passed on via the hemes to the outer membrane, where they will reduce molecular oxygen to superoxide (5) (Fig. 2). It is unresolved how hydrogen peroxide can be produced directly by Nox4 and Duox enzymes. Some Nox/Duox family members display distinct features, such as calcium sensitivity, due to the presence of calcium-binding EF hands, a motif commonly found in calcium-binding proteins, in their amino termini (Nox5, Duox1–2) or by being apparently constitutively active (Nox4).
The Contribution of Nox Enzymes to Cellular Signaling
Although the best characterized oxidase, the phagocyte oxidase (Nox2), is known for its critical role in host defense, the other members of the Nox/Duox family seem to be more involved in cell signaling. Several publications have placed Nox4 within the TGF-β signaling cascade and Nox4-deficient mice are resistant to TGF-β-driven, bleomycin-induced lung fibrosis (9, 27, 65). In vascular smooth muscle cells, Nox1 and Nox4 are the predominant Nox. They exhibit unique responses to stimulation with the platelet-derived growth factor or with angiotensin II. These stimuli upregulate Nox1, but not Nox4, resulting in a brief burst of superoxide (36). Reducing Nox1 expression downregulated the redox-sensitive p38 mitogen-activated protein kinase (MAPK) and Akt signaling pathways (36). These signaling pathways have also been associated with Nox1 in human and rat hepatoma cells (61). Herein, the increased signaling through the epidermal growth factor receptor correlated with increased Nox1 expression, again acting upstream of p38 MAPK and Akt. Nox4, on the other hand, can regulate the expression of antioxidant enzymes through activation of the Nrf2 transcription factor (8). Duox1-derived ROS modulated the expression of the mucin protein, MUC5A, in airway epithelia through a TGF-α signaling axis (64). Additionally, cross talk between the different Nox enzymes has emerged, with increased expression and activity of one oxidase being responsible for upregulation of another oxidase (14, 44). Considering the well-documented correlation between increased ROS generation and cancer, it is not surprising that dysregulation of Nox has been linked to tumorigenesis (54, 67). Although most of our current knowledge focuses on upregulation of activated NADPH oxidases (Nox1, Nox4) in cancer, examples exist where epigenetic silencing of oxidases (Duox1, Duox2) and thus diminished ROS generation appears to be an early event during tumor development and progression.
The Involvement of Nox Enzymes in Carcinogenesis
In a physiological cellular setting, evidence suggests that Nox1 and Nox4 may be necessary for replicative senescence in tissues, where they are highly expressed such as the lining of the blood vessels, the stomach, and the colon (4, 35). In these tissues, a state of senescence may be triggered by ROS as part of an antiangiogenic program. This enforced growth arrest may act as protection from transformation. This observation has been reported for both Nox1 and Nox4 in endothelial cells and in thyrocytes transduced with oncogenic H-Ras (62, 72). However, attempting to attach a candidate tumor suppressor designation to these proteins seems too facile. Clearly, these proteins exert cell signaling functions related and necessary to normal cell physiology and in some context these functions coincide with the terminal differentiated cell phenotype. However, it appears that Nox enzymes can find themselves co-opted in pursuit of a tumorigenic phenotype in various stages of transformation.
The contribution of Nox enzymes to carcinogenesis has been described for several types of cancer. For instance, Nox4 was reported to be at least partially responsible for the transformation of melanoma cells, and inhibition of Nox4 prevented the development of endothelial tumors in nude mice and restricted the invasiveness of gliomablastoma in rats (6, 48, 75). The p22 phox subunit, required for Nox1–4 activity, has been implicated in the inactivation of the tumor suppressor tuberin in renal carcinoma cells (7). Fibroblasts, engineered to overexpress Nox1, produced tumors when introduced into nude mice (66), suggesting a mitogenic function for Nox1. A close relationship between increased expression of Nox1 and activating mutations of K-Ras was uncovered in a screen of human colon cancer tissues (37, 62). The same report detected increased levels of Nox1 in a transgenic mouse expressing oncogenic K-RasG12V in the intestinal epithelium. While this may appear to contradict the H-Ras-induced senescence associated with Nox1 and Nox4 mentioned earlier (62, 72), H-Ras and K-Ras seem to exert distinct effects with H-Ras inducing growth arrest in lung and colon and K-Ras promoting increased proliferation (46, 57). In general, excessive ROS generation is thought to increase the mitogenic potential of tumors transformed with oncogenic Ras (46). Knockdown of Nox1 in rat kidney cells expressing constitutively active K-Ras resulted in a change from a transformed phenotype toward a more normal cell behavior. As mainly Nox1 and Nox4 have been connected to enhanced ROS production in tumors, these oxidases are likely the source for ROS as a means to produce epigenetic changes in other tumor-associated genes.
Duox enzymes are known for their involvement in thyroid hormone biosynthesis. Genetic mutations, such as substitutions and deletions causing frame shifts often cause loss of Duox2-DuoxA2 activity, leading to hypothyroidism (25, 52). An inverse correlation between Duox expression and thyroid cancer was detected. While the mRNA levels of Duox1 and Duox2 displayed no change between normal tissue and thyroid tumors, over half of the tissues showed decreased Duox protein expression in thyrocytes (34). A report evaluating Duox activity in thyroid nodular lesions found a decrease in Duox activity in these tumor tissues (22). Similarly, Duox1–2 enzymes are highly expressed in healthy airway epithelia (18, 21), but their expression is epigenetically silenced in lung cancer (42). The frequent and early downregulation of Duox in diverse lung tumors suggests an important physiological role for Duox in maintaining a fully functional lung epithelial barrier as discussed in following chapters. In contrast to these observations, colon adenoma microarray analysis demonstrated significantly upregulated expression of Duox2 message in tumor versus adjacent normal tissue (31). Elevated expression of Duox1 and Duox2 mRNA in hepatocellular carcinoma specimen was linked to poor prognosis (41).
This review is focused on Nox as targets of epigenetic changes in cancer and on the emerging role of ROS as inducers of epigenetic changes. As the examples demonstrate, localized ROS within the cellular environment are powerful signaling molecules, and one can assume that increased concentrations of ROS will have effects upon redox-sensitive epigenetic modifications.
A Brief Introduction to Epigenetic Modifications
Epigenetic modifications comprise methylation of cytosines (at sites referred to as CpG islands) and a range of modifications of the amino terminal tails of histones, including phosphorylation, acetylation, methylation, ubiquitination, and sumoylation.
Methylation of DNA does not occur uniformly across the genome and is typically found to favor promoter regions of the genome with approximately half of all promoters containing CpG islands (12). Retrotransposons and repeated sequences account for the majority of the remainder. However, the epigenome is not fixed in place and is responsive to environmental cues. In fact, the communication between these two systems can be as rapid as 4 h after exposure to airborne particulate matter (3), where long interspersed elements (LINE) were found to undergo a decrease in methylation. In fact, the same study found a correlation between methylation patterns and seasonal environmental changes for LINEs, thus indicating a high level of responsiveness in the epigenome.
Another level of modulating gene expression is through the acetylation of histones, which bind to and wrap the DNA into conformations that facilitate or inhibit gene expression. Typically, the acetylation of the N-terminal tails of the histone proteins will cause an increase in gene expression as the DNA is in a looser state called euchromatin, where transcription can proceed with more ease. The enzymes responsible for the addition of the acetyl residues to the histones are called histone acetyl transferases (HATs). Another family of enzymes termed histone deacetylases (HDACs) removes these acetyl residues, causing remodeling of the chromatin to an inactive state.
Epigenetic modifications are reversible and thus the epigenome is not static; rather it is dynamic and this plasticity relays information between the environment and the genome. Epigenetic changes may occur as a result of Nox/Duox-regulated signaling pathways. Alternatively, the Nox/Duox enzymes may themselves be subject to epigenetic regulation. Our work as discussed here focuses on epigenetic changes occurring in Duox enzymes.
Epigenetic Silencing of Duox Enzymes
As mentioned earlier, Duox1–2 enzymes are not only expressed in the thyroid, but also in mucosal barrier tissues and, in particular, on the apical side of the lung epithelium. Each Duox subunit associates with a particular partner protein, DuoxA1 and DuoxA2, to form a functional Duox complex. These complexes mature via the endoplasmic reticulum–Golgi pathway and translocate to the plasma membrane (24, 43, 47). The maturation factor DuoxA1 can be alternatively spliced to yield at least three different isoforms. These isoforms result from alternative splicing at two splice sites. These sites are to be found in the intron preceding exon 3 and within exon 6 (47). Variants lacking exon 3 are incapable of restoring oxidase activity when coexpressed with Duox1 (43, 47). The most active isoform, DuoxA1–2 (also termed DuoxA1α) is also capable of cross-functionality with Duox2 to produce ROS, but this association alters the cellular location of the active Duox2 enzyme (43).
The gene arrangement of the two Duox proteins and their respective maturation factors warrants discussion (Fig. 3). All four genes are found clustered within the same compact locus on chromosome 15 with Duox1 and Duox2 facing away from each other on the genome. This organization is conserved across a wide variety of species. However, the maturation factors are also arranged in a head to head configuration with their cognate Duox. It has been suggested that the intergenic regions between these pairs have a bidirectional promoter organization (11, 74). Linking the Duox1 and DuoxA1 genes is a highly GC-rich region containing many CpG islands. There is no TATA box, but the intergenic region contains binding sites for the transcription factor Sp1. The intergenic region between Duox2 and DuoxA2 is configured differently, displaying normal GC content. Only the DuoxA2 promoter region features a TATA box. As a result, any loss of expression occurring as a result of promoter hypermethylation is likely to affect all Duox-related proteins.

Loss of Duox1, DuoxA1, and Duox2 expression in lung cancer cell lines and lung tumor tissues by epigenetic silencing occurs frequently (42). In 9 out of 12 lung cancer lines and in ∼60% of the analyzed lung tumor samples, hypermethylation and silencing of Duox1–2 and DuoxA1 was observed (Fig. 4). Hypermethylation seems to take place throughout the bidirectional promoter regions. The expression and function of Duox1 and Duox2 was recovered in response to treatment with 5′-aza-2′-deoxycytidine. Often several modifications, methylation and histone deacetylation, are found layered upon the genome, acting as reinforcement for inhibition of expression. Interestingly, the sensitivity to DNA methylation appears to be the only epigenetic modification regulating Duox expression in lung tumors as treatment of lung cancer cell lines with trichostatin A, an inhibitor of histone deacetylation, did not restore Duox protein expression.

Given the localization of the Duox enzymes to barrier tissues and mucosal epithelial surfaces, it is important to consider their regulation in response to environmental factors. Downregulation of Duox enzymes was observed in smokers and patients with chronic obstructive pulmonary disease, conditions in which hypoxia is common (49). Epigenetic regulation is often a means of communication and interaction between the environment and the genome, and the presence or absence of Duox1–2 may represent an ideal model for further study of the mechanics of this interaction.
Epigenetic Modifications Affecting Nox in General
The current paucity of published information regarding epigenetic modification of Nox/Duox enzymes is not indicative of a lack of such regulation of these genes. On the contrary, vast amounts of CHiP data have been logged in databases through the ENCODE project and are available for perusal through sites, such as the human histone modification database (76). For instance, the histone modification pattern of Duox1 in lymph tissue has been logged into this database, originating from a large-scale screening (69). Close examination of the genes included in these screens will likely yield useful information about the modes of regulation these genes are subject to.
DNA methylation of the Nox5 gene has been linked to downregulation of Nox5 mRNA expression. In lung cancer cell lines, treatment with 5′-aza-2′-deoxycytidine caused at least a fourfold increase in Nox5 mRNA expression in four of seven lung cancer lines tested (63). Immortalized bronchial epithelial cells derived from a healthy donor showed comparable upregulation of Nox5 mRNA. However, the origin of these cells was an aged smoker. Thus, epigenetic silencing of Nox5 in lung cancer is still inconclusive and awaits more extensive studies. It is also worth mentioning that there are multiple isoforms of Nox5, including a short form reminiscent of Nox1–4 (20). The study mentioned here investigated the Nox5 short form, which is often found in cultured cell lines, but this Nox5 enzyme is not characterized well in respect of regulation and function. Considering the proximity of the Nox5 locus (15q23) to the Duox chromosomal arrangement (15q15–15q21) and the observed methylation-mediated transcriptional silencing of Duox, the implications regarding Nox5 silencing are interesting. It has been suggested that the Nox5 short isoform can bind to and inactivate the longer functional variants (53). Recently, hypermethylation of the Nox5 promoter was also observed in fetal ventricular septal defect (77).
The expression of the phagocyte oxidase (Nox2) appears to be subject to epigenetic regulation during the IFN-γ-mediated immune response. Treatment of myelomonocytic cells with this cytokine caused increased expression of both, Nox2 and p67 phox (17). This coordinated response ensures that a functioning oxidase complex will be produced, and arises from the fact that the promoters of these genes share a unique cis element that is necessary for the attachment of CREB binding protein (CBP), a HAT. HAT binding promotes the recruitment of other transcription factors to the promoters of these genes, allowing their increased expression (17).
The apparent effect of the environment on epigenetic changes is also reflected in increased cellular oxidative stress as a result of harmful environmental conditions. Diesel exhaust and cigarette smoke are well-known epigenetic modulators. In fact, cigarette smoke is capable of upregulating the expression of cytosolic phospholipase A2 via a Nox-dependent pathway that relies on p300/CBP HAT proteins (10). The effect of p300/CBP recruitment on Nox2 gene expression is sensitive to the effects of TNFα (38). Both of these effects were observed in human tracheal smooth muscle cells.
ROS Produced by Nox/Duox as Mediator of Epigenetic Changes
Excessive ROS production can alter gene expression by generating toxic DNA metabolites. Further, dependent on the concentration and species produced, protein function will be altered via post-translational modifications (oxidation, S-glutathionylation), demonstrating the far-reaching effects of these chemical moieties on gene/protein expression and functionality. It is not surprising therefore to discover that ROS exert changes on the epigenome (Fig. 5).
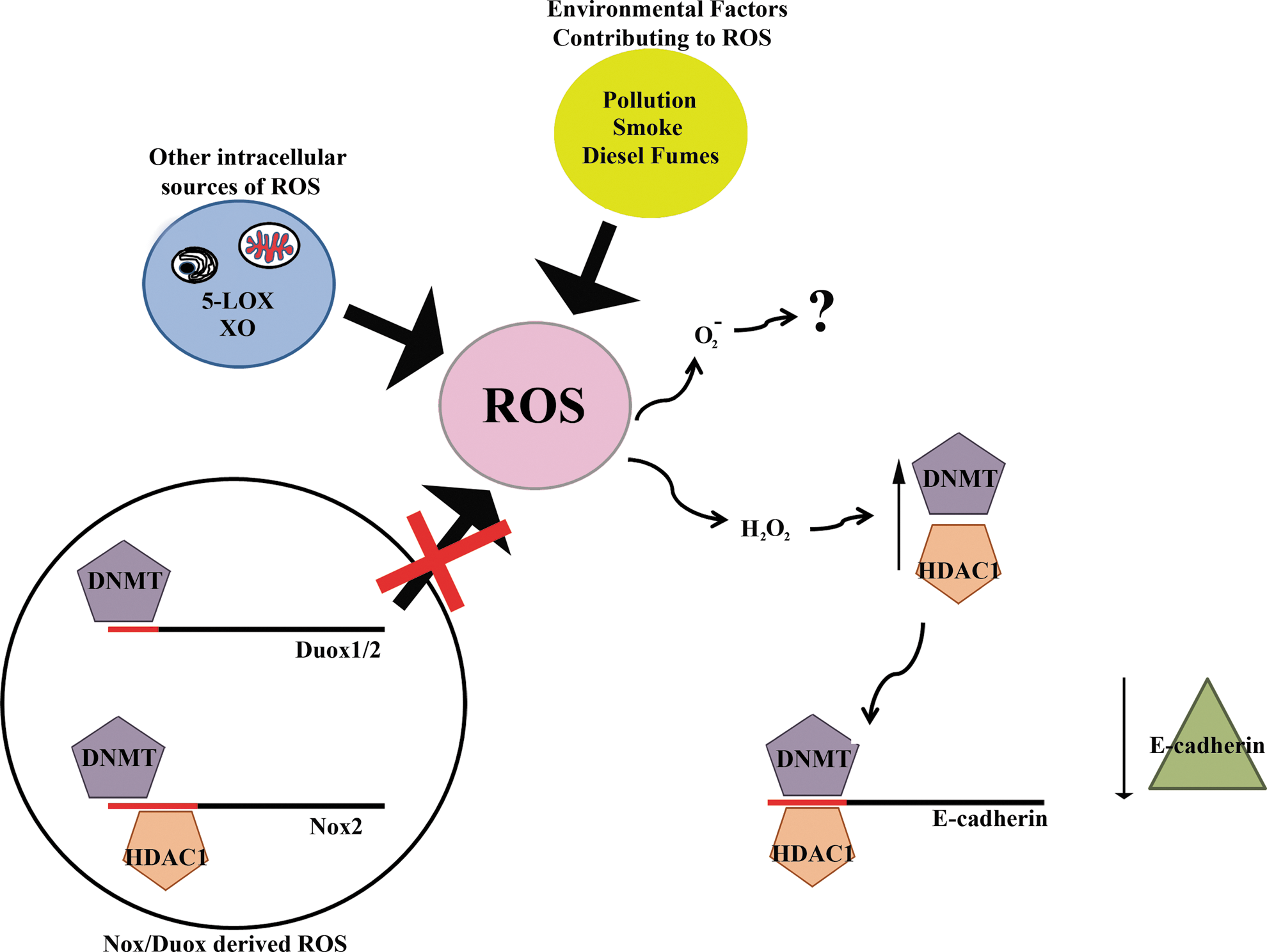
Lim and coworkers reported that ROS are capable of inducing changes in the methylation status of the E-cadherin promoter in hepatocellular carcinoma (40). E-cadherin is classified as a tumor suppressor and its silencing is a common carcinogenic event. This silencing is mediated through epigenetic modification of its promoter as opposed to inactivating mutations. Promoter hypermethylation and subsequent downregulation of E-cadherin expression was observed when hepatocellular carcinoma cells were exposed to H2O2, leading to the binding of the transcription factor SNAIL to regions within the E-cadherin promoter. The attachment of SNAIL appeared to act as a signal for recruitment of DNA methyltransferase 1 (DNMT1) and HDAC1 resulting in E-cadherin silencing. As the ROS source was either exposure to ROS or treatment with ROS-generating compounds, such as menadione or phenazine methosulfate, it is yet not apparent how this effect might be induced in a pathophysiological context. Although the expression of a range of antioxidant enzymes was measured in this study, including SOD1 and SOD2, only SOD1 protein expression was decreased. Hong and coworkers linked ROS generated by the Nox5 short isoform to the hypermethylation and subsequent suppression of the p16 tumor suppressor gene in oesophageal adenocarcinoma cells exposed to low pH (28). Another recent report examined the relationship between norepinephrine treatment and the repression of PKCɛ gene expression as a result of promoter hypermethylation (73). This link was found to be ROS-dependent, which the authors attributed to Nox1. The contribution of mitochondrial ROS was not sufficiently addressed, and the observed phenotype may, in fact, rely on more than one ROS generator. Indeed, there is evidence of cross talk between Nox and mitochondria (16, 70), where Nox-derived ROS seems to be partially responsible for the development of nitrate tolerance-associated endothelial dysfunction, a condition initiated by mitochondrial ROS
ROS-mediated epigenetic regulation can also be channeled through histone acetylation. Nickel ions are known to inhibit gene expression partly through inhibition of the acetylation of histone H4 through binding to an amino terminal histidine residue. Additionally, exposure of cells to nickel ions generates ROS, leading to inhibition of acetylation and subsequent gene suppression (29). However, the effect of ROS on epigenetic gene regulation is not straightforward. For example, thalidomide treatment increased acetylation of histone H4 via ROS (1). The study investigating the effects of nickel ions used human hepatoma cells, while the thalidomide experiments were performed in erythroid progenitor cells. The Nox expression profiles in these two cell types is likely quite different. This is an important point as under the ROS umbrella, a number of different species can be found. For example, cellular responses may differ depending whether superoxide or hydrogen peroxide is being generated. This question is especially relevant as some members of the Nox/Duox family are incapable of releasing superoxide, while others can generate both species. Recently, the effect of hydrogen peroxide on the expression of RUNX3, a transcription factor and known tumor suppressor, were investigated (30). Hydrogen peroxide induced increased expression and activity of DNMT1 and HDAC, thus causing epigenetic silencing of RUNX3. Analysis of expression and activation profiles of Nox together with probing carefully the type of ROS generated will give more insight in common principles of oxidative epigenome modulation.
The redox status of the cell is a finely tuned mechanism. In regard to ROS produced by an active Nox enzyme, a system of checks and balances exists to prevent or at least counteract nonspecific consequences. For instance, the expression of catalase is influenced by ROS through both direct and indirect means. The promoter regions of catalase and of Oct-1, one of catalase's transcription factors, contain CpG islands. Both genes are subject to hypermethylation upon exposure to H2O2, potentially a way to promote transformation by decreasing catalase expression during tumor development (45, 56).
While the examples given here suggest that ROS promote epigenetic changes, they do not address the mechanism(s) by which this occurs. How will oxidants whether Nox- or mitochondria-derived promote these changes? Redox regulation of DNMTs could explain ROS-induced silencing. In fact, O'Hagan and coworkers observed relocalization of a silencing complex that included DNMT1 and DNMT3b from non-GC- to GC-rich areas of the genome upon exposure of cells to exogenous hydrogen peroxide (51). Investigations into the redox-sensitive regulation of the FOXO4 transcription factor yields further clues (15). In response to ROS, the p300 HAT domain binds to FOXO4, thereby inhibiting its activity. This is achieved through specific cysteine residues distributed throughout the FOXO4 protein, which act as redox sensors. Upon oxidation, cysteine–thiol disulfide bridges facilitate the formation of a covalent complex between p300 and FOXO4. Although, p300 in this context is not acting in an epigenetic regulatory capacity, the fact that this protein is capable of association through disulfide bridges could have further, as yet, unknown implications for its role in histone acetylation and for ROS-induced epigenomic modifications in general.
Nox as Biomarkers in Cancer
Specific and sensitive markers for diagnosis, early detection, and risk assessment are acutely needed in cancers where late diagnosis is still common. Current strategies for detecting lung cancer, for example, are either too costly and invasive for screening programs or not specific and sensitive enough. Analysis of remote media, namely, blood and sputum, holds great promise, although specificity and sensitivity of existing approaches still requires improvement (2, 39, 59). To ensure tissue-specific, early detection of the different forms of nonsmall cell lung cancer, the most prevalent lung cancer, the recovery and testing of sputum in high-risk groups, such as smokers, seems the most promising avenue. It is now apparent that a panel of highly predictive biomarkers might be required. Such a panel may consist of a combination of DNA methylation-based and oncogenic markers. Several candidate genes have been identified, but more characterization and validation are necessary. The potential of Nox for inclusion in a biomarker panel is still uncertain. Oncogenic, activating mutations in Nox have not yet been observed. However, many cancers and, in particular, oncogenic Ras-driven tumors display increased oxidative stress due to upregulation of Nox/Duox enzymes (19). It seems rather unlikely that an increase in Nox expression can serve as a specific tumor biomarker as many other diseases, including acute hypoxia or asthma, also lead to upregulation of Nox/Duox (26, 58, 68). In contrast, Duox1 or DuoxA1 promoter hypermethylation might fit the bill in light of the predominantly lung-specific expression of these genes and their early silencing in a high percentage of lung cancers, although large-scale studies with well-defined tumor specimens are missing (Fig. 6). Duox enzymes are unlikely to fall into the category of tumor suppressors as absence of Duox function seems not to lead to a spontaneous tumor development and their effects on growth or transformation in cell culture are subtle (23, 42, 60). However, recent studies indicate an important role of Duox1 in wound healing (42, 50, 71), a process that when deregulated is now regarded as potential basis for oncogenesis (13). Thus, further studies linking Duox silencing in the lung epithelium with tissue injury and tumor development are warranted.

Innovation
The contribution of Nox/Duox enzymes to redox signaling is well established, but their dual role as target and source of epigenetic remodeling is only beginning to emerge. As a target of modifications, Duox enzymes are frequently epigenetically silenced in lung cancer via promoter hypermethylation. On the other hand, oxidative stress can initiate chromatin remodeling and aberrant DNA methylation, and Nox enzymes constitute a key ROS generator in cancer cells. As some anticancer therapeutics use selective oxidative stress as a proapoptotic approach, future efforts in deciphering the spectrum of beneficial versus detrimental ROS will be of particular interest.
Footnotes
Acknowledgments
The authors acknowledge support of Science Foundation Ireland (SFI Stokes award and SFI-PI award to U.G.K.).